

Abstract
In theory, pharmacological inhibition of oncogenic signaling is an effective strategy to halt cellular proliferation, induce apoptosis, and eliminate cancer cells. In practice, drugs (e.g., PLX-4032) that inhibit oncogenes like B-RAFV600E provide relatively short-term success in patients, due to a combination of incomplete cellular responses and the development of resistance. To define the relationship between PLX-4032 induced responses and resistance, we interrogated the contributions of anti-apoptotic BCL-2 proteins in determining the fate of B-RAFV600E inhibited melanoma cells. While PLX-4032 eliminated B-RAFV600E signaling leading to marked cell cycle arrest, only a fraction of cells eventually underwent apoptosis. These data proposed two hypotheses regarding B-RAFV600E inhibition: (1) only a few cells generate a pro-apoptotic signal, or (2) all the cells generate a pro-apoptotic signal but the majority silences this pathway to ensure survival. Indeed, the latter hypothesis is supported by our observations as the addition of ABT-737, an inhibitor to anti-apoptotic BCL-2 proteins, revealed massive apoptosis following PLX-4032 exposure. B-RAFV600E inhibition alone sensitized cells to the mitochondrial pathway of apoptosis characterized by the rapid accumulation of BIM on the outer mitochondrial membrane, which could be functionally revealed by ABT-737 to promote apoptosis and loss of clonogenic survival. Furthermore, PLX-4032 resistant cells demonstrated collateral resistance to conventional chemotherapy; yet could be re-sensitized to PLX-4032 by BCL-2 family inhibition in vivo and conventional chemotherapies in vitro. Our data suggest that inhibiting anti-apoptotic BCL-2 proteins will enhance primary responses to PLX-4032, along with reducing the development of resistance to both targeted and conventional therapies.
Keywords: Apoptosis, BCL-2 family, B-RAFV600E, Chemotherapy, Melanoma, Resistance
Introduction
In numerous tumor types, such as malignant melanoma, colorectal cancer, and adenocarcinoma of the lung, oncogenic signaling is required to maintain high levels of cellular proliferation and evade cell death (1, 2). A classic example of a cancer-associated oncogene is the mutated form of B-RAF, where a substitution of valine to glutamate at amino acid residue 600 (B-RAFV600E) renders the kinase hyperactive leading to constitutive MAPK signaling, unregulated cell cycle, and coordinated silencing of the cell death machinery (2–5). In the recent years, small molecules (e.g., PLX-4032) have been developed and approved for the treatment of B-RAFV600E positive cancers, and while oncogenic signaling is effectively blocked in these tumors, the death of tumor cells does not reflect similar rates of success (6–8). Moreover, B-RAFV600E positive patients are exposed to high concentrations of PLX-4032 for several weeks, leading to the rapid development of PLX-4032 resistant tumors and the subsequent requirement for alternative chemotherapeutic strategies (9, 10).
It is hypothesized that targeted (e.g., PLX-4032) and conventional (e.g., dacarbazine) chemotherapeutic strategies eliminate tumor cells by inducing a form of cell death called apoptosis, which is governed by the BCL-2 family of proteins and mitochondria (11). Apoptosis proceeds when the BCL-2 family compromises the outer mitochondrial membrane (OMM) allowing for pro-apoptotic factors (e.g., cytochrome c) within mitochondria to gain access to the cytoplasm, which leads to caspase activation and the apoptotic phenotype (12). The BCL-2 family is composed of two types of proteins: anti-apoptotic and pro-apoptotic (13). Anti-apoptotic proteins (e.g., BCL-2, BCL-xL, MCL-1) preserve survival by binding and inhibiting the pro-apoptotic proteins. The pro-apoptotic members are divided into ‘effectors’ and the ‘BH3-only proteins’. The effector proteins BAK and BAX homo-oligomerize into proteolipid pores at mitochondria to release cytochrome c; however, this requires an activation step mediated by the ‘direct activator’ BH3-only proteins (14–16). BID and BIM are the major direct activators, and function via their BH3 domains to induce BAK/BAX activation and apoptosis. In most cases, the other BH3-only proteins (e.g., BAD & PUMA; referred to as ‘sensitizer/de-repressor’ BH3-only proteins) only bind to anti-apoptotic BCL-2 proteins and establish the apoptotic threshold by promoting BID and/or BIM activities (17). Dynamic interactions between the pro- and anti-apoptotic proteins govern cellular fate: agents that promote BID/BIM activity enhance apoptosis, while mechanisms that block BID/BIM activity lead to cellular survival and chemotherapeutic resistance (11).
The clinical relevance of the BCL-2 family is highlighted by the development of small molecule BH3 mimetics (e.g., ABT-199, ABT-263, ABT-737) that have entered multiple clinical trials (18–21). These mimetics function by lowering the cellular threshold for apoptosis, presumably by decreasing the availability of anti-apoptotic BCL-2 family members which promotes pro-apoptotic protein function at the OMM. At present, there is minimal information on the application of these small molecules in melanoma treatment (19).
In the current study, we were interested in defining the mechanistic intersections between PLX-4032 induced cell death signaling and resistance, and interrogated the contributions of the BCL-2 proteins in determining the fate of B-RAFV600E inhibited cells. We utilized melanoma as a model system to explore the biology of B-RAFV600E, as this is the common oncogene associated with the majority of malignant melanoma cases (4, 5). Furthermore, B-RAFV600E positive melanoma patient responses to PLX-4032 have been encouraging, but require additional insights to improve primary treatment efficacy and reduce the likelihood of chemoresistance (6, 22, 23).
Materials and Methods
Reagents
All cell culture and transfection reagents were from Invitrogen; and standard reagents were from Sigma or Fisher Scientific. Drugs were from: ABT-737 (Abbott Pharmaceuticals), ABT-263/PLX-4032/GSK-110212 (Selleck), zVAD-fmk (Calbiochem), Hoechst 33342 (Anaspec), and staurosporine/cisplatin/dacarbazine/vinblastine (Sigma). Antibodies (clone): anti-actin (C4), anti-A1 (FL-175), anti-BCL-2 (100), anti-CD31 (BD Pharmingen #550274) anti-MCL-1 (Rockland), anti-BAD (C7), anti-BIM (22–40), anti-PUMA (CT; Sigma), anti-SMAC (H177), anti-BID (C20), anti-cytochrome c (7H8.2C12), anti-BAK (G23), anti-BAX (6A7 for IP; N20 for western blot), anti-BCL-xL (H5 for IP; S18 for western blot), anti-GAPDH (9B3), anti-HSP60 (B-9), anti-p44/p42 MAPK ERK1/2 (137F5), and anti-phospho p44/42 MAPK ERK1/2 (197G2). Caspase-8 cleaved mouse BID (C8-BID) was from R&D Systems. Full-length BAX was made as described (24). Human BCL-xLΔC, MCL-1ΔC, and PUMAβ were made as described (17). The human BIM BH3 domain peptide (> 98% purity, Abgent) was resuspended in anhydrous DMSO in a N2 environment, stored at −80°C, and thawed only once. All lipids for the LUVs studies were purchased from Avanti Polar Lipids. Statistical significance was evaluated by two tailed Student t test for p < 0.05.
Cell culture, transfection, apoptosis, and clonogenic survival assays
A375, MALME-3M, MeWo, MDA-MB-435, SK-MEL-28, WM266-4 lines were cultured in DMEM containing 10% FBS, 2 mM L-glutamine, and antibiotics. Cells were transfected (85,000 cells per well/12 well plate, cultured overnight before transfection) using Lipofectamine2000 according to the manufacturer’s instructions for 6 hours under serum-free conditions. Green fluorescent protein (pUS9-GFP, 100 ng) was co-transfected as an efficiency marker (30–50% of cells were usually transfected). For cell death studies, cells seeded 24 hours, treated as described, floating and attached cells were harvested, stained with AnnexinV-FITC in binding buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2), and analyzed by flow cytometry as indicated (25). For clonogenic survival studies, cells were treated with indicated drugs for 24 hours before changing the media. Colonies were stained with methylene blue 10 days after treatment and imaged. Colonies were then de-stained (20% methanol in 5% acetic acid), and the supernatant was measured for absorption at 520 nm for quantification of the wells. To generate PLX-4032 resistant lines, A375 and SK-MEL-28 were cultured in 1 µM and 5 µM PLX-4032, respectively, until proliferation rates returned to parental line’s rate, which was approximately 6 – 8 weeks. The PLX-4032 resistant lines were maintained in the above respective concentrations. For bim knockdown, HEK293T were transfected with pLKO.1 (Sigma-Aldrich; clones TRC1051 and TRC1054) using Lipofectamine2000; 24 and 48 hour lentiviral supernatants were 0.45 µm filtered, added to cells of interest, and Puromycin selection (0.5 µg/ml) was performed.
Results
Our experiments began by validating that the B-RAFV600E cellular model systems responded to PLX-4032 by decreasing the phosphorylation of ERK1/2 (p44/42). To address this, A375 (B-RAFV600E human malignant melanoma) and MeWo (B-RAFWT human malignant melanoma) were treated with PLX-4032 for 0 – 24 hours, and total cell lysates were analyzed by western blot. Indeed, PLX-4032 decreased the majority of phosphorylated ERK in the A375 cells, but MeWo remained unchanged (Fig. 1A). The cellular response to the decrease in ERK signaling reflected a marked inhibition of cellular proliferation as approximately 96% of A375 cells treated with PLX-4032 arrested in the G1 phase of cell cycle after 48 hours (Fig. 1B); while MeWo demonstrated no change in proliferation comparing DMSO (24.3% in S phase) to PLX-4032 (25.4% in S phase) treatments (Fig. 1C). Interestingly, when PLX-4032 exposure was extended to 72 hours, the treatment promoted a small percentage of cells to undergo cell death, as measured by Annexin V staining (Fig. 1D). The cell death phenotype was prevented by pre-treatment with zVAD-fmk, a pan-caspase inhibitor, which suggested the mechanism of cell death was apoptosis (Fig. 1E). Importantly, MeWo demonstrated no apoptotic response to PLX-4032 treatment (Fig. 1F). To ensure the effects of PLX-4032 were not specific to the A375 cell line, we also evaluated SK-MEL-28, another B-RAFV600E human malignant melanoma line, and while ERK phosphorylation and cell cycle were markedly inhibited, the cell death responses to PLX-4032 were minimal (Figs. S1A–C).
Figure 1.
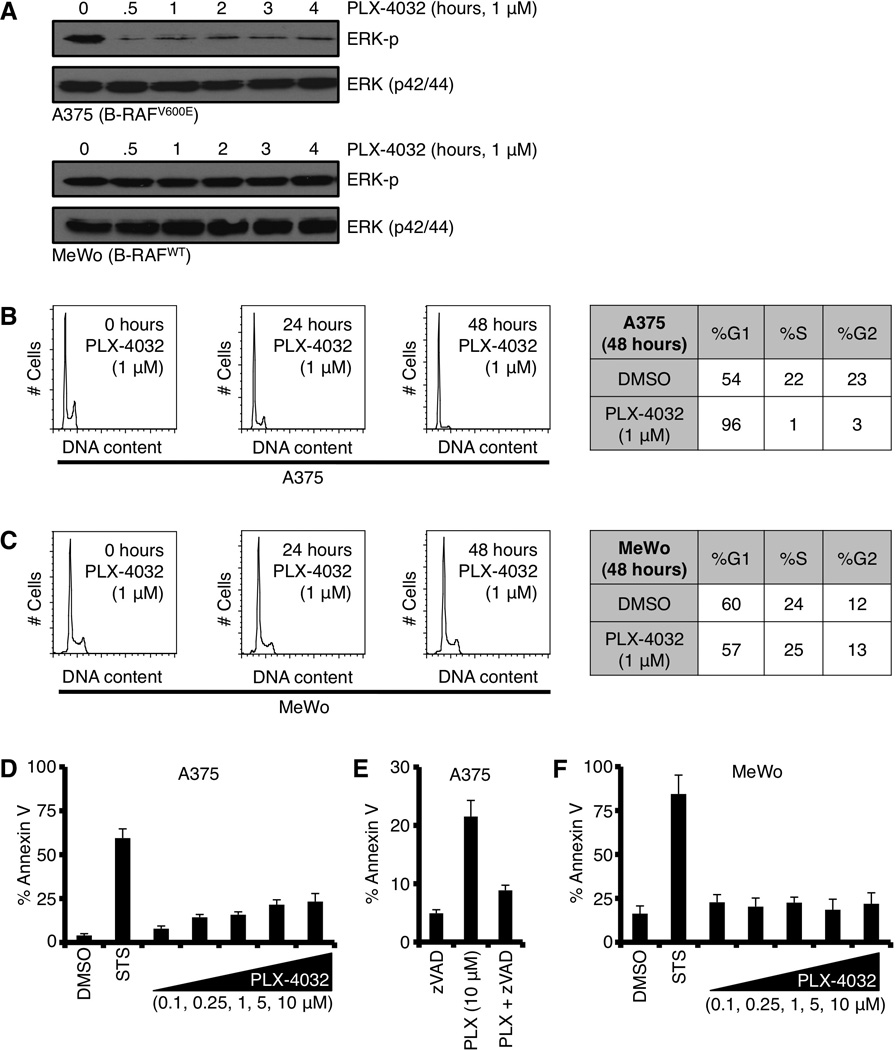
The majority of B-RAFV600E positive cells respond to PLX-4032 treatment by reducing phosphorylated ERK and G1 cell cycle arrest, but only a minority of cells undergoes apoptosis. (A) A375 (B-RAFV600E) and MeWo (B-RAFWT) were treated with PLX-4032 (1 µM) for indicated time points before western blot for phosphorylated ERK (ERK-p) and total ERK1/2 (p42/44). (B–C) A375 and MeWo were treated with PLX-4032 (1 µM) for indicated time points and analyzed for cell cycle distribution. Representative percentages are shown in charts. (D) A375 was treated with PLX-4032 (0.1, 0.25, 1, 5, 10 µM) for 72 hours before AnnexinV staining and flow cytometry. Staurosporine (50 nM, STS) is shown as a positive control. (E) A375 cells were pre-treated with zVAD-fmk (50 µM) for 1 hour before treatment with PLX-4032 (10 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. (F) MeWo was treated as in D. Staurosporine (50 nM, STS) is shown as a positive control. All data are representative of at least triplicate experiments, and reported as ± S.D., as required.
The results in figure 1D proposed two hypotheses regarding B-RAFV600E inhibition and cell death signaling: (1) only a few cells generate a pro-apoptotic signal and die following PLX-4032 treatment, or (2) all the cells generate a pro-apoptotic signal but the majority fail to respond ensuring survival. Pro-apoptotic signaling biochemically arises as the increased expression and/or activation of specific BH3-only proteins, and the major cellular mechanism to inhibit the BH3-only signal is by direct sequestration on the anti-apoptotic BCL-2 repertoire (e.g., BID directly inhibited by BCL-2) (13). To determine the contribution of the anti-apoptotic BCL-2 repertoire in the regulation of PLX-4032 induced pro-apoptotic signaling, we examined the effect of ABT-737, a small molecule inhibitor to several anti-apoptotic BCL-2 proteins (i.e., BCL-2, BCL-xL, and BCL-w; but not MCL-1) (21). ABT-737 is suggested to lower the threshold leading to apoptosis by preventing anti-apoptotic BCL-2 proteins from binding BH3-only proteins and/or by releasing BH3-only proteins from anti-apoptotic BCL-2 members to enhance BAK/BAX activation (21). Returning to our hypotheses, we next determined if ABT-737 pre-treatment revealed a pro-apoptotic signal in the majority of B-RAFV600E inhibited cells.
In mouse models and the clinic, ABT-737 as a single therapy is well tolerated and does not cause any marked apoptosis phenotype (21, 26). Indeed, the same is the case in A375 and SK-MEL-28 as ABT-737 treatment for up to 48 hours resulted in no apoptosis, which is likely explained by MCL-1 expression (Figs. 2A, 2B, S1C; n.b., ABT-737 does not inhibit MCL-1) (11). To directly examine our proposed above hypotheses, we co-treated cells with PLX-4032 and ABT-737 and measured apoptosis after 48 hours. Pretreatment with ABT-737 revealed dose-dependent apoptosis that was both rapid and robust (Figs. 2C, S1C), in several experiments nearly 100% of the population responded by inducing apoptosis. Similar to PLX-4032 alone (Fig. 1E), the marked cell death revealed by ABT-737 was prevented by zVAD-fmk pretreatment (Fig. 2D); and characterized by nuclear fragmentation, a hallmark feature of apoptosis (Figs. 2E–F).
Figure 2.
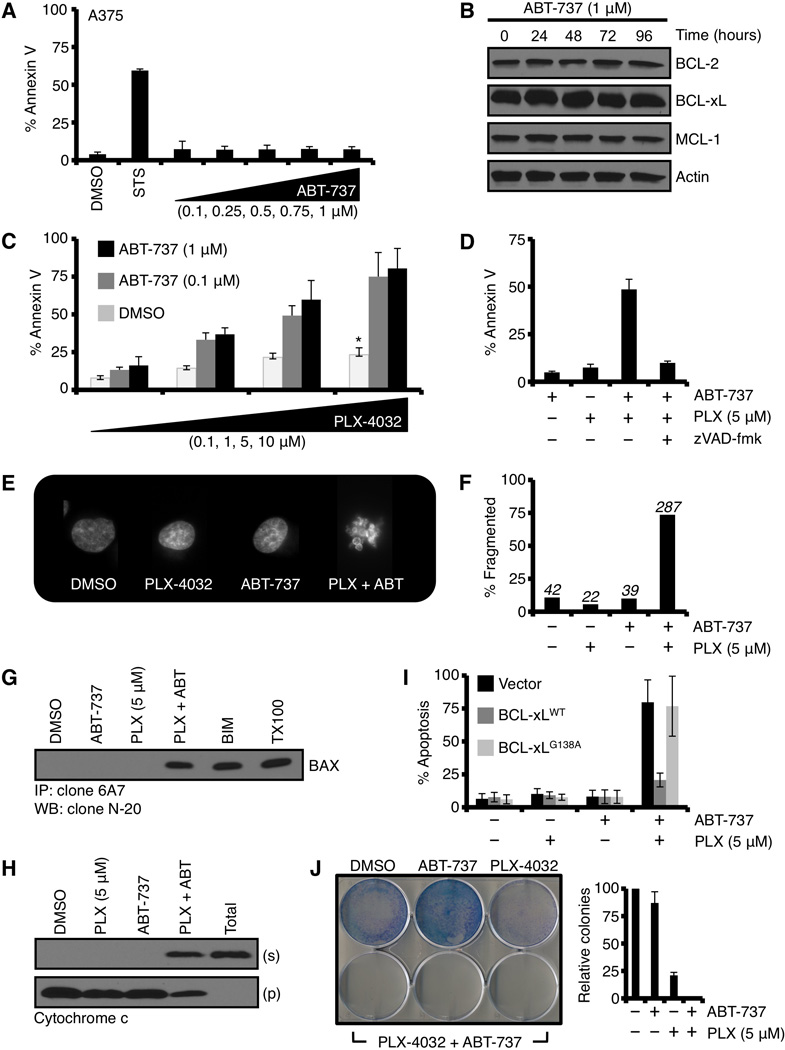
Collateral inhibition of B-RAFV600E signaling and anti-apoptotic BCL-2 protein function reveals marked commitment to the mitochondrial pathway of apoptosis and loss of clonogenic survival. (A) A375 was treated with ABT-737 (0.1, 0.25, 0.5, 0.75, 1 µM) for 48 hours before AnnexinV staining and flow cytometry. Staurosporine (50 nM, STS) is shown as a positive control. (B) A375 was treated with ABT-737 (1 µM) for indicated time points before western blotting for BCL-2, BCL-xL, and MCL-1. Actin is shown as a loading control. (C) A375 cells were pre-treated with ABT-737 (0.1, 1.0 µM) for 1 hour before treatment with PLX-4032 (0.1, 1, 5, 10 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. *p value = < 0.005. (D) A375 cells were pre-treated with ABT-737 (1.0 µM) and zVAD-fmk (50 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. (E) A375 cells were pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 48 hours. The cells were stained with Hoechst 33342 (20 µM) and imaged by fluorescent microscopy. (F) The data obtained in E were quantified. ~ 400 cells per condition were evaluated. (G) A375 cells were pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 48 hours. The cells were lysed in CHAPS buffer, and the lysates were subjected to anti-BAX immunoprecipitation using clone 6A7 and analyzed by western blot for BAX. BIM-transfected cells and TritonX-100 (0.25%, TX100) are positive controls for 6A7 positive BAX. (H) A375 cells were pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 24 hours. The cells were then subjected to fractionation for cytosol and heavy membranes (i.e., mitochondria), and these fractions were subjected to western blot for cytochrome c. Supernatants (s) and pellets (p) are shown. “Total” is a detergent (0.25% TX100) solubilized sample. (I) A375 were transiently transfected with empty vector, pcDNA3.1-BCL-xLWT, or pcDNA3.1-BCL-xLG138A. The next day, cells were pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. (J) A375 were pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 48 hours, the media was replaced, and the cells were cultured for an additional 10 days. The resulting colonies were stained (left panel) and quantified (right panel). The PLX-4032 + ABT-737 combination is shown in triplicate. All data are representative of at least triplicate experiments, and reported as ± S.D., as required.
In order to better understand the relationship between cellular responses to B-RAFV600E inhibition and the anti-apoptotic BCL-2 proteins, we next implicated the mitochondrial pathway of apoptosis as a target downstream of PLX-4032 treatment. Firstly, the pro-apoptotic protein BAX is often engaged following cellular stress leading to apoptosis, therefore we examined if BAX became activated by the use of an active conformation specific antibody (clone 6A7) following PLX-4032 and ABT-737 co-treatment (27). A375 cells were treated with PLX-4032 and ABT-737 for 24 hours, and whole cell lysates were subjected to 6A7 immunoprecipitation and western blot. Indeed, the combination led to a marked increase in 6A7-recognized BAX, whereas neither treatment alone resulted in significant recognition (Fig 2G). Secondly, as PLX-4032 and ABT-737 co-treatment results in BAX activation, we examined if the anti-apoptotic protein BCL-xL could block the death response. Indeed, transient transfection of wild type BCL-xL, but not a BAX-binding mutant (i.e., BCL-xL G138A), prevented the majority of cell death induced by PLX-4032 and ABT-737 co-treatment (Fig. 2I). Thirdly, BCL-xL inhibited BAX activation suggesting that our co-treatment results in cytochrome c release from mitochondria to the cytosol to initiate apoptosis. To directly examine this, we treated cells with PLX-4032 ± ABT-737 for 24 hours, fractionated the cells into pellet (“p”, contains mitochondria) and supernatant (“s”, cytosolic proteins), and subjected these fractions to western blot analysis for cytochrome c (Fig. 2H). As shown in figure 2H, the combination of PLX-4032 and ABT-737 resulted in marked cytochrome c redistribution from the pellet to the supernatant, whereas neither drug alone influenced cytochrome c localization. Finally, the combination of drugs led to a marked decrease in clonogenic survival in A375 and SK-MEL-28 (Figs. 2J, S1D). To broaden the relevance of these studies, we tested additional melanoma models: MALME-3E, MDA-MB-435, and WM266-4; and ABT-737 revealed dose-dependent apoptosis in all B-RAFV600E positive lines (Figs. S1E–G).
The above data suggest that the anti-apoptotic BCL-2 family members are responsible for silencing the pro-apoptotic signal induced by the inhibition of oncogenic B-RAFV600E. We hypothesized that the anti-apoptotic proteins were directly inhibiting a PLX-4032 induced pro-apoptotic BCL-2 family member, and therefore screened through all the pro-apoptotic BCL-2 proteins by preparing whole cell lysates and western blot studies. The majority of BCL-2 family proteins remained unchanged, except for BIM, which demonstrated a marked dose-dependent accumulation of two isoforms, BIM-EL and BIM-L following PLX-4032 treatment (Fig. 3A). BH3-only proteins bind with high affinity to anti-apoptotic BCL-2 proteins localized to the OMM, so we prepared heavy membrane fractions (i.e., commonly referred to as mitochondria) from PLX-4032 treated cells, and were able to observe marked PLX-4032 dependent accumulation of all three BIM isoforms, including the potent pro-apoptotic form, BIM-S (Figs. 3B, S1H). No other mitochondrial-associated BCL-2 family members demonstrated altered expression. We noted that the level of each BIM isoform was relatively modest in relationship to the anti-apoptotic BCL-2 proteins, and all BIM isoforms were exclusively localized to the heavy membrane fractions (i.e., no unbound cytosolic BIM protein was detected), suggesting that the availability of anti-apoptotic BCL-2 proteins superseded the levels of expressed BIM (Fig. 3C, S1H), and this may be the mechanism explaining minimal pro-apoptotic responses following PLX-4032 treatment (Figs 1D, 2C, S1C). As control, ABT-737 did not regulate the expression of BIM in any cell line (Figs. 3B, S1H), and MeWo did not demonstrate PLX-4032 regulated BIM expression (Fig. 3D). To ensure BIM was critical to PLX-4032 induced apoptosis, we stably silenced bim expression in A375 and SK-MEL-28 (Fig. 3E), and examined cell death following PLX-4032 and ABT-737 co-treatment (Fig. 3F). Indeed, silencing bim eliminated the majority of cell death induced by the PLX-4032 and ABT-737 combination treatment.
Figure 3.
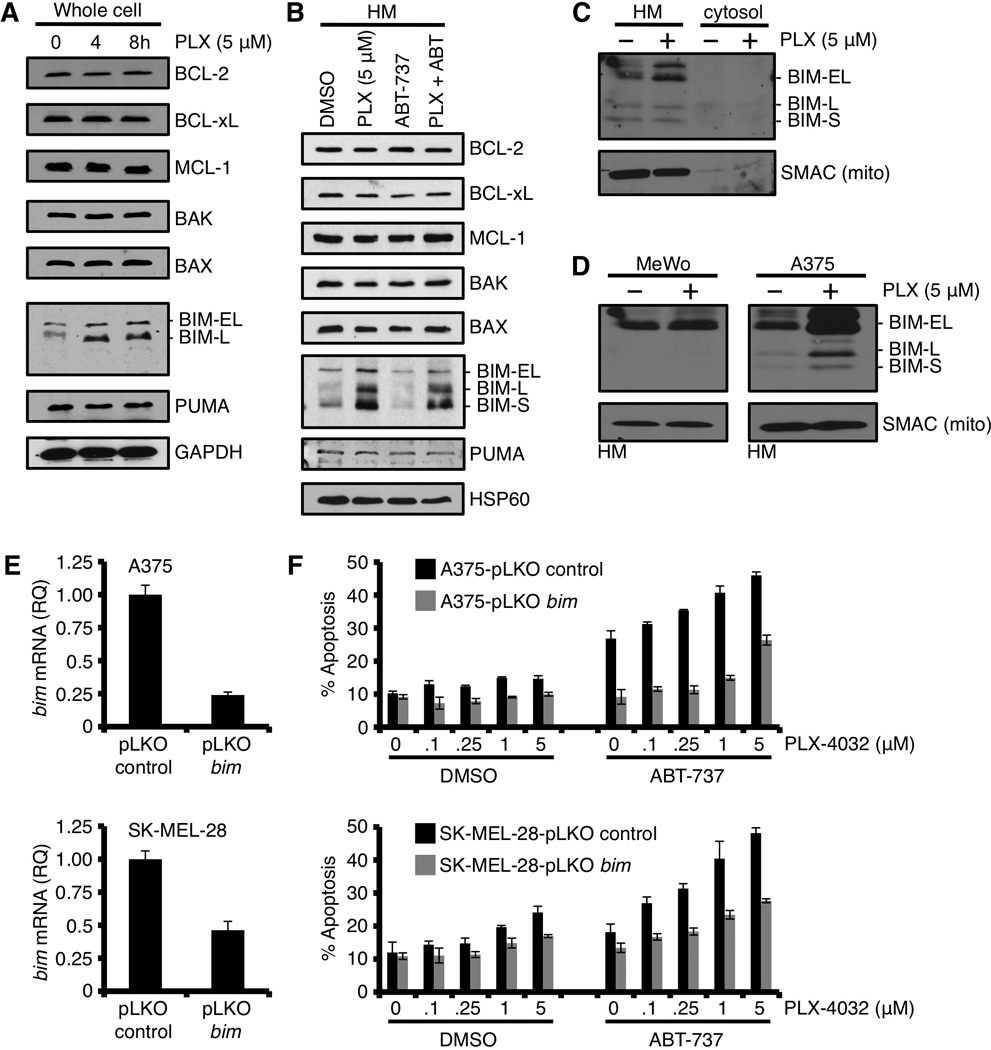
PLX-4032 induced inhibition of B-RAFV600E promotes marked expression and rapid mitochondrial accumulation of BIM; however, this pro-apoptotic signal remains silenced. (A) A375 was treated with PLX-4032 (5 µM) for indicated time points before western blot for the indicated BCL-2 family members. GAPDH is shown as a loading control. (B) A375 cells were pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (5 µM) for 24 hours. The heavy membrane fractions (“HM”, i.e., mitochondria) were then analyzed for the indicated BCL-2 family members. HSP60 is shown as a loading control. (C) A375 cells were treated with PLX-4032 (5 µM) for 24 hours, and fractionated into cytosol and heavy membranes. These fractions were subjected to western blot for BIM. SMAC and actin are shown as mitochondrial and cytosolic fractionation controls. (D) A375 and MeWo were treated with PLX-4032 (5 µM) for 24 hours, the HM fractions were isolated and subjected to western blot analysis for BIM. SMAC is shown as a mitochondrial loading control. (E) A375 and SK-MEL-28 cells stably expressing bim shRNA (or the control vector, pLKO) were quantified for bim knock-down by qPCR. Expression was normalized with β-actin and gapdh. (F) The cells in E were treated with PLX-4032 (0, 0.1, 0.25, 1, 5 µM) ± ABT-737 (0.5 µM) for 48 hours before AnnexinV staining and flow cytometry. All data are representative of at least triplicate experiments, and reported as ± S.D., as required.
We next hypothesized that ABT-737 could de-repress BIM from the inhibitory interaction with anti-apoptotic BCL-2 proteins at the OMM to promote cytochrome c release (Fig. 2H) and apoptosis (Figs. 2C, S1C). To biochemically examine this hypothesis, we prepared heavy membrane fractions from PLX-4032 treated cells and tested for ABT-737 induced cytochrome c release in vitro. As shown in figure 4A, heavy membranes from PLX-4032 treated A375 cells demonstrated dose-dependent cytochrome c release in response to ABT-737 addition; in contrast, mitochondria from A375 cells treated with DMSO elicited no response to ABT-737 (Fig. 4A). C8-BID treatment is a positive control for BH3-only protein induced cytochrome c release to ensure all mitochondrial isolations respond in vitro (Fig. 4A); and mitochondria isolated from PLX-4032 treated MeWo are a negative control and demonstrated no response to ABT-737 (Fig. 4B). The above observations demonstrate that: (1) the PLX-4032 mediated inhibition of oncogenic B-RAFV600E induces a pro-apoptotic signal that accumulates on mitochondria; (2) the pro-apoptotic signal remains silenced within B-RAFV600E cells via an interaction with anti-apoptotic proteins on the OMM; and (3) ABT-737 can reveal the PLX-4032 induced pro-apoptotic signal to engage cytochrome c release and cell death.
Figure 4.
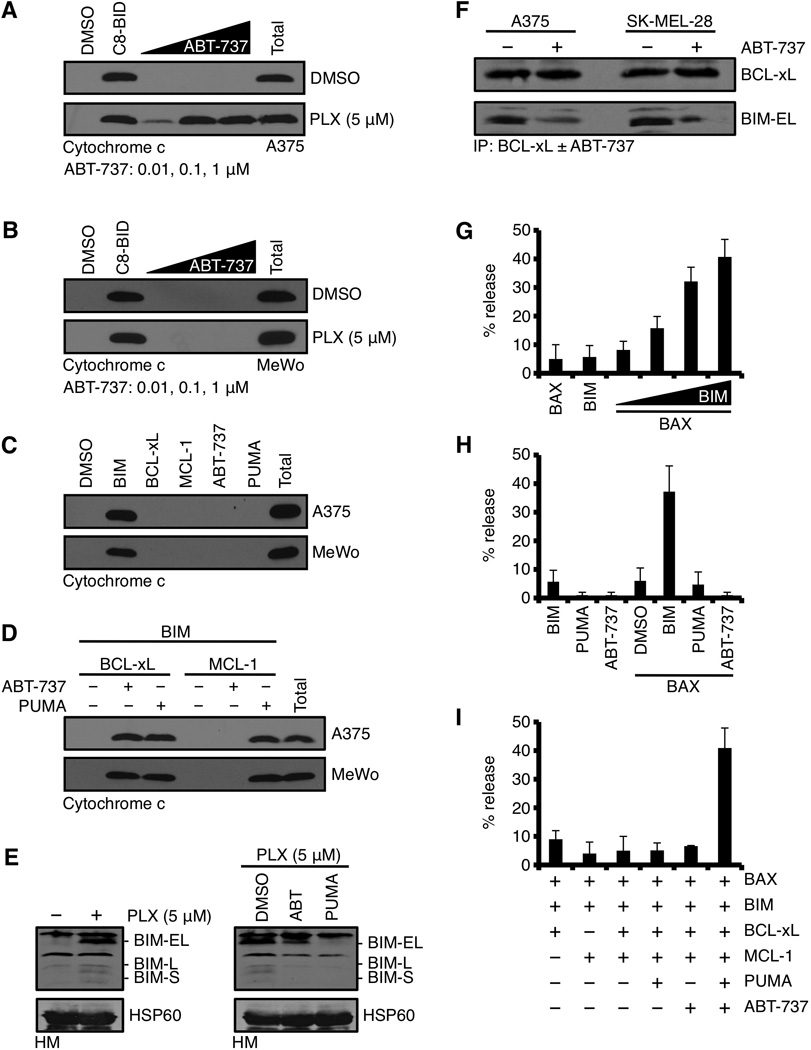
Despite the presence of numerous pro-apoptotic BH3-only proteins, ABT-737 is required to reveal PLX-4032 induced BIM-dependent mitochondrial outer membrane permeabilization. (A) A375 were treated with DMSO or PLX-4032 (5 µM) for 24 hours, the HM fractions isolated, treated with ABT-737 (0.01, 0.1, 1 µM) for 30 minutes at 37°C, centrifuged, and the supernatants were subjected to western blot for cytochrome c. Caspase-8 cleaved BID (C8-BID, 10 nM) is a positive control for physiological cytochrome c release; “Total” is a detergent solubilized sample and is the maximal cytochrome c per sample. (B) The same assay as in A, but with MeWo. (C) HM fractions from untreated A375 or MeWo were incubated with the BIM BH3 domain peptide (5 µM), BCL-xL (50 nM), MCL-1 (50 nM), ABT-737 (1 µM), or PUMA (1 µM) for 30 minutes at 37°C, centrifuged, and the supernatants were subjected to western blot for cytochrome c. (D) HM fractions from untreated A375 or MeWo were incubated with the indicated combinations of BCL-2 family reagents and analyzed as in C. (E) A375 were treated with PLX-4032 (5 µM) for 24 hours, the HM fractions were isolated and subjected to western blot analysis for BIM (left panel). Isolated mitochondria were treated with DMSO, ABT-737 (100 nM), PUMA (100 nM) for 1 hour at 37°C, centrifuged, and the pellets were subjected to western blot for BIM (right panel). HSP60 is shown as a mitochondrial loading control. (F) A375 and SK-MEL-28 were treated with PLX-4032 (5 µM) for 24 hours, the HM fractions were isolated, lysed, and subjected to immunoprecipitation with an anti-BCL-xL antibody (1 µg) ± ABT-737 (100 nM). BCL-xL and BIM were detected by western blot. (G) LUVs were treated with combinations of BAX (100 nM) and the BIM BH3 (5 µM alone, or 0.1, 0.5, 1, 5 µM) for 30 minutes at 37°C. 100% release was determined by a detergent treated sample. (H) LUVs were treated with combinations of BIM BH3 (5 µM), PUMA (1 µM), ABT-737 (1 µM), and BAX (100 nM) for 30 minutes at 37°C. (I) LUVs were treated with combinations of reagents described in E, including BCL-xL (50 nM) and MCL-1 (50 nM) for 30 minutes at 37°C. All data are representative of at least triplicate experiments, and reported as ± S.D., as required.
The data in figures 3 and 4A suggest that BIM is involved in PLX-4032/ABT-737 induced cytochrome c release; therefore, we directly tested this hypothesis by examining the cytochrome c releasing activity of BIM on purified mitochondria from A375 and MeWo. Mitochondria were isolated, treated with BIM, fractionated into pellet and supernatant, and the fractions were analyzed by western blot for cytochrome c. Indeed, treatment with the direct activator BIM was sufficient to promote rapid and complete cytochrome c release, while treatment with a sensitizer/de-repressor BH3-only protein (e.g., PUMA) or ABT-737 resulted in no cytochrome c release (Fig. 4C). We interpret these results to suggest that independent of B-RAF status, our model systems sustain the ability to induce cytochrome c release and apoptosis, and that pro-apoptotic signaling upstream of BIM and mitochondria dictates cellular fate.
Our previous results suggested that BCL-xL could block PLX-4032 induced pro-apoptotic signaling (Fig. 2I), therefore we examined if BIM-mediated cytochrome c release in vitro was also blocked by BCL-xL. The addition of BCL-xL completely inhibited BIM-induced cytochrome c release from both A375 and MeWo mitochondria; and furthermore, this inhibition was reversed by the addition of ABT-737 (Fig. 4D), which parallels our cellular results (Figs. 2C, S1C). As control for our protein and mitochondrial purifications, we also provide evidence that MCL-1 mediated inhibition of BIM-induced cytochrome c release is only reversed by the addition of PUMA (n.b., ABT-737 does not bind MCL-1), whereas PUMA or ABT-737 could de-repress the inhibitory activity of BCL-xL, (i.e., PUMA and ABT-737 bind BCL-xL) (Fig. 4D) (11, 13, 17, 21).
In our experiments, we show that ABT-737 can promote PLX-4032 mediated apoptosis, presumably by releasing pro-apoptotic BIM from anti-apoptotic BCL-2 proteins localized to the OMM. We formally examined this hypothesis by determining if BIM could be displaced from the OMM by the addition of ABT-737 in vitro. Mitochondria from PLX-4032 treated A375 cells were isolated, incubated with ABT-737 for 1 hour prior to centrifugation, and subjected to western blot for BIM. PLX-4032 treatment promoted the accumulation of multiple BIM isoforms on the OMM (Fig. 4E, left panel); and the addition of ABT-737 to these mitochondria resulted in a decreased presence of BIM isoforms (Fig. 4E, right panel). For control, we also examined the effect of PUMA on the binding of BIM to these mitochondria. PUMA binds with high affinity to all anti-apoptotic BCL-2 proteins, and this should eliminate the majority of BIM associated with the OMM. Indeed, PUMA addition reduced BIM levels to mitochondria isolated from untreated A375 cells (Fig. 4E). To ensure the release of BIM from the OMM was mediated by anti-apoptotic BCL-2 proteins, we immunoprecipitated BCL-xL from PLX-4032 treated mitochondria ± ABT-737. As shown in figure 4F, BCL-xL·BIM complexes were observed in PLX-4032 treated cells, but the addition of ABT-737 markedly reduced the level of BIM associated with BCL-xL. All together, these data suggest that ABT-737 can efficiently displace BIM from anti-apoptotic BCL-2 proteins on the OMM to promote apoptosis (Figs. 4E–F).
Figures 3A and 3B show that multiple anti-apoptotic (i.e., BCL-2/BCL-xL and MCL-1) and pro-apoptotic (i.e., BAK/BAX, BIM, and PUMA) BCL-2 family members likely regulate cellular sensitivity to cytochrome c release following PLX-4032 treatment. Furthermore, the requirement for ABT-737 within this complex BCL-2 family network reveals a unique dependence on BCL-2/BCL-xL to maintain survival following the inhibition of B-RAFV600E. To examine the exquisite dependency on ABT-737 to promote cytochrome c release, we modeled the BCL-2 family network observed in A375 cells using biochemically defined large unilamellar vesicles (LUVs) and recombinant BCL-2 family members (14, 28). The LUVs are composed of the major lipid species that comprise mitochondrial membranes, and treatment of mitochondrial LUVs with BAX or BIM alone did not cause LUV permeabilization; but when combined, resulted in BIM mediated dose-dependent BAX activation and LUV permeabilization (Fig. 4G). As suggested in figures 3A–D and 4E–F, BIM is the pro-apoptotic protein directly responsible for cytochrome c release, and similar results were obtained using the LUV system, as PUMA did not cause permeabilization alone or in the presence of BAX (Figure 4H). Finally, we examined the integration of BAX, BCL-xL, BIM, MCL-1, and PUMA in the LUV system, which revealed that this unique combination of BCL-2 family proteins did not promote LUV permeabilization unless ABT-737 was added, suggesting that ABT-737 could specifically trigger BIM-mediated BAX activation in this setting (Fig. 4I).
From these data, we propose: (1) the pro-apoptotic signal mediated by the inhibition of oncogenic B-RAFV600E is the mitochondrial accumulation of multiple BIM isoforms; (2) the majority of pro-apoptotic BIM is silenced by the anti-apoptotic BCL-2 family leading to sustained survival; (3) ABT-737 reveals the pro-apoptotic activity of BIM to induce cytochrome c release and marked apoptosis; and (4) orally available derivates of ABT-737 (i.e., ABT-263) may be a useful combination strategy with PLX-4032 to boost cell death responses early in chemotherapeutic regimens (20).
Patients treated with targeted therapies like PLX-4032 often experience prolonged survival, yet succumb to the rapid development of drug-resistant populations leading to few clinical options (10). To evaluate the utility of ABT-737 in treating PLX-4032 resistant populations, we created several PLX-4032 resistant B-RAFV600E cell models using parental A375 and SK-MEL-28 cells, and refer to these lines as ‘A375R’ and ‘SK-MEL-28R’, respectively. To characterize the lines, we removed PLX-4032 for 24 hours prior to re-treating and subsequent analyses. A375R and SK-MEL-28R cells were treated with PLX-4032 for 24 hours and we analyzed responses to ERK phosphorylation status and cell cycle distribution. PLX-4032 treatment did not markedly change ERK signaling in A375R (Fig. 5A) or SK-MEL-28R (Fig. S3A), and there were minimal changes to cell cycle distribution compared to the parental lines (Figs. 5B, S3B), both of which suggest bone fide PLX-4032 resistance. We next compared the apoptotic responses in the parental and resistant lines following PLX-4032 exposure. As shown in figures 5C and S3C, A375R and SK-MEL-28R demonstrated markedly less cell death compared to the parental lines. Furthermore, A375R also displayed parallel resistance to common inducers of apoptosis, e.g., staurosporine and cycloheximide, but not VP16. This suggested to us that the resistant lines selectively silenced pro-apoptotic signaling following certain stresses, but did not have a mutation in the core apoptotic signaling cascade (e.g., APAF-1 or caspases-9/-3) or marked changes within the BCL-2 family (Figs. 5D, S2A–B).
Figure 5.
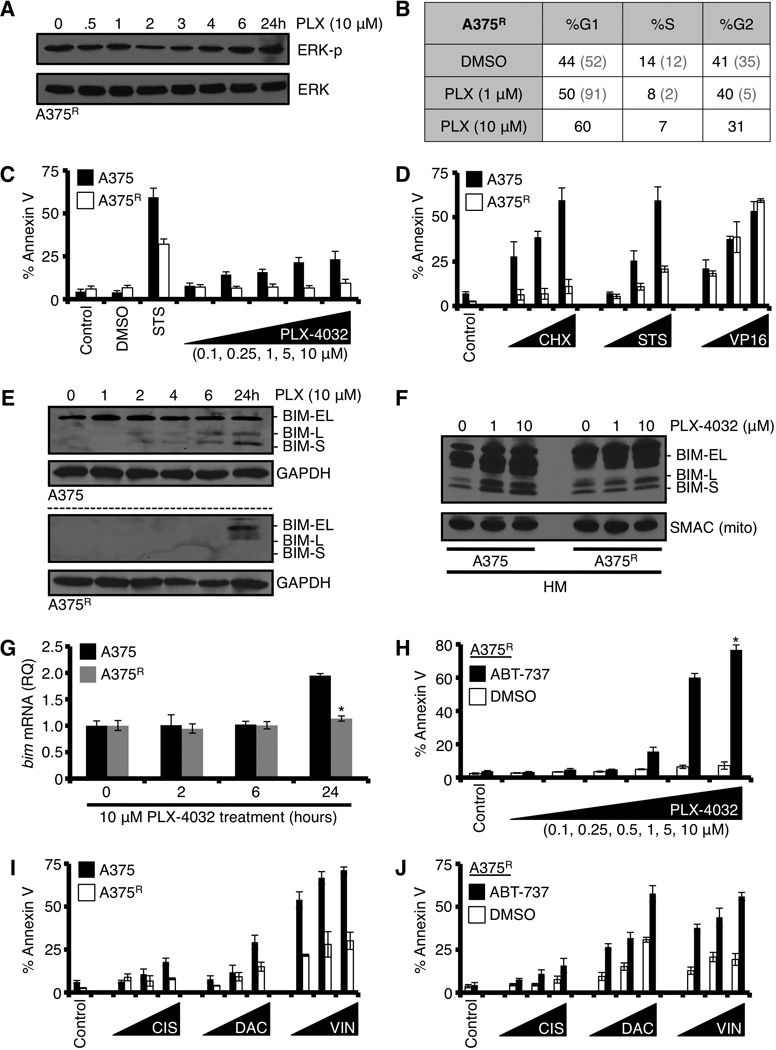
Inhibition of anti-apoptotic BCL-2 family members with ABT-737 sensitizes PLX-4032 resistant A375 cells to undergo apoptosis following targeted or conventional chemotherapeutics. (A) PLX-4032 resistant A375 ‘A375R’ cells were treated with PLX-4032 (10 µM) for indicated time points before western blot for phosphorylated ERK (ERK-p) and total ERK1/2 (p42/44). (B) A375R was treated with PLX-4032 (1, 10 µM) for 24 hours and analyzed for cell cycle distribution. Representative percentages are shown in charts. The grey numbers within parentheses are the parental A375 responses, for comparison. The parental line was treated with only the lower dose of PLX-4032 to avoid apoptosis. (C) A375 and A375R were treated with PLX-4032 (0.1, 0.25, 1, 5, 10 µM) for 72 hours before AnnexinV staining and flow cytometry. Staurosporine (50 nM, STS) is shown as a positive control. *p value = < 0.05. (D) A375 and A375R were treated with cycloheximide (CHX; 10, 25, 100 µg/ml), STS (1, 10, 50 nM), or VP16 (5, 10, 50 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. (E) A375 and A375R were treated with 10 µM PLX-4032 for indicated times before western blot for BIM. GAPDH is shown as a loading control. (F) A375 and A375R were treated with PLX-4032 (1, 10 µM) for 24 hours, the HM fractions were isolated and subjected to western blot analysis for BIM. SMAC is shown as a mitochondrial loading control. (G) A375 and A375R were treated with PLX-4032 (10 µM) for indicated times, and bim mRNA was measured by qPCR. Data are normalized to gapdh and 18S. *p value = < 0.005. (H) A375R was pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with PLX-4032 (0.1, 0.25, 0.5, 1, 5, 10 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. (I) A375 and A375R were treated with cisplatin (CIS; 10, 50, 100 µM), dacarbazine (DAC; 100, 500, 1000 µM), or vinblastine (VIN; 1, 10, 50 nM) for 48 hours, and analyzed by AnnexinV and flow cytometry. (J) A375R was pre-treated with ABT-737 (1.0 µM) for 1 hour before treatment with cisplatin (CIS; 10, 50, 100 µM), dacarbazine (DAC; 100, 500, 1000 µM), or vinblastine (VIN; 1, 10, 50 nM) for 48 hours, and analyzed by AnnexinV and flow cytometry. All data are representative of at least triplicate experiments, and reported as ± S.D., as required.
To investigate a mechanism of selective apoptosis-insensitivity in A375R and SK-MEL-28R, we next compared BIM expression and accumulation on mitochondria in the parental and PLX-4032 resistant lines following PLX-4032 exposure. PLX-4032 treatment induced marked BIM expression (Fig. 5E) and mitochondrial accumulation (Fig. 5F) in the parental A375 line, but these responses were severely blunted in A375R, which also paralleled the lack of apoptosis (Figs. 5C). Likewise, A375R failed to induce bim mRNA compared to the parental A375 line after PLX-4032 treatment (Fig. 5G). Despite not being able to detect significant BIM mRNA or protein accumulation in A375R, ABT-737 pre-treatment was able to reveal a pro-apoptotic signal following PLX-4032 treatment, suggesting that either minimal BIM induction or an unimplicated BH3-only protein was functional to promote apoptosis (Figs 5H). Importantly, the above observations were reproducible in SK-MEL-28R (Fig S3D–E).
In addition to B-RAFV600E specific therapies, drugs inhibiting other components of the MAPK signaling pathway (i.e., MEK) and conventional chemotherapeutic agents are often used to treat metastatic melanoma (19, 29). For this reason, and the observed apoptotic resistance presented in figures 5C–G and S3C–E, we were interested in investigating if the A375R and SK-MEL-29R lines maintained sensitivity to another targeted therapy (i.e., GSK-110212, a MEK inhibitor) and/or conventional chemotherapeutic agents used to treat metastatic disease (e.g., cisplatin, dacarbazine, and vinblastine). A375R and SK-MEL-28R lines displayed marked resistance to GSK-110212 induced apoptosis, compared to the parental lines (Figs. S4A–C). Furthermore, A375R were also resistant to the cisplatin, dacarbazine, and vinblastine-induced apoptosis (Fig. 5I); but this phenotype did not extend to the SK-MEL-28R line (Figs. S3F–G). To determine the role of the anti-apoptotic BCL-2 repertoire in mediating the marked resistance in A375R, we pre-treated the resistant line with ABT-737, and then exposed the cells to GSK-1101212, cisplatin, dacarbazine, or vinblastine. Interestingly, re-sensitization of A375R to cell death induced by all of these drugs was achieved with ABT-737 in a manner similar to parental A375 (Figs. 5J, S4A–B, S4D), indicating that the anti-apoptotic BCL-2 repertoire may be of significant clinical utility following acquired drug resistance.
To extend our findings that anti-apoptotic BCL-2 family inhibition may enhance treatment success when cancer cells display resistant to PLX-4032, as suggested by in vitro experiments using A375R and SK-MEL-29R (Figs. 5H and S3E), we next examined SK-MEL-28R melanoma xenografts. Instead of using ABT-737 for these experiments, we switched to ABT-263, a small molecule inhibitor that displays nearly identical binding properties to anti-apoptotic BCL-2 proteins compared to ABT-737, but ABT-263 is orally bioavailable and administered to patients (20). First, we compared ABT-263 and ABT-737 for the ability to enhance PLX-4032 induced apoptosis in SK-MEL-28 and SK-MEL-28R. As shown in figures 6A–B, ABT-263 and ABT-737 functioned equivalently to sensitize both cell lines to PLX-4032 induced apoptosis. Next, we generated SK-MEL-28R xenografts (Fig. 6C), and then treated these tumor-bearing mice with a daily oral gavage of PLX-4032 (100 mg/kg) ± ABT-263 (10 mg/kg) for one week, which is half the dose and average treatment duration for PLX-4032 regimens in melanoma patients. Control (vehicle injected) or ABT-263 treatment alone resulted in minimal changes in tumor growth; however, we noted PLX-4032 treatment resulted in larger tumors compared to control (Fig. 6D). Despite the increase in tumor volume induced by PLX-4032 treatment, the combination of PLX-4032 and ABT-263 resulted in a marked decrease in tumor growth and subsequent increases in TUNEL positivity, a marker of apoptosis (Figs. 6D–F). Importantly, there were no marked changes to tumor cell proliferation (Figs. 6G–H, S5A) or tumor vascularization (Fig. S5B) amongst treatment groups. It should be noted that we observed the SK-MEL-28R tumors grew with greater size heterogeneity compared to the parental SK-MEL-28 line (Figs. 6C and S6A). SKMEL-28R average tumor volumes before and after treatment, along with the range in tumor sizes, are provided (Fig. S6B). Finally, we ensured that the PLX-4032 in the in vivo treatments was effective against parental SK-MEL-28 tumors, so we evaluated SK-MEL-28 xenografts for PLX-4032 responses. Indeed, PLX-4032 treated SK-MEL-28 tumors responded as there was an observable decrease in tumor volume (Fig. S6C).
Figure 6.
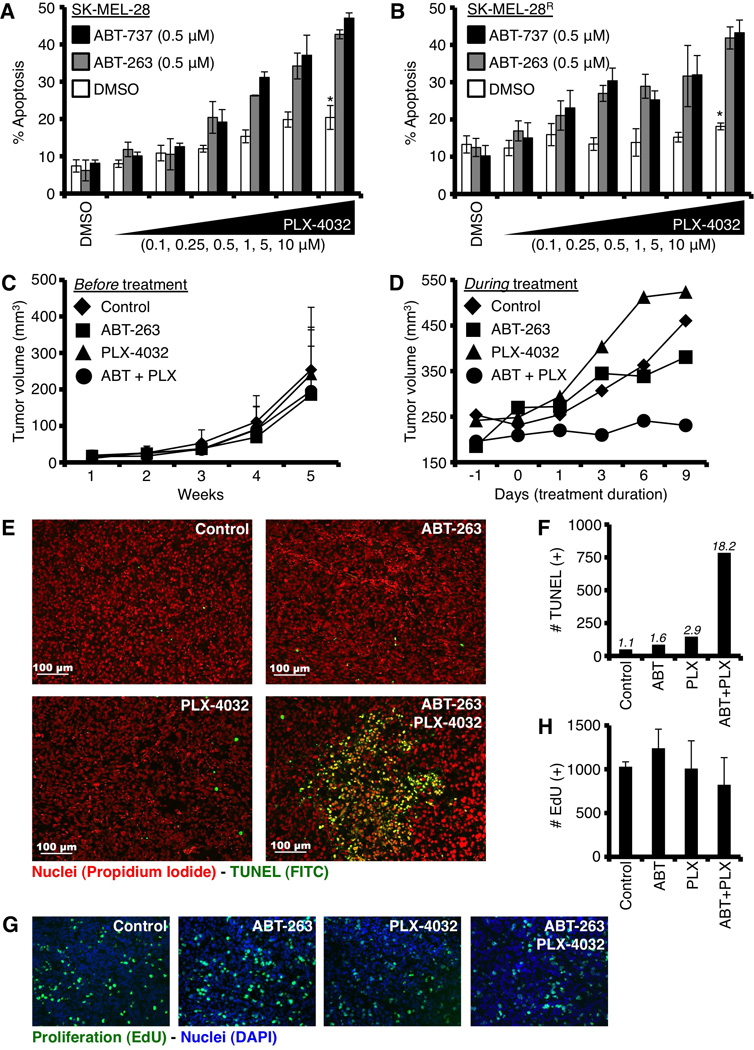
Inhibition of anti-apoptotic BCL-2 family members with ABT-263 sensitizes SK-MEL-28R to undergo apoptosis following PLX-4032 treatment. (A–B) SK-MEL-28 and SM-MEL-28R were pre-treated with ABT-737 or ABT-263 (0.5 µM) for 1 hour before treatment with PLX-4032 (0.1, 0.25, 0.5, 1, 5, 10 µM) for 48 hours, and analyzed by AnnexinV and flow cytometry. *p value = < 0.005. (C) SK-MEL-28R tumors were monitored for tumor growth for up to 5 weeks prior to treatment. (D) Tumor-bearing mice in C were treated as indicated (10 mg/kg ABT-263 ± 100 mg/kg PLX-4032, or control) for one week, and final tumor volumes were recorded. (E) Tumor sections were stained with TUNEL and propidium iodide for apoptosis and total cell number, respectively. Bars indicate 100 µM. (F) Representative tissue sections from each treatment were quantified to determine the total number of TUNEL positive cells. Approximately 5000 cells per treatment were evaluated; and the percentages of TUNEL (+) cells per treatment are indicated. (G) Tumor sections were stained for EdU and DAPI to determine the number of proliferating cells (EdU) per unit (DAPI) area. Individual colors are shown in figure S5A. (H) Quantification of EdU positive cells is shown; values are adjusted using ~ 1 × 106 µm2 DAPI positive regions for normalization. Two tumors per treatment group were analyzed in three regions. All figures are representative of at least triplicate data points, and reported as ± S.D. Xenograft data represent at least 8 tumors for each treatment group.
Discussion
The conclusions from our work reveal the mechanism of action for synergy between the parallel inhibition of B-RAFV600E signaling and the anti-apoptotic BCL-2 family repertoire (Figs. 2 – 6). Furthermore, our data suggest that collateral inhibition of the anti-apoptotic BCL-2 repertoire with ABT-737 (or the orally available form, ABT-263) may provide a much-needed clinical opportunity to increase primary response rates to targeted therapies like PLX-4032 and GSK-110212. This combination may decrease the chances of chemoresistance as the concentrations of drugs required to achieve a durable response will likely be lower (Fig. 6D), and many more tumor cells will be eliminated (Figs. 2C, 5H, 6A–B, S1C, S1E, S3E, S4A–B, S4D, S6C). In addition, if PLX-4032 resistance is unavoidable, ABT-737/263 may assist in re-sensitizing patients to additional targeted therapies as suggested by our data and other publications (Figs. S4A–B) (30), and may increase the response rates to conventional chemotherapeutic strategies (Figs. 5J, S3F–G, S4D). While our data have been generated in several commonly used laboratory models of B-RAFV600E and melanoma biology, the utility of our biochemical and promising cellular results must be explored using in vivo genetic model systems to fully appreciate their likelihood of benefiting patients.
Supplementary Material
Acknowledgements
We would like to thank everyone in the Chipuk Laboratory for their assistance and support; and Drs. Stuart Aaronson, Mark Lebwohl, Emily Bernstein, and Poulikos Poulikakos for mentorship, guidance, and/or discussion about the melanoma field; and Dr. Suvendu Das, Eliana Sarrou, Rana Elkholi, and Andrew Cruz for technical assistance. This work was supported by: NIH CA157740 (to J.E.C.), NIH KL2TR000069 (to S.I.), the JJR Foundation (to J.E.C.), the William A. Spivak Fund (to J.E.C.), and the Fridolin Charitable Trust (to J.E.C.). This work was also supported in part by a Research Grant FY13-238 from the March of Dimes Foundation (to J.E.C.), an Einstein Research Fellowship (to S.Y.W.), and an American Skin Association Medical Students Grant (to S.Y.W.).
Footnotes
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2(35):35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- 3.Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283(2):125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107(33):14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011;19(1):11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8(2):121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 12.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37(3):299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111(3):331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 15.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 16.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17(4):525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Chipuk JE, Fisher JC, Dillon CP, Kriwacki RW, Kuwana T, Green DR. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc Natl Acad Sci U S A. 2008;105(51):20327–20332. doi: 10.1073/pnas.0808036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 19.Anvekar RA, Asciolla JJ, Missert DJ, Chipuk JE. Born to be alive: a role for the BCL-2 family in melanoma tumor cell survival, apoptosis, and treatment. Front Oncol. 2011;1(34) doi: 10.3389/fonc.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 21.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 22.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, et al. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012;30(14):1628–1634. doi: 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103(4):645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 25.Logue SE, Elgendy M, Martin SJ. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoc. 2009;4(9):1383–1395. doi: 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- 26.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 27.Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272(21):13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 28.Asciolla JJ, Renault TT, Chipuk JE. Examining BCL-2 Family Function with Large Unilamellar Vesicles. J Vis Exp. 2012;(68) doi: 10.3791/4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anvekar RA, Asciolla JJ, Lopez-Rivera E, Floros KV, Izadmehr S, Elkholi R, et al. Sensitization to the mitochondrial pathway of apoptosis augments melanoma tumor cell responses to conventional chemotherapeutic regimens. Cell Death Dis. 2012;3:e420. doi: 10.1038/cddis.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118(11):3651–3659. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.