

Abstract
Bicarbonate is one of the major anions in mammalian tissues and extracellular fluids. Along with accompanying H+, HCO3- is generated from CO2 and H2O, either spontaneously or via the catalytic activity of carbonic anhydrase. It serves as a component of the major buffer system, thereby playing a critical role in pH homeostasis. Bicarbonate can also be utilized by a variety of ion transporters, often working in coupled systems, to transport other ions and organic substrates across cell membranes. The functions of HCO3- and HCO3--transporters in epithelial tissues have been studied extensively, but their functions in heart are less well understood. Here we review studies of the identities and physiological functions of Cl-/HCO3- exchangers and Na+/HCO3- cotransporters of the SLC4A and SLC26A families in heart. We also present RNA Seq analysis of their cardiac mRNA expression levels. These studies indicate that slc4a3 (AE3) is the major Cl-/HCO3- exchanger and plays a protective role in heart failure, and that Slc4a4 (NBCe1) is the major Na+/HCO3- cotransporter and affects action potential duration. In addition, previous studies show that HCO3- has a positive inotropic effect in the perfused heart that is largely independent of effects on intracellular Ca2+. The importance of HCO3- in the regulation of contractility is supported by experiments showing that isolated cardiomyocytes exhibit sharply enhanced contractility, with no change in Ca2+ transients, when switched from Hepes-buffered to HCO3-- buffered solutions. These studies demonstrate that HCO3- and HCO3--handling proteins play important roles in the regulation of cardiac function.
Keywords: SLC4, SLC26, Slc26a6, AE1, AE2, NBCn1
Core tip: Bicarbonate is one of the major anions in mammalian tissues and fluids. It plays a critical role in pH homeostasis and is utilized by various transporters to transport other ions and organic substrates across cell membranes. Here we review studies of the physiological functions of Cl-/HCO3- exchangers and Na+/HCO3- cotransporters in heart, present RNA Seq analysis of their cardiac mRNA expression levels, and show that bicarbonate is required for optimal contractility in isolated cardiac myocytes. These studies demonstrate that HCO3- and HCO3- handling proteins are abundant in heart and play important roles in the regulation of cardiac function.
INTRODUCTION
In mammalian tissues, bicarbonate/CO2 is the major extrinsic buffer system of both extracellular and intracellular fluids. HCO3-/CO2 are likely to play a particularly important role in cardiac muscle[1,2], which relies almost entirely on oxidative metabolism and continuously converts large quantities of O2 to CO2. In vivo, HCO3- is usually formed by carbonic anhydrase-mediated hydration of CO2[2,3], in a reaction that also generates H+ (CO2 + H2O → H+ + HCO3-). As might be expected, cardiac myocytes express a variety of ion transporters that mediate extrusion of H+ and either extrusion or uptake of HCO3-. This allows fine control of intracellular pH (pHi) and coupling of H+ and HCO3- transport to the transport of other ions, thereby affecting not only pHi, but also cell volume and both cellular and systemic ion homeostasis[4-6].
H+ and HCO3- are, in effect, transient ions[2] that can be used to transport other ions and organic substrates across cell membranes, both directly and by secondary active transport. These ion transporters include Cl-/HCO3- exchangers, Na+/HCO3- cotransporters (NBCs), and Na+/H+ exchangers (NHEs). The functions of the various acid-base transporters have been studied most extensively in epithelial tissues; however, they exhibit a surprising abundance and diversity in cardiac tissues. In this paper we review studies describing the identities, membrane locations, and functions of the major HCO3- transporters in heart. In addition, we report relative mRNA expression levels in mouse heart for members of the SLC4A and SLC26A anion transporter families[7-9], which include all of the known Cl-/HCO3- exchangers and Na+/HCO3- cotransporters. Finally, we discuss previous studies of the effects of HCO3- on the isolated heart[10] and correlate those results with new data using isolated cardiac myocytes. The available evidence shows that a diverse group of transporters are responsible for movements of HCO3- into and out of the heart and demonstrate that the presence of HCO3- has a major stimulatory effect on contractility that is, at least in part, independent of changes in intracellular Ca2+.
IDENTIFICATION AND LOCATIONS OF CARDIAC HCO3- TRANSPORTERS
Cloning and hybridization analyses have led to the identification of three Cl-/HCO3- exchangers of the SLC4A family in heart. These anion exchangers are termed AE1, AE2, and AE3 (Anion Exchanger 1, 2, and 3; gene symbols, Slc4a1-3), and one Cl-/HCO3- exchanger of the SLC26A family, termed Slc26a6 or PAT1 (putative anion transporter 1). Among the known Na+/HCO3- cotransporters (NBCs), which are members of the SLC4A family, the electrogenic NBCe1 and electroneutral NBCn1 (gene symbols, Slc4a4 and Slc4a7) have been identified in heart. Excellent reviews of the SLC4A and SLC26A[7-9,11,12] families of transporters have been published recently and provide detailed information about specific isoforms, including their ion transport specificities and their physiological functions in various tissues.
AE1 is the band 3 Cl-/HCO3- exchanger that is expressed most prominently in red blood cells[13] and also includes a kidney variant[14] derived from an alternative promoter in intron 4 of the erythrocyte transcription unit[15]. Erythrocyte AE1 plays major roles in maintaining the stability of the cytoskeleton[16] and in gas exchange[17]. Cardiac AE1 mRNA identified in rat heart is smaller than that of the erythroid and kidney variants[18]. It encodes a truncated protein based on immunoblot analyses, although the exact identity of the cardiac AE1 protein in rat heart has not been determined[19]. Immunofluorescence studies of rat heart suggest that truncated cardiac AE1 protein is restricted to intercalated discs[20]. Expression of AE1 is sharply reduced in adult mouse heart compared with its levels in fetal heart[21], consistent with the RNA Seq data discussed below. AE2 is expressed at low levels in heart[18,21,22], and AE2a, one of 4 variants derived from the use of alternative promoters[23], was the only variant detected[22]. The membrane location of AE2 in heart has not been determined. AE3 mRNAs are expressed at very high levels in heart[18,24] and encode both a full-length variant (AE3fl) that is expressed in brain and other tissues and a much more abundant cardiac variant[25-27]. The cardiac AE3 (AE3c) mRNA is derived from an alternative promoter located in intron 6 of the longer transcription unit and has a unique 73-amino acid sequence that replaces the first 270 amino acids of AE3fl[25]. In fetal mouse heart, AE3fl is the predominant form; however, in adult heart, AE3fl is largely restricted to the atria, while AE3c is the predominant form in ventricles[27]. In cardiac myocytes, AE3 protein has been localized to t-tubules and the sarcolemma, with apparent foci of expression at costameres[28].
The SLC26A family transports a broad range of anions, including sulfate, chloride, iodide, bicarbonate, oxalate, and formate, and some isoforms can function as anion channels[9]. The first member of this family shown to function as a Cl-/HCO3- exchanger was Slc26a3[29]; however, it is primarily an epithelial transporter and is expressed at only low levels in adult heart[21]. Slc26a6 can mediate Cl-/HCO3- exchange[30-32], which appears to be its major function in apical membranes of the intestine[33]. In the renal proximal tubule, however, Slc26a6 functions primarily as a Cl-/formate and Cl-/oxalate exchanger[34,35]. Slc26a6 also mediates Cl-/OH- exchange and has been proposed to serve both as a Cl-/HCO3- exchanger and as a Cl-/OH- exchanger in heart[21,36]. Slc26a6 protein has been localized to the t-tubules and sarcolemma[28].
Prior to the molecular cloning of Na+/HCO3- cotransporter (NBC) isoforms, both electroneutral[37,38] and electrogenic[39,40] Na+/HCO3- cotransport activities had been identified in cardiac muscle. NBCe1 (gene symbol, Slc4a4), the first NBC to be cloned[41,42], is electrogenic. In kidney, NBCe1 mediates outward transport of 1 Na+ and 3 HCO3- across the basolateral membrane of proximal tubule epithelial cells[41,42]. In most other tissues, including heart, regulation via phosphorylation results in a stoichiometry of 1:2[43]. Cloning of the cardiac form of NBCe1 revealed that it has a different N-terminus than the kidney variant[44], which is derived from an alternative promoter and first exon. NBCn1 is electroneutral and transports Na+ and HCO3- in a 1:1 ratio. It was cloned from rat smooth muscle[45] and skeletal muscle[46] and shown to be expressed in heart. A study using an antibody to the N-terminal sequence of an NBCn1 variant indicated that expression in heart was restricted to endothelial and smooth muscle cells[47]. However, NBCn1 transcripts undergo extensive alternative splicing, including a relatively cardiac-specific exon[48], and the use of alternative promoters that yield alternative N-termini[49]. Both NBCe1 and NBCn1 have been shown by Western blots to be expressed in cardiac myocytes[50]. Immunolocalization studies demonstrated that both isoforms are localized to t-tubules, lateral sarcolemma, and intercalated discs[50].
EXPRESSION LEVELS OF HCO3- TRANSPORTERS IN HEART
As discussed above, the major HCO3- transporters in the mammalian heart include both Cl-/HCO3- exchangers and Na+/HCO3- cotransporters of the SLC4A gene family and at least one Cl-/HCO3- exchanger of the SLC26A family. Gene expression data for hearts of wild-type FVBN mice were determined by RNA Seq analysis[51], a powerful method for determining the expression levels of all known mRNAs in a tissue of interest[52,53]. Relative mRNA expression levels for the SLC4A and SLC26A families are shown in Table 1. Transcript levels are expressed as RPKM values (reads per kilobase of exon per million mapped reads), which normalizes expression to the length of the mRNA. Graphical representations of mRNA expression levels of the HCO3- transporters that have been identified in heart and appear to be expressed at physiologically relevant levels are shown in Figure 1.
Table 1.
Relative mRNA levels for the Slc4a and Slc26a anion transporters in mouse heart
| SLC4A family | SLC26A family | ||||
|
Gene symbol |
Transporter name and major function(s);
alternate names |
Average ± SE |
Gene symbol |
Transporter name and major function(s);
alternate names |
Average ± SE |
| Slc4a1 | AE1 Cl-/HCO3- exchanger; Band 3 | 0.2 ± 0.05 | Slc26a1 | SAT1 sulfate/anion exchanger; Slc26a1 | 0.34 ± 0.07 |
| Slc4a2 | AE2 Cl-/HCO3- exchanger | 12.57 ± 0.28 | Slc26a2 | DTDST sulfate/anion exchanger; Slc26a2 | 2.98 ± 0.17 |
| Slc4a3 | AE3 Cl-/HCO3- exchanger | 85.7 ± 0.64 | Slc26a3 | DRA Cl-/HCO3-, exchanger; Slc26a3 | 0.64 ± 0.05 |
| Slc4a4 | NBCe1 Na+/HCO3- cotransporter; NBC1 | 9.45 ± 0.25 | Slc26a4 | Pendrin Cl-/HCO3- exchanger; Slc26a4 | 0.02 ± 0.01 |
| Slc4a5 | NBCe2 Na+/HCO3- cotransporter; NBC4 | 0.003 ± 0.002 | Slc26a6 | PAT1 Cl-/HCO3-, Cl-/formate exchanger; Slc26a6 | 4.56 ± 0.26 |
| Slc4a7 | NBCn1 Na+/HCO3- cotransporter; NBC2; NBC3 | 4.24 ± 0.05 | Slc26a7 | Slc26a7 Cl-/HCO3- exchanger, Cl- channel; TAT1 | 0.15 ± 0.04 |
| Slc4a8 | NDCBE Na+-driven Cl-/HCO3- exchanger; Slc4a8 | 0.73 ± 0.04 | Slc26a9 | Slc26a9 Cl-/HCO3- exchanger, Cl- channel | 0.01 ± 0.01 |
| Slc4a9 | AE4 Cl-/HCO3- exchanger | 0.02 ± 0.01 | Slc26a10 | Slc26a10, transporter function unknown | 31.66 ± 2.55 |
| Slc4a10 | NBCn2 Na+/HCO3- cotransporter | 0.02 ± 0.003 | Slc26a11 | Slc26a11 anion exchanger, Cl- channel; KBAT | 1.15 ± 0.14 |
Relative mRNA expression levels were determined using RNA from 4-month-old male FVB/N mouse hearts (n = 4) as described previously[51]. Values are RPKM (reads per kilobase per million mapped reads) ± SE and are a measure of the relative abundance of specific gene transcripts[53]. For some Slc26a transporters, ion transport specificities are more complex than indicated; see Alper and Sharma[9].
Figure 1.
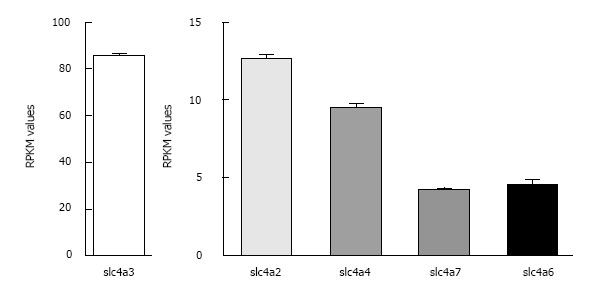
Relative expression levels of the major Cl-/HCO3- exchangers and Na+/HCO3- cotransporters in mouse heart. RPKM values ± SE as determined by RNA Seq analysis (see Table 1 legend) are shown for the most abundant known HCO3- transporters in wild-type FVB/N mouse hearts (n = 4). Note the difference in scale for AE3 and the other transporters.
Among the SLC4A transporters, the AE3 Cl-/HCO3- exchanger was expressed at very high levels (RPKM = 85.7 ± 0.6), suggesting that it serves as the major HCO3- efflux mechanism in mouse cardiac myocytes. AE2 (RPKM = 12.57 ± 0.28) was expressed at lower levels than AE3, but its levels of expression were still greater than some of the other transporters. Although NBC4 (Slc4a5 or NBCe2) was reported in human heart[54], it is not expressed at significant levels in mouse heart or in rat heart[55]. Of the two major Na+/HCO3- cotransporters in mouse heart, NBCe1 (RPKM = 9.45 ± 0.25) was more abundant than NBCn1 (RPKM = 4.24 ± 0.05). By comparison, RPKM values for the NHE1 Na+/H+ exchanger[51], recognized as the major Na+-dependent acid extruder in heart, were 9.10 ± 0.20.
Among the SLC26A transporters, Slc26a2, Slc26a6, and Slc26a10 were expressed most abundantly. However, among these three SLC26A transporters, the only known HCO3- transporter is Slc26a6 (RPKM = 4.56 ± 0.26). As discussed above, it has been shown to function as a Cl-/HCO3- exchanger, but it also mediates Cl-/OH-, Cl-/formate, and Cl-/oxalate exchange[9]. Slc26a2 is a sulfate transporter and also transports Cl- and oxalate[9]. The ion specificity of Slc26a10 has not been determined[9], but its high level of expression (RPKM = 31.66 ± 2.55) suggests that it plays an important role in mouse heart.
Some of the HCO3- transporters expressed at low levels (e.g., the Slc4a8 Na+-dependent Cl-/HCO3- exchanger and Slc26a3 Cl-/HCO3- exchanger) could still play important roles in heart, particularly if they were restricted to specialized regions of the heart or were expressed primarily in earlier stages of development or in cell-types other than cardiac myocytes. For example, Na+-dependent Cl-/HCO3- exchange, which could be due to the activity of Slc4a8[56], has been identified in both chicken embryonic cardiomyocytes[57] and in vascular endothelial cells[58].
PHYSIOLOGICAL FUNCTIONS OF CARDIAC HCO3- TRANSPORTERS
Cl-/HCO3- Exchangers
Because of the high Cl- concentrations of extracellular fluids, electroneutral Cl-/HCO3- exchangers mediate outward transport of HCO3- and inward transport of Cl-. The direct effect of this activity is to reduce pHi, thereby contributing to pHi regulation[59], and to enhance Cl--loading, which could affect Cl- currents that in turn could affect action potentials or rhythmicity[60,61]. Also, when coupled with Na+-dependent acid extrusion mechanisms, Cl-/HCO3- exchange facilitates Na+-loading, which can affect contractility as discussed below and may contribute to cardiac hypertrophy[2]. AE1, AE2, and AE3 are electroneutral, but Cl-/HCO3- exchangers of the SLC26A family are reported to support both electroneutral or electrogenic anion exchange[9,12].
The physiological functions of AE1 in heart are unclear. An AE1 global knockout mouse has been shown to develop cardiac hypertrophy[28]. The investigators noted, however, that the levels of AE1 in the adult heart are relatively low (confirmed by the data in Table 1) and that null mutants exhibit severe hemolytic anemia and spherocytosis. They attributed the hypertrophy to the blood defect and concluded that one of the more abundant Cl-/HCO3- exchangers, possibly AE3, was more likely to provide the HCO3- extrusion capability that has been proposed to balance Na+-dependent acid extrusion via transporters such as the NHE1 Na+/H+ exchanger (discussed below). AE2 is a potential candidate for this activity as it is known to operate in concert with NHE1 on basolateral membranes of colonic epithelial cells[62]. AE2 is much less abundant than AE3 and its functions in heart have not been determined.
It has been suggested that one of the major functions of Cl-/HCO3- exchange in heart is to counter the alkalinizing effects of Na+/H+ exchange. This would allow increased Na+/H+ exchange activity[2,63], which in turn would lead to increased Na+-loading and Ca2+-loading via reverse activity of the Na+/Ca2+ exchanger[64]. Inhibition of Na+/H+ exchange reduces cardiac hypertrophy[4,65] and overexpression of an activated NHE1 Na+/H+ exchanger induces hypertrophy and increases cytosolic Na+, Ca2+ transients, and contractility[66]. Studies have shown that the reduction in hypertrophy in spontaneously hypertensive rats in response to angiotensin II blockade involves reductions in both Na+/H+ exchange and Cl-/HCO3- exchange[67]. Intracellular pH in NHE1-overexpressing myocytes was significantly higher when they were maintained in Hepes-buffered media than in CO2/HCO3--buffered media[66]. This is consistent with the view[68,69] that Cl-/HCO3- exchange balances the alkalinizing effects of Na+/H+ exchange, which would be expected to facilitate pHi-neutral Na+-loading in vivo.
The Cl-/HCO3- exchanger that has been most heavily studied in heart is AE3. Its mRNA is expressed at much higher levels in heart than those of the other HCO3- transporters (see Table 1) and it has a cardiac specific variant[25-27], indicating that it serves a specialized function. Earlier studies showed that Cl-/HCO3- exchange and Na+/H+ exchange were increased in the hypertrophic heart of spontaneously hypertensive rats[67] and that AE3fl mRNA was upregulated[70]. Although this might suggest that AE3fl accounts for the increased anion exchange activity, the investigators cautioned against this interpretation as AE3fl is expressed at low levels in the adult rat and mouse heart[19,25,27]. Treatment of papillary muscles with an inhibitory anti-AE3 antibody led to an increase in the slow-force response to stretch, which is dependent on Na+/H+ exchange, and caused a substantial reduction in Cl-/HCO3- exchange, supporting the view that AE3 is the major Cl-/HCO3- exchanger in cardiac muscle[71]. Analysis of pHi in tissues treated with the anti-AE3 antibody indicated that AE3 is the major anion exchanger responsible for countering the alkalinizing effects of NHE1-mediated Na+/H+ exchange[71]. It has been suggested that activation of AE3 and NHE1 together might contribute to hypertrophy[72], but so far there is no direct proof of this hypothesis.
The initial studies of a gene-targeted AE3-null mouse showed that the loss of AE3 did not impair cardiovascular performance in vivo under basal conditions or after β-adrenergic stimulation, and it also had no effect on ischemia-reperfusion injury using the Langendorff-perfused heart[6]. The latter finding was surprising as there is evidence that Cl-/HCO3- exchange mediates some of the changes in pHi and intracellular Cl- that contribute to reperfusion injury[73]. Heart weight/body weight ratios were significantly reduced in null mutants relative to wild-type mice, consistent with the possibility that loss of AE3 activity might contribute to a reduction in hypertrophy. When AE3-null mice were crossed with an NKCC1 Na+-K+-2Cl- cotransporter-null mouse, which had normal contractility[74], the double mutant mice exhibited a contractility defect in vivo and in isolated myocytes, and Ca2+ extrusion mediated by the Na+/Ca2+ exchanger was increased[6]. NKCC1 has been shown to provide substantial Na+ influx in chick cardiac myocytes[75] and to affect Na+/Ca2+ exchange in mouse astrocytes[76]. Therefore, it is possible that the additional loss of NKCC1 caused a reduction in Na+-loading in AE3/NKCC1 double mutants, with subsequent effects on contractility.
The above studies were consistent with the possibility that loss of AE3 might protect against hypertrophy; however, they also showed that its absence can impair cardiac function under certain conditions. To test whether AE3-deficiency might protect against hypertrophy, AE3-null mice were crossed with a transgenic hypertrophic cardiomyopathy mouse model[77] carrying a Glu180Gly mutation in α-tropomyosin[78]. The additional loss of AE3 in the Glu180Gly mutant caused no decrease in the degree of hypertrophy and led to more rapid decompensation, heart failure, and death. Cardiac performance in response to β-adrenergic stimulation was severely impaired in double mutants. The double mutants exhibited more arrhythmic events as heart rates were increased by electrical pacing to assess force-frequency responses and Ca2+-handling was also impaired. It was concluded that AE3 activity is needed for better preservation of cardiac function during heart failure and that it would not be an appropriate therapeutic target for cardiac arrhythmias or hypertrophy. In a more recent study[79] it was shown that hearts of AE3-null mice exhibit blunting of the force-frequency response when they are paced to higher heart rates in vivo. Phosphorylation of Akt, which plays a central role in mechanosensory signaling, was increased in paced AE3-null hearts and phosphorylation of adenosine 5’-monophosphate-activated protein kinase (AMPK) was reduced[79]. These data suggest that the increased susceptibility of AE3-null mice to decompensation in heart failure might be due impaired rate dependent inotropy, an insufficient response to biomechanical stress, and metabolic perturbations.
The functions of Slc26a6 in heart have not yet been determined. As discussed by Alper and Sharma[9], there is controversy about the electrogenicity of the Cl-/HCO3- exchange activity of Slc26a6, which is the only known Cl-/HCO3- exchanger of the SLC26A family expressed at significant levels in heart[21]. Some studies[80,81] reported electrogenic Cl-/HCO3- exchange for Slc26a6 with a stoichiometry of 1Cl-/2HCO3- and others[82] reported electroneutral exchange with a 1:1 ratio. In the latter study[82], the investigators could detect electrogenic Cl-/oxalate transport mediated by Slc26a6 in oocytes but Cl-/HCO3- and Cl-/OH- exchange appeared to be electroneutral. Cl-/OH- exchange in cardiac myocytes, which has been attributed to Slc26a6[21], is electroneutral[36], and Slc26a6-mediated Cl-/formate exchange is electroneutral[83]. Regardless of whether Slc26a6-mediated Cl-/HCO3- exchange is electrogenic, during most of the excitation-relaxation cycle, when the membrane potential is negative, it would transport HCO3- out of the cell. However, it is possible that reversal of electrogenic Cl-/HCO3- exchange might occur at positive membrane potentials. The potential functions of electrogenic Slc26a6-mediated Cl-/HCO3- exchange in heart have not been studied. Furthermore, it is not clear which of the various transport functions of Slc26a6 is the most important in heart.
Na+/HCO3- Cotransporters
As discussed above, there is evidence for both electroneutral and electrogenic NBC activities in cardiac myocytes[37-40,84,85]. NBCe1 and NBCn1, along with NHE1, have been immunolocalized in rat myocytes[50]. These three transporters are the major Na+-dependent alkalinizing mechanisms in cardiac myocytes. In the isolated perfused ferret heart, NBC and NHE activities contributed equally to recovery of pHi, both after an acid load[86] and also during reperfusion following ischemia[87]. In the latter study, it was suggested that NBC-mediated Na+ influx might contribute to Ca2+ overload and injury after reperfusion. Later studies using an inhibitory antibody showed that inhibition of NBCe1 protected against ischemia-reperfusion injury in the isolated rat heart[88]. Similarly, in rat ventricular myocytes subjected to anoxic conditions, simultaneous inhibition of NBC and NHE1 activities prevented hypercontracture induced by Ca2+-overload during reoxygenation, whereas inhibition of either activity alone was insufficient[89]. This suggests that NBCe1, like the NHE1 Na+/H+ exchanger, can be a significant source of Na+-loading, although the magnitude of Na+-loading via NBCe1 has been estimated to be lower than that of Na+/H+ exchange[64]. Also, NHE1 has cardioprotective effects that appear to be independent of effects on Na+ and Ca2+ loading[51,90].
NBCe1 is localized to t-tubules[50], along with the L-type Ca channel (LTCC) and NCX1 Na+/Ca2+ exchanger[91], whereas NHE1 is expressed at highest levels in intercalated discs[50,92]. Thus, NBCe1 appears to be well situated to affect excitation-contraction coupling[93], particularly since it is electrogenic. In fact, a substantial NBC-mediated HCO3- current has been demonstrated beginning at -50 millivolts[84], and electrogenic NBC activity causes a shortening of the action potential duration (APD) and affects the resting membrane potential[40,94]. By shortening the APD, NBCe1 could reduce the open time of the LTCC and with its location in the t-tubule it could reduce intraluminal (extracellular) pH and increase pHi, both of which reduce LTCC-mediated Ca2+ currents[95]. Thus, while NBCe1 activity may serve as a Na+-loading mechanism that could, in principle, contribute to Ca2+-loading via reverse mode activity of the Na+/Ca2+ exchanger, its effects on the APD and on LTCC activity might counteract this tendency. NBCn1 is also in t-tubules of ventricular myocytes[50] and because it is electroneutral it could operate throughout the excitation-contraction cycle.
Both NBCe1 and NBCn1, along with NHE1, were induced in rat heart during pressure overload hypertrophy[55]. After an acid load, the rate of pHi recovery via NBC and NHE activities were increased accordingly, and NBC activity in the physiological pHi range was similar to that of NHE1[55]. Additional experiments[55] showed that when rats were subjected to pressure overload and then treated with losartan, an angiotensin II AT1 receptor antagonist, both hypertrophy and the induction of NBCe1 and NBCn1 were sharply reduced. The results suggest, but do not prove, that increased NBC activities contribute to the development of hypertrophy. NBCe1 mRNA and protein were also induced in human failing hearts[88] and in rat hearts following myocardial infarction[96]; however, in the latter study, treatment with an angiotensin II AT1 receptor antagonist had no effect on NBCe1 expression. The effects of angiotensin II on NBC activity in cardiac myocytes are complex as some studies report activation of NBC activity[97,98] and others report inhibition[99]. A more recent study showed that cardiac expression of both NBCn1 and NBCe1 were induced in spontaneously hypertensive rats in which angiotensin II plays a major role[100]. However, NBCe1 activity was reduced due to a reduction in its protein expression in t-tubule and sarcolemmal membranes; nevertheless, total NBC activity increased due to an increase in NBCn1 activity. The authors noted that a reduction in NBCe1 activity leads to an increase in APD, which is a common occurrence during cardiac hypertrophy[101], and that this would likely cause an increase in Ca2+-influx via LTCC[102]. Thus, NBCe1 activity, rather than inhibition of its activity, may be cardioprotective in some disease conditions.
EFFECTS OF HCO3- ON CONTRACTILITY AND Ca2+ IN ISOLATED HEARTS
Given the abundance and diversity of HCO3- transporters in heart and the fact that HCO3- is part of the major buffer system in biological systems, it is surprising that there has been little reported work on the specific effects of HCO3- on contractility and Ca2+-handling. In an interesting and important study, Fülöp et al[10] analyzed Langendorff-perfused guinea pig hearts in the presence of both Krebs solution buffered with CO2/HCO3- and Tyrode solution buffered with HEPES[10]. Contractility in isolated hearts was significantly greater in Krebs solution than in Tyrode solution. However, when Tyrode solution was supplemented with CO2/HCO3-, in the continuing presence of HEPES, contractility increased to the levels observed in Krebs solution. Changes in contractility were reversible as the buffers were switched between those containing CO2/HCO3- or HEPES alone. Despite increased contractility, both the amplitude and duration of the Ca2+ transients were lower in solutions containing CO2/HCO3- buffer, indicating that enhanced Ca2+ transients were not responsible for the enhanced contractility. Analyses of isolated trabeculae also revealed increased contractility in CO2/HCO3- buffer, with faster times to peak tension and shorter relaxation times[10]. In purkinje fibers and papillary muscles, the duration of the action potential was reduced in the presence of CO2/HCO3- buffer[10]. This finding is consistent with the proposed effects of NBCe1 activity on the action potential[103].
The reduced contractility in the isolated heart and isolated tissues in response to the absence of HCO3- may have been due to reduced pHi or buffering power[10]. However, in wild-type myocytes used in a study of the effects of Na+/H+ exchange on Ca2+ and contractility, pHi was the same in HEPES buffer as in buffer containing CO2/HCO3-[66]. Also, in myocytes overexpressing an activated NHE1, the absence of CO2/HCO3- led to an increase in pHi[66]. These results suggest that a HCO3--dependent transport mechanisms, i.e., Cl-/HCO3- exchange, is needed to counter the alkalinizing effects of NHE1.
EFFECTS OF HCO3- ON CONTRACTILITY AND Ca2+ IN ISOLATED MYOCYTES
Although studies of cardiac myocyte mechanics and Ca2+ handling are commonly performed in HEPES buffered solution (Tyrode’s solution), we are unaware of studies directly comparing the effects of the two buffer conditions. Therefore, we performed experiments to assess the effects of HEPES-buffered and HCO3--buffered solutions on both contraction of rat ventricular myocytes and Ca2+ transients. The concentrations of cations were identical for the two solutions, and their osmolarities were the same.
Switching from HEPES-buffered solution to HCO3--buffered solution had a bi-phasic effect on myocyte contraction, determined by measurements of cell shortening as described previously[104]. It first resulted in transient suppression of myocyte contraction, followed by reversal of suppression, and enhancement of contraction (Figure 2A). On average, the downward suppressive phase lasted about 50 s (Figure 2B), resulting in a peak suppression of cell shortening from 7.04% in control (i.e., HEPES) to 4.35% (Figure 2C). This was followed by gradual stimulation of contraction; at steady-state, contraction was significantly increased to 11.39% (Figure 2C). Switching from HEPES-buffered Tyrode’s solution to an isosmotic solution that contained both HEPES and HCO3- produced the same bi-phasic effect and reached the same steady-state increase in contractility. Thus, the stimulation of contraction by HCO3- in isolated cardiac myocytes was fully reversible, as observed previously in the isolated heart[10].
Figure 2.
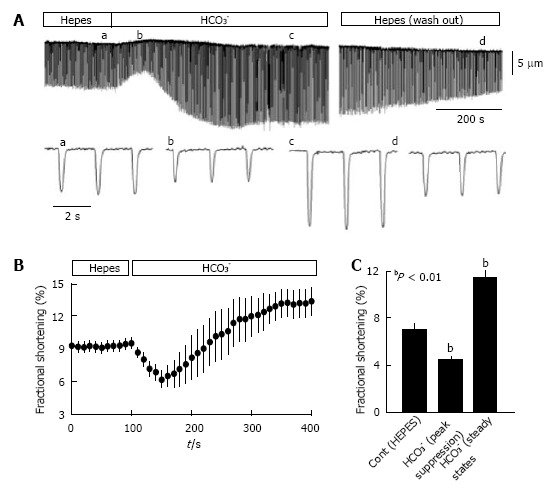
Isolated myocytes exhibit greater contractility in CO2/HCO3- buffer than in HEPES buffer. Ventricular myocytes from rat hearts were enzymatically dissociated using Langendorff perfusion[104] and myocyte mechanics were analyzed at room temperature (24 °C), with stimulation at 0.5 Hz as described previously[107]. Myocytes were switched between HEPES-buffered Tyrode’s solution (in mmol/L: NaCl 140, KCl 5.4, MgCl2 1, CaCl2 1.8, glucose 10, and Na-HEPES 5; pH = 7.4; bubbled with 100% O2) and HCO3--buffered Krebs solution (in mmol/L: NaCl 120, NaHCO3- 25, KCl 4.2, KH2PO4 1.2, MgCl2 1, CaCl2 1.8, and glucose 10; pH = 7.4 when bubbled with 95% O2 and 5% CO2). A: Representative contraction tracing of a myocyte bathed in HEPES buffer, then switched to HCO3- buffer, and then returned to HEPES buffer; the lower tracings show an expanded scale for the indicated (a-d) time points. The lower panels show the time course of fractional shortening (B) and average fractional shortening (C) of myocytes (n = 19) in HEPES buffer and then switched to HCO3--containing buffer. Experiments were performed using myocytes from 3 hearts and statistical analysis was conducted using a paired t-test. Values are means ± SE.
In addition to affecting myocyte contractility, HCO3- also had a small but significant effect on myocyte length. Switching from HEPES to the HCO3--buffered solution transiently increased myocyte length by approximately 1% (data not shown). Such transient lengthening was independent of myocyte contraction, and was also observed in unpaced, quiescent myocytes. This suggests the possibility that cell volume was increased by the addition of HCO3-, which is reasonable given the role of Cl-/HCO3- exchange in cell volume regulation[105,106].
Interestingly, the marked effect of HCO3- on myocyte contraction was not accompanied by any detectable change in the Ca2+ transient. Figure 3A shows Ca2+ transients from the same myocyte in the presence of HEPES or HCO3- solutions. Switching from HEPES-buffered solutions to HCO3--buffered solutions altered neither the amplitude nor the time constant of the Ca2+ transient (Figure 3B). These data using isolated myocytes correlate well with the previous studies using isolated hearts[10]. Both sets of data show that HCO3- has a major effect on contractility, without any major effects on the amplitude of the Ca2+ transient.
Figure 3.
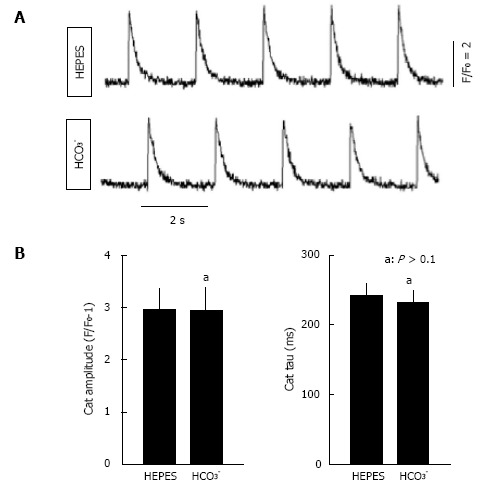
Ca2+ transient analysis in isolated rat myocytes bathed in CO2/HCO3- buffer and in HEPES buffer. For recording of Ca2+ transients, isolated ventricular myocytes were loaded with fluo-4 acetoxymethyl ester (5 μmol/L, Molecular Probes, Eugene, OR) and activated with field stimulation at 0.5 Hz. Fluorescence signals were measured using a Nikon TE 2000 microscope and an InCyt Standard PM photometry system (Intracellular Imaging, Cincinnati, OH) as described previously[104]. A: Representative Ca2+ transients in HEPES and HCO3--containing buffers; B: Average Ca2+ transient (CaT) amplitudes and tau values, a measure of the rate of decay of the Ca2+ transient in HEPES and HCO3--containing buffers (n = 12). The same cells were imaged in both buffers. Myocytes were from the same preparations used in Figure 2, with statistical analyses performed using a paired t-test. No significant differences were observed.
CONCLUSION
The studies reviewed here show that the mammalian heart contains an abundance of HCO3- transporters, which include both Cl-/HCO3- exchangers and Na+/HCO3- cotransporters. Their most obvious function is regulation of pHi, although it is possible that this is not their major function. This is particularly apparent in the case of the Cl-/HCO3- exchangers because at the high frequencies occurring in vivo, cardiac myocytes generate a substantial acid load. Thus, there would appear to be little need to maintain a robust capacity for recovery from an alkaline load. Nevertheless, it is possible that the cardiac anion exchangers regulate the pH or electrolyte concentrations of sub-sarcolemmal or t-tubule microdomains. With regard to electrolyte homeostasis, coupling of Cl-/HCO3- exchange and Na+/HCO3- cotransport (or Na+/H+ exchange) can serve as a pHi-neutral Na+- and Cl--loading mechanism, with Na+ affecting Ca2+-loading via Na+/Ca2+ exchange. In addition to effects on Na+-loading, electrogenic Na+/HCO3- cotransport can affect the duration of the action potential[40,84,94] and, by affecting subsarcolemmal and t-tubular pH, it might also affect the activity of L-type Ca2+ channels[95]. Finally, the available data show that the presence of CO2/HCO3- buffer has a major effect on contractility that cannot be readily explained by effects on Ca2+-handling, thus suggesting that HCO3- homeostasis plays an important role in the regulation of cardiac contractility. The mechanism is not known, but it is conceivable that intracellular HCO3- concentrations affect myofibrillar function and that dynamic transporter-mediated HCO3- fluxes have a major effect on electrical and ionic properties of the myocyte. Further studies of the effects of HCO3- and the cardiac functions of each of the major HCO3- transporters will be needed to resolve these issues.
Footnotes
Supported by National Institutes of Health Grants HL061974 to Shull GE; and ES017263 to Wang HS
P- Reviewer: Bies J, Laghmani K, Tatulian S S- Editor: Wen LL L- Editor: A E- Editor: Lu YJ
References
- 1.Vaughan-Jones RD, Spitzer KW. Role of bicarbonate in the regulation of intracellular pH in the mammalian ventricular myocyte. Biochem Cell Biol. 2002;80:579–596. doi: 10.1139/o02-157. [DOI] [PubMed] [Google Scholar]
- 2.Casey JR. Why bicarbonate? Biochem Cell Biol. 2006;84:930–939. doi: 10.1139/o06-184. [DOI] [PubMed] [Google Scholar]
- 3.Spitzer KW, Skolnick RL, Peercy BE, Keener JP, Vaughan-Jones RD. Facilitation of intracellular H(+) ion mobility by CO(2)/HCO(3)(-) in rabbit ventricular myocytes is regulated by carbonic anhydrase. J Physiol. 2002;541:159–167. doi: 10.1113/jphysiol.2001.013268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karmazyn M, Kilić A, Javadov S. The role of NHE-1 in myocardial hypertrophy and remodelling. J Mol Cell Cardiol. 2008;44:647–653. doi: 10.1016/j.yjmcc.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. doi: 10.1016/j.yjmcc.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 6.Prasad V, Bodi I, Meyer JW, Wang Y, Ashraf M, Engle SJ, Doetschman T, Sisco K, Nieman ML, Miller ML, et al. Impaired cardiac contractility in mice lacking both the AE3 Cl-/HCO3- exchanger and the NKCC1 Na+-K+-2Cl- cotransporter: effects on Ca2+ handling and protein phosphatases. J Biol Chem. 2008;283:31303–31314. doi: 10.1074/jbc.M803706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009;212:1672–1683. doi: 10.1242/jeb.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero MF, Chen AP, Parker MD, Boron WF. The SLC4 family of bicarbonate (HCO₃⁻) transporters. Mol Aspects Med. 2013;34:159–182. doi: 10.1016/j.mam.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. 2013;34:494–515. doi: 10.1016/j.mam.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fülöp L, Szigeti G, Magyar J, Szentandrássy N, Ivanics T, Miklós Z, Ligeti L, Kovács A, Szénási G, Csernoch L, et al. Differences in electrophysiological and contractile properties of mammalian cardiac tissues bathed in bicarbonate - and HEPES-buffered solutions. Acta Physiol Scand. 2003;178:11–18. doi: 10.1046/j.1365-201X.2003.01114.x. [DOI] [PubMed] [Google Scholar]
- 11.Soleimani M. SLC26 Cl-/HCO3- exchangers in the kidney: roles in health and disease. Kidney Int. 2013;84:657–666. doi: 10.1038/ki.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonar PT, Casey JR. Plasma membrane Cl⁻/HCO₃⁻ exchangers: structure, mechanism and physiology. Channels (Austin) 2008;2:337–345. doi: 10.4161/chan.2.5.6899. [DOI] [PubMed] [Google Scholar]
- 13.Kopito RR, Lodish HF. Primary structure and transmembrane orientation of the murine anion exchange protein. Nature. 1985;316:234–238. doi: 10.1038/316234a0. [DOI] [PubMed] [Google Scholar]
- 14.Kudrycki KE, Shull GE. Primary structure of the rat kidney band 3 anion exchange protein deduced from a cDNA. J Biol Chem. 1989;264:8185–8192. [PubMed] [Google Scholar]
- 15.Kudrycki KE, Shull GE. Rat kidney band 3 Cl-/HCO3- exchanger mRNA is transcribed from an alternative promoter. Am J Physiol. 1993;264:F540–F547. doi: 10.1152/ajprenal.1993.264.3.F540. [DOI] [PubMed] [Google Scholar]
- 16.Peters LL, Shivdasani RA, Liu SC, Hanspal M, John KM, Gonzalez JM, Brugnara C, Gwynn B, Mohandas N, Alper SL, et al. Anion exchanger 1 (band 3) is required to prevent erythrocyte membrane surface loss but not to form the membrane skeleton. Cell. 1996;86:917–927. doi: 10.1016/s0092-8674(00)80167-1. [DOI] [PubMed] [Google Scholar]
- 17.Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, Mohandas N, Anstee DJ, Tanner MJ. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101:4180–4188. doi: 10.1182/blood-2002-09-2824. [DOI] [PubMed] [Google Scholar]
- 18.Kudrycki KE, Newman PR, Shull GE. cDNA cloning and tissue distribution of mRNAs for two proteins that are related to the band 3 Cl-/HCO3- exchanger. J Biol Chem. 1990;265:462–471. [PubMed] [Google Scholar]
- 19.Richards SM, Jaconi ME, Vassort G, Pucéat M. A spliced variant of AE1 gene encodes a truncated form of Band 3 in heart: the predominant anion exchanger in ventricular myocytes. J Cell Sci. 1999;112(Pt 10):1519–1528. doi: 10.1242/jcs.112.10.1519. [DOI] [PubMed] [Google Scholar]
- 20.Moura Lima PR, Salles TS, Costa FF, Saad ST. alpha-cardiac actin (ACTC) binds to the band 3 (AE1) cardiac isoform. J Cell Biochem. 2003;89:1215–1221. doi: 10.1002/jcb.10561. [DOI] [PubMed] [Google Scholar]
- 21.Alvarez BV, Kieller DM, Quon AL, Markovich D, Casey JR. Slc26a6: a cardiac chloride-hydroxyl exchanger and predominant chloride-bicarbonate exchanger of the mouse heart. J Physiol. 2004;561:721–734. doi: 10.1113/jphysiol.2004.077339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Schultheis PJ, Shull GE. Three N-terminal variants of the AE2 Cl-/HCO3- exchanger are encoded by mRNAs transcribed from alternative promoters. J Biol Chem. 1996;271:7835–7843. doi: 10.1074/jbc.271.13.7835. [DOI] [PubMed] [Google Scholar]
- 23.Medina JF, Lecanda J, Acín A, Ciesielczyk P, Prieto J. Tissue-specific N-terminal isoforms from overlapping alternate promoters of the human AE2 anion exchanger gene. Biochem Biophys Res Commun. 2000;267:228–235. doi: 10.1006/bbrc.1999.1951. [DOI] [PubMed] [Google Scholar]
- 24.Kopito RR, Lee BS, Simmons DM, Lindsey AE, Morgans CW, Schneider K. Regulation of intracellular pH by a neuronal homolog of the erythrocyte anion exchanger. Cell. 1989;59:927–937. doi: 10.1016/0092-8674(89)90615-6. [DOI] [PubMed] [Google Scholar]
- 25.Linn SC, Kudrycki KE, Shull GE. The predicted translation product of a cardiac AE3 mRNA contains an N terminus distinct from that of the brain AE3 Cl-/HCO3- exchanger. Cloning of a cardiac AE3 cDNA, organization of the AE3 gene, and identification of an alternative transcription initiation site. J Biol Chem. 1992;267:7927–7935. [PubMed] [Google Scholar]
- 26.Yannoukakos D, Stuart-Tilley A, Fernandez HA, Fey P, Duyk G, Alper SL. Molecular cloning, expression, and chromosomal localization of two isoforms of the AE3 anion exchanger from human heart. Circ Res. 1994;75:603–614. doi: 10.1161/01.res.75.4.603. [DOI] [PubMed] [Google Scholar]
- 27.Linn SC, Askew GR, Menon AG, Shull GE. Conservation of an AE3 Cl-/HCO3- exchanger cardiac-specific exon and promoter region and AE3 mRNA expression patterns in murine and human hearts. Circ Res. 1995;76:584–591. doi: 10.1161/01.res.76.4.584. [DOI] [PubMed] [Google Scholar]
- 28.Alvarez BV, Kieller DM, Quon AL, Robertson M, Casey JR. Cardiac hypertrophy in anion exchanger 1-null mutant mice with severe hemolytic anemia. Am J Physiol Heart Circ Physiol. 2007;292:H1301–H1312. doi: 10.1152/ajpheart.00449.2006. [DOI] [PubMed] [Google Scholar]
- 29.Melvin JE, Park K, Richardson L, Schultheis PJ, Shull GE. Mouse down-regulated in adenoma (DRA) is an intestinal Cl(-)/HCO(3)(-) exchanger and is up-regulated in colon of mice lacking the NHE3 Na(+)/H(+) exchanger. J Biol Chem. 1999;274:22855–22861. doi: 10.1074/jbc.274.32.22855. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Petrovic S, Mann E, Soleimani M. Identification of an apical Cl(-)/HCO3(-) exchanger in the small intestine. Am J Physiol Gastrointest Liver Physiol. 2002;282:G573–G579. doi: 10.1152/ajpgi.00338.2001. [DOI] [PubMed] [Google Scholar]
- 31.Petrovic S, Ma L, Wang Z, Soleimani M. Identification of an apical Cl-/HCO-3 exchanger in rat kidney proximal tubule. Am J Physiol Cell Physiol. 2003;285:C608–C617. doi: 10.1152/ajpcell.00084.2003. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Z, Grichtchenko II, Boron WF, Aronson PS. Specificity of anion exchange mediated by mouse Slc26a6. J Biol Chem. 2002;277:33963–33967. doi: 10.1074/jbc.M202660200. [DOI] [PubMed] [Google Scholar]
- 33.Simpson JE, Schweinfest CW, Shull GE, Gawenis LR, Walker NM, Boyle KT, Soleimani M, Clarke LL. PAT-1 (Slc26a6) is the predominant apical membrane Cl-/HCO3- exchanger in the upper villous epithelium of the murine duodenum. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1079–G1088. doi: 10.1152/ajpgi.00354.2006. [DOI] [PubMed] [Google Scholar]
- 34.Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci USA. 2001;98:9425–9430. doi: 10.1073/pnas.141241098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aronson PS. Ion exchangers mediating Na+, HCO3 - and Cl- transport in the renal proximal tubule. J Nephrol. 2006;19 Suppl 9:S3–S10. [PubMed] [Google Scholar]
- 36.Niederer SA, Swietach P, Wilson DA, Smith NP, Vaughan-Jones RD. Measuring and modeling chloride-hydroxyl exchange in the Guinea-pig ventricular myocyte. Biophys J. 2008;94:2385–2403. doi: 10.1529/biophysj.107.118885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dart C, Vaughan-Jones RD. Na(+)-HCO3- symport in the sheep cardiac Purkinje fibre. J Physiol. 1992;451:365–385. doi: 10.1113/jphysiol.1992.sp019169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagadic-Gossmann D, Buckler KJ, Vaughan-Jones RD. Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. J Physiol. 1992;458:361–384. doi: 10.1113/jphysiol.1992.sp019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camilión de Hurtado MC, Alvarez BV, Pérez NG, Cingolani HE. Role of an electrogenic Na(+)-HCO3- cotransport in determining myocardial pHi after an increase in heart rate. Circ Res. 1996;79:698–704. doi: 10.1161/01.res.79.4.698. [DOI] [PubMed] [Google Scholar]
- 40.Aiello EA, Petroff MG, Mattiazzi AR, Cingolani HE. Evidence for an electrogenic Na+-HCO3- symport in rat cardiac myocytes. J Physiol. 1998;512(Pt 1):137–148. doi: 10.1111/j.1469-7793.1998.137bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero MF, Hediger MA, Boulpaep EL, Boron WF. Expression cloning and characterization of a renal electrogenic Na+/HCO3- cotransporter. Nature. 1997;387:409–413. doi: 10.1038/387409a0. [DOI] [PubMed] [Google Scholar]
- 42.Burnham CE, Amlal H, Wang Z, Shull GE, Soleimani M. Cloning and functional expression of a human kidney Na+: HCO3- cotransporter. J Biol Chem. 1997;272:19111–19114. doi: 10.1074/jbc.272.31.19111. [DOI] [PubMed] [Google Scholar]
- 43.Gross E, Kurtz I. Structural determinants and significance of regulation of electrogenic Na(+)-HCO(3)(-) cotransporter stoichiometry. Am J Physiol Renal Physiol. 2002;283:F876–F887. doi: 10.1152/ajprenal.00148.2002. [DOI] [PubMed] [Google Scholar]
- 44.Choi I, Romero MF, Khandoudi N, Bril A, Boron WF. Cloning and characterization of a human electrogenic Na+-HCO-3 cotransporter isoform (hhNBC) Am J Physiol. 1999;276:C576–C584. doi: 10.1152/ajpcell.1999.276.3.C576. [DOI] [PubMed] [Google Scholar]
- 45.Choi I, Aalkjaer C, Boulpaep EL, Boron WF. An electroneutral sodium/bicarbonate cotransporter NBCn1 and associated sodium channel. Nature. 2000;405:571–575. doi: 10.1038/35014615. [DOI] [PubMed] [Google Scholar]
- 46.Pushkin A, Abuladze N, Lee I, Newman D, Hwang J, Kurtz I. Cloning, tissue distribution, genomic organization, and functional characterization of NBC3, a new member of the sodium bicarbonate cotransporter family. J Biol Chem. 1999;274:16569–16575. doi: 10.1074/jbc.274.23.16569. [DOI] [PubMed] [Google Scholar]
- 47.Damkier HH, Nielsen S, Praetorius J. An anti-NH2-terminal antibody localizes NBCn1 to heart endothelia and skeletal and vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H172–H180. doi: 10.1152/ajpheart.00713.2005. [DOI] [PubMed] [Google Scholar]
- 48.Cooper DS, Lee HJ, Yang HS, Kippen J, Yun CC, Choi I. The electroneutral sodium/bicarbonate cotransporter containing an amino terminal 123-amino-acid cassette is expressed predominantly in the heart. J Biomed Sci. 2006;13:593–595. doi: 10.1007/s11373-006-9078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Qin X, Wang DK, Guo YM, Gill HS, Morris N, Parker MD, Chen LM, Boron WF. Effects of optional structural elements, including two alternative amino termini and a new splicing cassette IV, on the function of the sodium-bicarbonate cotransporter NBCn1 (SLC4A7) J Physiol. 2013;591:4983–5004. doi: 10.1113/jphysiol.2013.258673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garciarena CD, Ma YL, Swietach P, Huc L, Vaughan-Jones RD. Sarcolemmal localisation of Na+/H+ exchange and Na+-HCO3- co-transport influences the spatial regulation of intracellular pH in rat ventricular myocytes. J Physiol. 2013;591:2287–2306. doi: 10.1113/jphysiol.2012.249664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prasad V, Lorenz JN, Miller ML, Vairamani K, Nieman ML, Wang Y, Shull GE. Loss of NHE1 activity leads to reduced oxidative stress in heart and mitigates high-fat diet-induced myocardial stress. J Mol Cell Cardiol. 2013;65:33–42. doi: 10.1016/j.yjmcc.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 53.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pushkin A, Abuladze N, Newman D, Lee I, Xu G, Kurtz I. Cloning, characterization and chromosomal assignment of NBC4, a new member of the sodium bicarbonate cotransporter family. Biochim Biophys Acta. 2000;1493:215–218. doi: 10.1016/s0167-4781(00)00149-4. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto T, Shirayama T, Sakatani T, Takahashi T, Tanaka H, Takamatsu T, Spitzer KW, Matsubara H. Enhanced activity of ventricular Na+-HCO3- cotransport in pressure overload hypertrophy. Am J Physiol Heart Circ Physiol. 2007;293:H1254–H1264. doi: 10.1152/ajpheart.00964.2006. [DOI] [PubMed] [Google Scholar]
- 56.Grichtchenko II, Choi I, Zhong X, Bray-Ward P, Russell JM, Boron WF. Cloning, characterization, and chromosomal mapping of a human electroneutral Na(+)-driven Cl-HCO3 exchanger. J Biol Chem. 2001;276:8358–8363. doi: 10.1074/jbc.C000716200. [DOI] [PubMed] [Google Scholar]
- 57.Liu S, Piwnica-Worms D, Lieberman M. Intracellular pH regulation in cultured embryonic chick heart cells. Na(+)-dependent Cl-/HCO3- exchange. J Gen Physiol. 1990;96:1247–1269. doi: 10.1085/jgp.96.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun B, Vaughan-Jones RD, Kambayashi J. Two distinct HCO(-)(3)-dependent H(+) efflux pathways in human vascular endothelial cells. Am J Physiol. 1999;277:H28–H32. doi: 10.1152/ajpheart.1999.277.1.H28. [DOI] [PubMed] [Google Scholar]
- 59.Chiappe de Cingolani GE, Ennis IL, Morgan PE, Alvarez BV, Casey JR, Camilión de Hurtado MC. Involvement of AE3 isoform of Na(+)-independent Cl(-)/HCO(3)(-) exchanger in myocardial pH(i) recovery from intracellular alkalization. Life Sci. 2006;78:3018–3026. doi: 10.1016/j.lfs.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 60.Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- 61.Vaughan-Jones RD. Chloride activity and its control in skeletal and cardiac muscle. Philos Trans R Soc Lond B Biol Sci. 1982;299:537–548. doi: 10.1098/rstb.1982.0150. [DOI] [PubMed] [Google Scholar]
- 62.Gawenis LR, Bradford EM, Alper SL, Prasad V, Shull GE. AE2 Cl-/HCO3- exchanger is required for normal cAMP-stimulated anion secretion in murine proximal colon. Am J Physiol Gastrointest Liver Physiol. 2010;298:G493–G503. doi: 10.1152/ajpgi.00178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pérez NG, Alvarez BV, Camilión de Hurtado MC, Cingolani HE. pHi regulation in myocardium of the spontaneously hypertensive rat. Compensated enhanced activity of the Na(+)-H+ exchanger. Circ Res. 1995;77:1192–1200. doi: 10.1161/01.res.77.6.1192. [DOI] [PubMed] [Google Scholar]
- 64.Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H⁺-activated Na⁺ influx in the ventricular myocyte couples Ca²⁺-signalling to intracellular pH. J Mol Cell Cardiol. 2013;61:51–59. doi: 10.1016/j.yjmcc.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida H, Karmazyn M. Na(+)/H(+) exchange inhibition attenuates hypertrophy and heart failure in 1-wk postinfarction rat myocardium. Am J Physiol Heart Circ Physiol. 2000;278:H300–H304. doi: 10.1152/ajpheart.2000.278.1.H300. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res. 2008;103:891–899. doi: 10.1161/CIRCRESAHA.108.175141. [DOI] [PubMed] [Google Scholar]
- 67.Ennis IL, Alvarez BV, Camilión de Hurtado MC, Cingolani HE. Enalapril induces regression of cardiac hypertrophy and normalization of pHi regulatory mechanisms. Hypertension. 1998;31:961–967. doi: 10.1161/01.hyp.31.4.961. [DOI] [PubMed] [Google Scholar]
- 68.Camilión de Hurtado MC, Alvarez BV, Pérez NG, Ennis IL, Cingolani HE. Angiotensin II activates Na+-independent Cl--HCO3- exchange in ventricular myocardium. Circ Res. 1998;82:473–481. doi: 10.1161/01.res.82.4.473. [DOI] [PubMed] [Google Scholar]
- 69.Alvarez BV, Fujinaga J, Casey JR. Molecular basis for angiotensin II-induced increase of chloride/bicarbonate exchange in the myocardium. Circ Res. 2001;89:1246–1253. doi: 10.1161/hh2401.101907. [DOI] [PubMed] [Google Scholar]
- 70.Chiappe de Cingolani G, Morgan P, Mundiña-Weilenmann C, Casey J, Fujinaga J, Camilión de Hurtado M, Cingolani H. Hyperactivity and altered mRNA isoform expression of the Cl(-)/HCO(3)(-) anion-exchanger in the hypertrophied myocardium. Cardiovasc Res. 2001;51:71–79. doi: 10.1016/s0008-6363(01)00276-0. [DOI] [PubMed] [Google Scholar]
- 71.Cingolani HE, Chiappe GE, Ennis IL, Morgan PG, Alvarez BV, Casey JR, Dulce RA, Pérez NG, Camilión de Hurtado MC. Influence of Na+-independent Cl--HCO3- exchange on the slow force response to myocardial stretch. Circ Res. 2003;93:1082–1088. doi: 10.1161/01.RES.0000102408.25664.01. [DOI] [PubMed] [Google Scholar]
- 72.Alvarez BV, Johnson DE, Sowah D, Soliman D, Light PE, Xia Y, Karmazyn M, Casey JR. Carbonic anhydrase inhibition prevents and reverts cardiomyocyte hypertrophy. J Physiol. 2007;579:127–145. doi: 10.1113/jphysiol.2006.123638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lai ZF, Nishi K. Intracellular chloride activity increases in guinea pig ventricular muscle during simulated ischemia. Am J Physiol. 1998;275:H1613–H1619. doi: 10.1152/ajpheart.1998.275.5.H1613. [DOI] [PubMed] [Google Scholar]
- 74.Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber CS, Paul RJ, Shull GE. Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(-) cotransporter. Am J Physiol Heart Circ Physiol. 2002;283:H1846–H1855. doi: 10.1152/ajpheart.00083.2002. [DOI] [PubMed] [Google Scholar]
- 75.Frelin C, Chassande O, Lazdunski M. Biochemical characterization of the Na+/K+/Cl- co-transport in chick cardiac cells. Biochem Biophys Res Commun. 1986;134:326–331. doi: 10.1016/0006-291x(86)90566-8. [DOI] [PubMed] [Google Scholar]
- 76.Lenart B, Kintner DB, Shull GE, Sun D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004;24:9585–9597. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Prabhakar R, Boivin GP, Grupp IL, Hoit B, Arteaga G, Solaro RJ, Wieczorek DF. A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33:1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 78.Al Moamen NJ, Prasad V, Bodi I, Miller ML, Neiman ML, Lasko VM, Alper SL, Wieczorek DF, Lorenz JN, Shull GE. Loss of the AE3 anion exchanger in a hypertrophic cardiomyopathy model causes rapid decompensation and heart failure. J Mol Cell Cardiol. 2011;50:137–146. doi: 10.1016/j.yjmcc.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prasad V, Lorenz JN, Lasko VM, Nieman ML, Al Moamen NJ, Shull GE. Loss of the AE3 Cl(-)/HCO(-) 3 exchanger in mice affects rate-dependent inotropy and stress-related AKT signaling in heart. Front Physiol. 2013;4:399. doi: 10.3389/fphys.2013.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim KH, Lee MG, Naruse S, et al. A molecular mechanism for aberrant CFTR-dependent HCO(3)(-) transport in cystic fibrosis. EMBO J. 2002;21:5662–5672. doi: 10.1093/emboj/cdf580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shcheynikov N, Wang Y, Park M, Ko SB, Dorwart M, Naruse S, Thomas PJ, Muallem S. Coupling modes and stoichiometry of Cl-/HCO3- exchange by slc26a3 and slc26a6. J Gen Physiol. 2006;127:511–524. doi: 10.1085/jgp.200509392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, Vandorpe DH, Alper SL. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J Biol Chem. 2005;280:8564–8580. doi: 10.1074/jbc.M411703200. [DOI] [PubMed] [Google Scholar]
- 83.Ohana E, Shcheynikov N, Yang D, So I, Muallem S. Determinants of coupled transport and uncoupled current by the electrogenic SLC26 transporters. J Gen Physiol. 2011;137:239–251. doi: 10.1085/jgp.201010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamamoto T, Swietach P, Rossini A, Loh SH, Vaughan-Jones RD, Spitzer KW. Functional diversity of electrogenic Na+-HCO3- cotransport in ventricular myocytes from rat, rabbit and guinea pig. J Physiol. 2005;562:455–475. doi: 10.1113/jphysiol.2004.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vaughan-Jones RD, Villafuerte FC, Swietach P, Yamamoto T, Rossini A, Spitzer KW. pH-Regulated Na(+) influx into the mammalian ventricular myocyte: the relative role of Na(+)-H(+) exchange and Na(+)-HCO Co-transport. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S134–S140. doi: 10.1111/j.1540-8167.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- 86.Grace AA, Kirschenlohr HL, Metcalfe JC, Smith GA, Weissberg PL, Cragoe EJ, Vandenberg JI. Regulation of intracellular pH in the perfused heart by external HCO3- and Na(+)-H+ exchange. Am J Physiol. 1993;265:H289–H298. doi: 10.1152/ajpheart.1993.265.1.H289. [DOI] [PubMed] [Google Scholar]
- 87.Vandenberg JI, Metcalfe JC, Grace AA. Mechanisms of pHi recovery after global ischemia in the perfused heart. Circ Res. 1993;72:993–1003. doi: 10.1161/01.res.72.5.993. [DOI] [PubMed] [Google Scholar]
- 88.Khandoudi N, Albadine J, Robert P, Krief S, Berrebi-Bertrand I, Martin X, Bevensee MO, Boron WF, Bril A. Inhibition of the cardiac electrogenic sodium bicarbonate cotransporter reduces ischemic injury. Cardiovasc Res. 2001;52:387–396. doi: 10.1016/s0008-6363(01)00430-8. [DOI] [PubMed] [Google Scholar]
- 89.Schäfer C, Ladilov YV, Siegmund B, Piper HM. Importance of bicarbonate transport for protection of cardiomyocytes against reoxygenation injury. Am J Physiol Heart Circ Physiol. 2000;278:H1457–H1463. doi: 10.1152/ajpheart.2000.278.5.H1457. [DOI] [PubMed] [Google Scholar]
- 90.Schäfer C, Ladilov YV, Schäfer M, Piper HM. Inhibition of NHE protects reoxygenated cardiomyocytes independently of anoxic Ca(2+) overload and acidosis. Am J Physiol Heart Circ Physiol. 2000;279:H2143–H2150. doi: 10.1152/ajpheart.2000.279.5.H2143. [DOI] [PubMed] [Google Scholar]
- 91.Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM, Ottersen OP. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–1337. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 92.Petrecca K, Atanasiu R, Grinstein S, Orlowski J, Shrier A. Subcellular localization of the Na+/H+ exchanger NHE1 in rat myocardium. Am J Physiol. 1999;276:H709–H717. doi: 10.1152/ajpheart.1999.276.2.H709. [DOI] [PubMed] [Google Scholar]
- 93.Aronsen JM, Swift F, Sejersted OM. Cardiac sodium transport and excitation-contraction coupling. J Mol Cell Cardiol. 2013;61:11–19. doi: 10.1016/j.yjmcc.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 94.Villa-Abrille MC, Petroff MG, Aiello EA. The electrogenic Na+/HCO3- cotransport modulates resting membrane potential and action potential duration in cat ventricular myocytes. J Physiol. 2007;578:819–829. doi: 10.1113/jphysiol.2006.120170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saegusa N, Moorhouse E, Vaughan-Jones RD, Spitzer KW. Influence of pH on Ca²⁺ current and its control of electrical and Ca²⁺ signaling in ventricular myocytes. J Gen Physiol. 2011;138:537–559. doi: 10.1085/jgp.201110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sandmann S, Yu M, Kaschina E, Blume A, Bouzinova E, Aalkjaer C, Unger T. Differential effects of angiotensin AT1 and AT2 receptors on the expression, translation and function of the Na+-H+ exchanger and Na+-HCO3- symporter in the rat heart after myocardial infarction. J Am Coll Cardiol. 2001;37:2154–2165. doi: 10.1016/s0735-1097(01)01287-6. [DOI] [PubMed] [Google Scholar]
- 97.Kohout TA, Rogers TB. Angiotensin II activates the Na+/HCO3- symport through a phosphoinositide-independent mechanism in cardiac cells. J Biol Chem. 1995;270:20432–20438. doi: 10.1074/jbc.270.35.20432. [DOI] [PubMed] [Google Scholar]
- 98.De Giusti VC, Garciarena CD, Aiello EA. Role of reactive oxygen species (ROS) in angiotensin II-induced stimulation of the cardiac Na+/HCO3- cotransport. J Mol Cell Cardiol. 2009;47:716–722. doi: 10.1016/j.yjmcc.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 99.De Giusti VC, Orlowski A, Aiello EA. Angiotensin II inhibits the electrogenic Na+/HCO3- cotransport of cat cardiac myocytes. J Mol Cell Cardiol. 2010;49:812–818. doi: 10.1016/j.yjmcc.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 100.Orlowski A, Ciancio MC, Caldiz CI, De Giusti VC, Aiello EA. Reduced sarcolemmal expression and function of the NBCe1 isoform of the Na⁺-HCO₃⁻ cotransporter in hypertrophied cardiomyocytes of spontaneously hypertensive rats: role of the renin-angiotensin system. Cardiovasc Res. 2014;101:211–219. doi: 10.1093/cvr/cvt255. [DOI] [PubMed] [Google Scholar]
- 101.Lebeche D, Kaprielian R, Hajjar R. Modulation of action potential duration on myocyte hypertrophic pathways. J Mol Cell Cardiol. 2006;40:725–735. doi: 10.1016/j.yjmcc.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 102.Carmeliet E. Action potential duration, rate of stimulation, and intracellular sodium. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S2–S7. doi: 10.1111/j.1540-8167.2006.00378.x. [DOI] [PubMed] [Google Scholar]
- 103.De Giusti VC, Orlowski A, Villa-Abrille MC, de Cingolani GE, Casey JR, Alvarez BV, Aiello EA. Antibodies against the cardiac sodium/bicarbonate co-transporter (NBCe1) as pharmacological tools. Br J Pharmacol. 2011;164:1976–1989. doi: 10.1111/j.1476-5381.2011.01496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yan S, Chen Y, Dong M, Song W, Belcher SM, Wang HS. Bisphenol A and 17β-estradiol promote arrhythmia in the female heart via alteration of calcium handling. PLoS One. 2011;6:e25455. doi: 10.1371/journal.pone.0025455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mason MJ, Smith JD, Garcia-Soto JJ, Grinstein S. Internal pH-sensitive site couples Cl-(-)HCO3- exchange to Na+-H+ antiport in lymphocytes. Am J Physiol. 1989;256:C428–C433. doi: 10.1152/ajpcell.1989.256.2.C428. [DOI] [PubMed] [Google Scholar]
- 106.Jiang L, Chernova MN, Alper SL. Secondary regulatory volume increase conferred on Xenopus oocytes by expression of AE2 anion exchanger. Am J Physiol. 1997;272:C191–C202. doi: 10.1152/ajpcell.1997.272.1.C191. [DOI] [PubMed] [Google Scholar]
- 107.Belcher SM, Chen Y, Yan S, Wang HS. Rapid estrogen receptor-mediated mechanisms determine the sexually dimorphic sensitivity of ventricular myocytes to 17β-estradiol and the environmental endocrine disruptor bisphenol A. Endocrinology. 2012;153:712–720. doi: 10.1210/en.2011-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]