

Abstract
Obesity and insulin resistance are closely associated with the development of low-grade inflammation. Interleukin 6 (IL-6) is linked to obesity-associated inflammation, however its role in this context remains controversial. Here, we show that mice with inactivated Il6ra gene in myeloid cells (Il6raΔmyel) displayed exaggerated deterioration of glucose homeostasis upon diet-induced obesity due to enhanced insulin resistance. Insulin target tissues showed increased inflammation and a shift in macrophage polarization. IL-6 induced IL-4-receptor expression and augmented the response to IL-4 in macrophages in a cell-autonomous manner. Il6raΔmyel mice were resistant to IL-4-mediated alternative macrophage polarization and exhibited increased susceptibility to LPS-induced endotoxemia. These results reveal IL-6 signaling as an important determinant for alternative macrophage-activation and assign IL-6 an unexpected homeostatic role to limit inflammation.
During the last decades, western societies have experienced a lifestyle shift towards reduced physical activity and increased consumption of energy dense food, changes that have led to a dramatic increase of overweight and obese individuals1. Obesity is associated with a chronic, low-grade inflammatory state, reflected by immune cell infiltration and activation in adipose tissue as well as systemic elevation of pro-inflammatory cytokines2–4. These cytokines in turn activate intracellular stress-signaling cascades, namely transcription factor NF-kB and JNK-associated pathways, which subsequently inhibit insulin action in target tissues such as adipose tissue, liver, skeletal muscle, and even in the central nervous system5–7. Prolonged inhibition of insulin signaling eventually leads to the development of insulin resistance and ultimately progression to overt type 2 diabetes mellitus (T2DM)8,9.
Interleukin 6 (IL-6) is a cytokine linked to obesity and insulin resistance, however it is still heavily debated whether IL-6 plays a harmful or protective role in this context10. IL-6 increases in the circulation with the degree of obesity11 and acute IL-6 infusion impairs insulin sensitivity in mice12. However, IL-6 deficiency leads to adult onset obesity13 and hepatic disruption of IL-6 signaling causes insulin resistance14.
Because macrophages are central mediators of obesity-associated inflammation, we aimed to directly investigate IL-6 signaling in these cells. In the current study, we found that IL-6 primes macrophages for IL-4-dependent M2 polarization by inducing IL-4 receptor expression. In mice with conditional inactivation of the Il6ra-gene in myeloid cells, we observed an increased propensity to develop obesity-induced inflammation and glucose intolerance as well as an exaggerated response to LPS-induced endotoxemia. These results identify IL-6 as an important regulator of alternative macrophage activation during inflammatory conditions such as obesity and endotoxemia.
Results
IL-6 signaling in myeloid cells regulates glucose homeostasis
To investigate the role of IL-6 signaling in macrophages in obesity, we conditionally inactivated the Il6ra gene in myeloid cells in mice. To this end, we generated mice heterozygous for the LysM-Cre transgene, which is specifically expressed in myeloid lineage cells15, and homozygous for the Il6ra-loxP-flanked allele (Il6rafl/fl, LysM-CreTg/wt; called Il6raΔmyel throughout). Littermates not carrying the LysM-Cre transgene (Il6rafl/fl) served as controls. To verify efficient disruption of IL-6 signaling in these mice, we isolated bone marrow-derived macrophages (BMDM) from control and Il6raΔmyel mice and stimulated them with IL-6. Immunoblot analysis revealed robust phosphorylation of STAT3 (p-STAT3) in BMDM derived from control mice upon IL-6-stimulation, whereas p-STAT3 was undetectable in protein lysates of IL-6-stimulated BMDM derived from Il6raΔmyel mice (called Il6ra−/− throughout) (Supplementary Fig. 1a).
Next, we exposed control and Il6raΔmyel mice to either normal chow diet (NCD) or high fat diet (HFD) and monitored body weight from 4 until 19 weeks of age. HFD-feeding induced a pronounced weight gain when compared with NCD, however body weight was similar between both genotypes (Fig. 1a). In line with unaltered obesity development, NCD- and HFD control and Il6raΔmyel mice exhibited similar body composition, fat pad weight and, as an indirect measure of body fat content, serum leptin concentration (Supplementary Fig. 1b–d). Moreover, Il6raΔmyel mice showed no difference in oxygen consumption or daily caloric intake compared to control mice (Supplementary Fig. 1e, f). To analyze possible alterations in glucose homeostasis, we subjected control and Il6raΔmyel mice to glucose tolerance tests (GTT). Whereas NCD-fed control and Il6raΔmyel mice had comparable blood glucose concentrations during the GTT (Supplementary Fig. 2a), HFD Il6raΔmyel mice were significantly more glucose intolerant than HFD control animals (Fig. 1b). Similarly, while NCD-fed animals showed no alterations in insulin tolerance tests between genotypes (ITT) (Supplementary Fig. 2b), HFD Il6raΔmyel mice were more insulin resistant compared to HFD control mice (Fig. 1c). Moreover, a deterioration of glucose homeostasis in HFD Il6raΔmyel compared to control mice was also reflected by increased fasting serum insulin concentrations and an elevated homeostatic model assessment index of insulin resistance (HOMA-IR) in HFD Il6raΔmyel compared to control mice (Fig. 1d, e).
Figure 1. IL-6 signaling in myeloid cells regulates glucose homeostasis.

(a) Body weight of control (Ctrl) and Il6raΔmyel mice (n=12 Ctrl NCD; n=32 Ctrl HFD; n=16 Il6raΔmyel NCD; n=18 Il6raΔmyel HFD). (b) Glucose tolerance tests (GTT; n=32 vs 31; *p≤0.05; **p≤0.01) or (c) insulin tolerance tests (ITT; n=27 vs 17; *p≤0.05; **p≤0.01; Data are expressed as % of initial blood glucose) of HFD Ctrl and Il6raΔmyel mice. (d) Fasted serum insulin concentrations (n=9 in each group; *p≤0.05) and (e) homeostatic model assessment of insulin resistance (HOMA-IR; n=9 in each group; *p≤0.01)) indices were determined in HFD Ctrl and Il6raΔmyel mice. (f) Glucose infusion rate (GIR; n=8 vs 7; *p≤0.001) and (g) glucose uptake rate (GUR; n=8 vs 7; *p≤0.05) in skeletal muscle (SM), brown adipose tissue (BAT) and white adipose tissue (WAT; p=0.06) during euglycemic-hyperinsulinemic clamp analyses of HFD Ctrl and Il6raΔmyel mice. (h) Suppression of free fatty acid (FFA) release in the serum of HFD Ctrl and Il6raΔmyel mice during clamp analyses (n=8 vs 7; *p≤0.05). (i) qRT-PCR analyses of Lipe expression in WAT from Ctrl and Il6raΔmyel mice at the end of the clamp analyses (n=8 vs 7; *p≤0.01; Data are expressed as % of Ctrl). (j) Suppression of hepatic glucose production (HGP) during clamp analyses of HFD Ctrl and Il6raΔmyel mice (n=8 vs 7; *p≤0.05). (k) qRT-PCR analyses of G6pc expression in livers from Ctrl and Il6raΔmyel mice at the end of the clamp analyses (n=8 vs 7; *p≤0.05; Data are expressed as % of Ctrl). (Values are expressed as mean ± sem).
Insulin exerts its blood glucose-lowering effects by promoting glucose uptake in skeletal muscle and adipose tissue and via the suppression of de novo gluconeogenesis, predominantly in liver16. To determine which of these processes is impaired in HFD Il6raΔmyel mice, we performed euglycemic-hyperinsulinemic clamps. Il6raΔmyel mice had a 50% reduction in glucose infusion rate during the steady-state, further supporting the notion that these animals exhibit a profound degree of insulin resistance (Fig. 1f and Supplementary Fig. 2c, d). Moreover, we determined insulin-stimulated glucose uptake in skeletal muscle (SM), brown adipose tissue (BAT) and white adipose tissue (WAT). While we observed no difference in insulin-induced glucose uptake in the skeletal muscle between HFD control and HFD Il6raΔmyel mice, insulin-stimulated glucose uptake was reduced by 25% in WAT and 40% in BAT of HFD Il6raΔmyel mice compared to HFD controls (Fig. 1g). Moreover, the ability of insulin to suppress lipolysis and limit the expression of hormone sensitive lipase (Lipe) in WAT was significantly impaired in Il6raΔmyel mice compared to controls (Fig. 1h, i), indicating insulin resistance in WAT and BAT of Il6raΔmyel mice on HFD. Finally, insulin-mediated suppression of hepatic glucose production (HGP) in HFD Il6raΔmyel mice was reduced by 50% compared to HFD control mice (Fig. 1j). Consistent with this finding, expression of glucose 6 phosphatase (G6pc), a rate-limiting enzyme of gluconeogenesis, was significantly increased in livers of HFD Il6raΔmyel mice compared to control mice during clamps and in the fasted state (Fig. 1k and Supplementary Fig. 2e). Furthermore, hepatic insulin resistance was accompanied by increased liver triglyceride content and increased triglyceride concentrations in serum of 6 hour-fasted, HFD Il6raΔmyel mice (Supplementary Fig. 2f, g). Thus, IL-6 signaling in myeloid cells limits obesity-associated deterioration of glucose metabolism by promoting insulin sensitivity in WAT, BAT and the liver.
HFD Il6raΔmyel mice have increased systemic inflammation
Obesity induces a pro-inflammatory state and macrophage accumulation in the adipose tissue2,3. To investigate possible inflammatory changes in HFD Il6raΔmyel, we performed gene expression analysis in WAT by quantitative real-time PCR (qRT-PCR). These analyses revealed increased expression of genes typically linked to the development of insulin resistance, namely tumor necrosis factor (Tnf), interleukin-1β (Il1b), Il6, Il12a and inducible nitric oxide synthase (Nos2) in WAT of HFD Il6raΔmyel mice compared with HFD controls (Fig. 2a). We also observed increased expression of the M1 macrophage marker mineralocorticoid receptor (Nr3c2) together with a reduction of the IL-4R α chain (Il4ra), mannose receptor 1 (Mrc1) and resistin-like α (Retnla) expression, all M2 macrophage-associated genes (Fig. 2a). Additionally, immunohistochemical analysis of F4/80- and MAC-2-positive cells, revealed a 2-fold increase of crown-like structures (CLS) in the adipose tissue of HFD Il6raΔmyel compared with HFD control mice, indicating increased numbers of activated macrophages in these animals (Fig. 2b and Supplementary Fig. 3a). Similarly, expression of pro-inflammatory genes and M1 polarization marker genes was increased in BAT of HFD Il6raΔmyel mice, while also expression of M2 marker genes was significantly reduced (Fig. 2c). Because HFD Il6raΔmyel mice displayed marked hepatic insulin resistance, we next assessed inflammatory gene expression in liver. Consistent with the observed inflammatory changes in WAT and BAT, livers of HFD Il6raΔmyel mice exhibited elevated expression of Il6, Il12a, Ccl3, Nr3c2 and Nos2 (Fig. 2d). We did not observe alterations in expression of pro-inflammatory cytokines or M1-M2 polarization marker genes in any of the analyzed tissues obtained from NCD-fed Il6raΔmyel mice (Supplementary Fig. 3b–d). Moreover, we observed similar changes in pro-inflammatory and M2 polarization marker gene expression in WAT and liver of HFD conventional Il6−/− mice (Supplementary Fig. 4a, b), which exhibit an exaggerated propensity to diet-induced insulin resistance17. No alteration of inflammatory marker gene expression in WAT, BAT or liver was observed in HFD LysM-CreTg/wt control mice when compared to HFD wild-type mice, excluding the possibility that the observed alterations in Il6raΔmyel mice might be dependent on expression of Cre alone (Supplementary Fig. 4c–e).
Figure 2. HFD Il6raΔmyel mice have increased systemic inflammation.

(a) qRT-PCR analyses of white adipose tissue (WAT) from HFD control (Ctrl) and Il6raΔmyel mice (n=8–9 per genotype; *p≤0.05; **p≤0.01; ***p≤0.001; Data are expressed as % of Ctrl). (b) Representative immunohistochemical staining of F4/80-positive cells in WAT from HFD Ctrl and Il6raΔmyel mice and quantification of F4/80-positive crown-like structures (CLS) in WAT from HFD Ctrl and Il6raΔmyel mice (n=7 per genotype;; *p≤0.05; Data are expressed as % CLS of adipocytes; scale bars represent 100μm). (c) qRT-PCR analyses of brown adipose tissue (BAT) from HFD Ctrl and Il6raΔmyel mice (n=8–9 per genotype; *p≤0.05; **p≤0.01; Data are expressed as % of Ctrl). (d) qRT-PCR analyses of liver from HFD Ctrl and Il6raΔmyel mice (n=8–9 per genotype; *p≤0.05; **p≤0.01; Data are expressed as % of Ctrl). (e) C-Jun N-terminal kinase (JNK) activity in WAT, BAT and liver of HFD Ctrl and Il6raΔmyel mice was measured by performing immunoprecipitation (IP) of p-JNK and after subsequent in vitro phosphorylation assay, recombinant c-Jun was detected by immunoblot (IB). Total JNK and calnexin loading was used as input control; Representative immunoblots are shown (upper panel); Quantification of JNK activity in WAT, BAT and liver of HFD Ctrl and Il6raΔmyel mice (lower panel; n=5–9 per genotype; *p≤0.05; **p≤0.01; ***p≤0.001; Data are expressed as % of Ctrl). (Values are expressed as mean ± sem;).
To further assess how changes in pro-inflammatory gene expression might promote insulin resistance, we assessed JNK activation, a critical regulator of obesity-associated insulin resistance18, in WAT, BAT, and liver of HFD Il6raΔmyel and HFD control mice. Immunoprecipitation of phosphorylated JNK and subsequent in vitro kinase assay with recombinant c-Jun fusion protein revealed a 6–10-fold increase in JNK activation in WAT, BAT, and liver of HFD Il6raΔmyel mice compared with HFD controls (Fig. 2e). These experiments indicate that IL-6 acts in myeloid lineage cells to promote M2 polarization and to limit pro-inflammatory gene expression, activation of JNK and insulin resistance in WAT, BAT and liver of obese animals.
IL-6 signaling promotes IL-4R expression in macrophages
Because HFD Il6raΔmyel mice exhibited a shift towards M1 macrophage polarization in WAT, BAT and liver, we next investigated the underlying macrophage-autonomous changes that depend on IL-6 signaling. To this end, we stimulated control and Il6ra−/− BMDM with IL-6 and performed gene expression analysis via microarray hybridization. Of 35518 annotated transcripts, 1487 were differentially regulated between IL-6-stimulated control and Il6ra−/− macrophages (p≤0.05). Ingenuity pathway analyses of gene-expression patterns revealed differential regulation of Nos2, Il10, Il6 and Ppar -signaling, among others (Supplementary Fig. 4f). Applying a cut-off at ± 1.25 fold-change yielded 91 regulated transcripts, 31 up- and 60 down-regulated in Il6ra−/− cells compared to controls (Fig. 3a). Along with Il6ra, one of the transcripts whose expression was most prominently reduced in the absence of functional IL-6 signaling was the Il4ra gene (Fig. 3a). To verify these findings, we performed qRT-PCR analyses and observed that IL-6-stimulation increased Il4ra mRNA expression in control BMDM (Fig. 3b and Supplementary Fig. 4g) and that this response was blunted in Il6ra-deficient cells, which exhibited a ~95% reduction of Il6ra mRNA expression (Fig. 3b). We next analyzed IL-6-stimulated control BMDM by flow cytometry and observed increased IL-4Rα abundance compared to unstimulated cells (Fig. 3c and Supplementary Fig. 4h).
Figure 3. IL-6 signaling promotes IL-4R expression in macrophages.

(a) Heatmap of differentially regulated transcripts between IL-6-stimulated (4 hours) bone marrow-derived macrophages (BMDM) either from control (Ctrl) or Il6raΔmyel (Il6ra−/−) mice. (n=4 per group; fold-change cut off ± 1.25; p≤0.05). (b) qRT-PCR analyses of Il6ra and Il4ra expression in untreated (NT) and IL-6-stimulated (12 hour) Ctrl and Il6ra−/− BMDM (n=6 per group; *p≤0.001 vs Ctrl; **p≤0.001 vs NT; Data are expressed as % of NT Ctrl). (c) FACS analysis of untreated (NT) and IL-6-stimulated (24 hour) Ctrl BMDM (n=5; *p≤0.001 vs NT). (d) Ctrl BMDM were left untreated (NT) or stimulated with IL-6 (12 hours) in the absence (IgG) or presence of an IL-10-neutralizing antibody (αIL-10) and qRT-PCR analysis of Il4ra expression was performed (Data is representative of two independent experiments; Data are expressed as % of NT Ctrl). (e) Immunoblot of Ctrl BMDM that were left untreated (NT) or stimulated with IL-6 (12 hours) in the presence of a control (IgG) or IL-10-neutralizing antibody (αIL-10) (n=3). (f) qRT-PCR analyses of Stat3 and Il6ra expression in siRNA-transfected Ctrl BMDM (n=3; *p≤0.001; Data are expressed as % of Ctrl siRNA). (g) qRT-PCR analyses of Il4ra expression in untreated (NT) or IL-6 stimulated (4h) siRNA-transfected Ctrl BMDM (n=3; *p≤0.001 vs IL-6 stimulated Ctrl siRNA; Data are expressed as % of NT Ctrl siRNA). (Values are expressed as mean ± sem)
Because IL-10 can directly induce Il4ra gene expression in macrophages19 and IL-6 can stimulate IL-10 expression and release, we next investigated whether the IL-6-mediated modulatory effects on Il4ra expression were mediated via IL-10 release. Gene expression analyses in IL-6-stimulated macrophages showed a robust increase of Il10 mRNA in control macrophages (Supplementary Fig. 4i). While Il6ra−/− BMDM exhibited slightly decreased levels of Il10 mRNA in the basal state, the IL-6-induced upregulation of Il10 was blunted in these cells compared to control cells (Supplementary Fig. 4i), suggesting that the observed IL-6-mediated increase of Il4ra expression could be dependent on autocrine IL-10 signaling. To further pursue this possible mode of action, we next treated control BMDM IL-6 in the presence or absence of a neutralizing antibody directed against IL-10, which efficiently blocked IL-10-mediated STAT3 phosphorylation (Supplementary Fig. 4j). However, IL-6 induced Il4ra mRNA and IL-4Rα protein expression in IL10-blocked or control IgG-blocked BMDM to the same extent, indicating that IL-6-dependent upregulation of IL-4Rα expression occurred independently of IL-10 expression and signaling (Fig. 3d, e).
We next tested if IL-6-mediated IL-4Rα expression is dependent on STAT3. siRNA-mediated knockdown of Stat3 and, as a positive control, Il6ra reduced expression of their respective target genes by ~80% when compared to control siRNA-transfected BMDM (Fig. 3f). Knockdown of either Stat3 or Il6ra in BMDM potently blunted the IL-6-stimulated induction of Il4ra mRNA by ~70% and ~50%, respectively (Fig. 3g). Socs3 expression showed a similar reduction as Il4ra upon Stat3 or Il6ra knockdown (Supplementary Fig. 4k).
In summary, we demonstrate that IL-6 signaling promotes IL-4Rα expression in macrophages in an IL-10-independent manner and that the transcription factor STAT3 is essential for mediating this effect.
STAT3 binds to distinct motifs in the Il4ra promoter
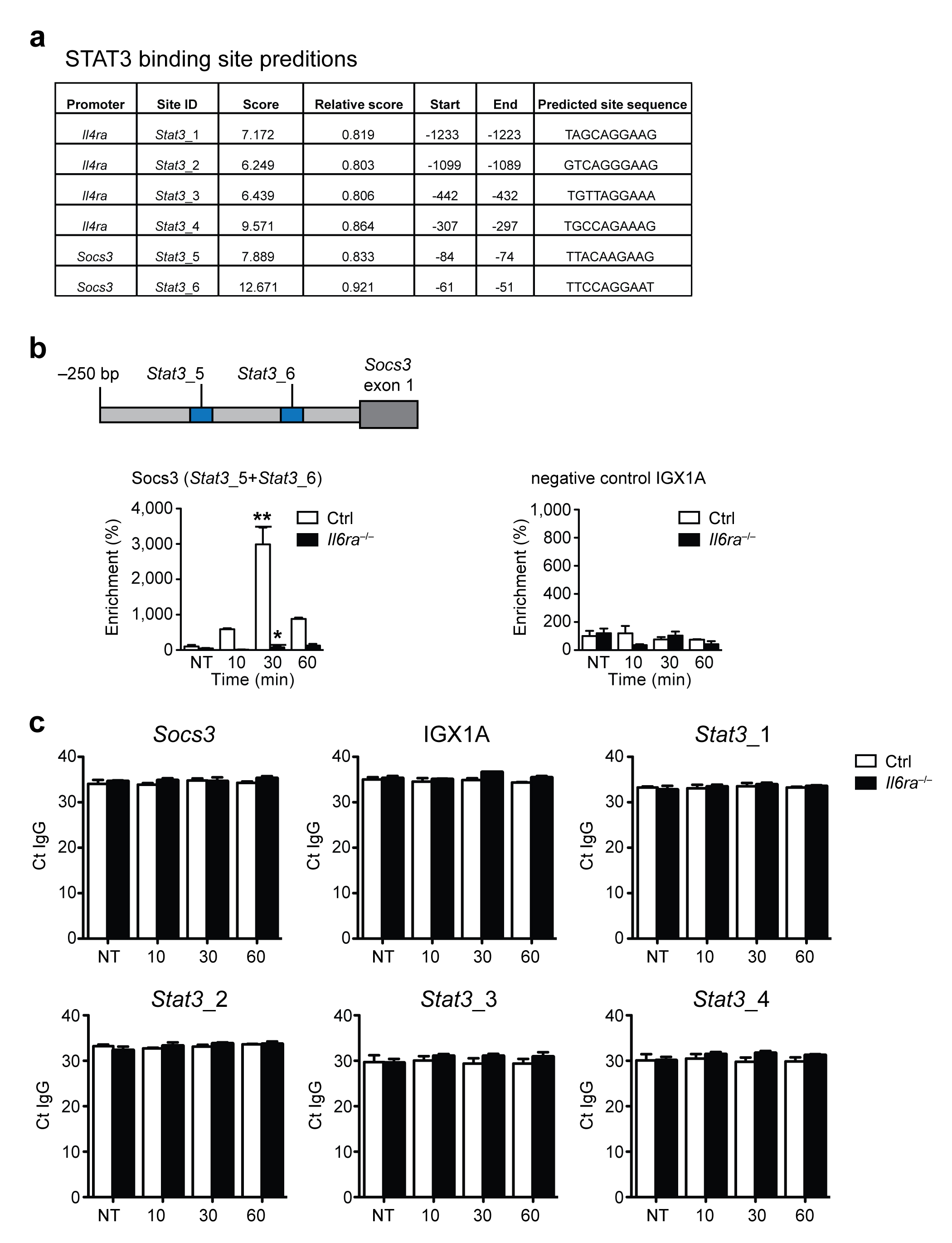
To further investigate the role of IL-6-STAT3 signaling in the regulation of IL-4Rα expression, we looked for STAT3-binding sites in the Il4ra promoter. JASPAR analysis20 revealed four putative STAT3-sites located within 1.3 kb upstream of the first exon of the Il4ra promoter and two putative STAT3-sites in the Socs3 promoter (Supplementary Fig. 5a). To determine which of these sites are necessary for IL-6-STAT3-mediated IL-4Rα expression, we cloned different segments of the Il4ra promoter upstream of Il4ra exon 1 in a firefly luciferase reporter and transfected these constructs into immortalized macrophages21 (Fig. 4a). Only the full length fragment, containing the two distal sites (Stat3_1 and Stat3_2), resulted in robust luciferase expression and this expression was substantially increased after stimulation with IL-6 (Fig. 4a). Deletion of these sites almost completely abolished luciferase expression, whereas deletion of the two proximal STAT3-binding sites (Stat3_3 and Stat3_4) didn’t further reduce luciferase activity (Fig. 4a), indicating that those sites appear to be dispensable for IL-6-dependent IL-4Rα expression.
Figure 4. IL-6-induced STAT3 binds to distinct motifs in the Il4ra promoter.

(a) Luciferase activity of the indicated reporter constructs in untreated (NT) or IL-6-treated (12h) immortalized macrophages. (n=3; *p≤0.001 vs NT −1,300 bp fragment). (b) qRT-PCR analyses of ChIP assay showing occupancy of p-STAT3 over the Il4ra promoter in control (Ctrl) and Il6ra−/− BMDM stimulated with IL-6 for the indicated time points (n=3 vs 3; *p≤0.001 vs Ctrl; **p≤0.05, ***p≤0.001 vs NT; Data are expressed as % of NT Ctrl). (Values are expressed as mean ± sem)
To determine whether STAT3 regulated Il4ra expression directly, we performed chromatin immunoprecipitation (ChIP) analyses with a p-STAT3 antibody. Transient but robust Socs3 promoter occupation was observed following stimulation with IL-6 in control but not in Il6ra−/− BMDM (Supplementary Fig. 5b, c). Additionally, neither control nor Il6ra−/− macrophages showed enrichment of a non-open reading frame region upon p-STAT3 ChIP (negative control IGX1A) (Supplementary Fig. 5b, c). Thus, we tested whether the identified putative STAT3-binding sites showed specific enrichment after exposure of BMDM to IL-6. In line with the luciferase expression pattern, the two distal (Stat3_1, Stat3_2) but not the two proximal (Stat3_3, Stat3_4) STAT3-sites in the Il4ra promotor were specifically enriched upon p-STAT3 ChIP after IL-6 stimulation in control BMDM (Fig. 4b and Supplementary Fig. 5c). No significant enrichment at these sites was detected in Il6ra−/− BMDM (Fig. 4b and Supplementary Fig. 5c). Thus, IL-6 induces STAT3-binding in the Il4ra-promoter.
IL-6 signaling in myeloid cells augments the IL-4 response
We next investigated if IL-4Rα upregulation renders macrophages more sensitive to IL-4 stimulation. Immunoblot analyses showed that IL-6 stimulation potently increased p-STAT3 and IL-4Rα protein expression in control but not in Il6ra−/− BMDM (Fig. 5a). When we assessed IL-4-stimulated phosphorylation of STAT6, pre-incubation with IL-6 augmented the IL-4-response in control BMDM, an effect that was abrogated in Il6ra−/− BMDM (Fig. 5a). This suggests that IL-6 not only promotes IL-4Rα expression in macrophages but also augments the IL-4-mediated phosphorylation of STAT6.
Figure 5. IL-6 signaling in myeloid cells augments the IL-4 response.

(a) Immunoblot of control (Ctrl) and Il6ra−/− BMDM that were left untreated or pre-incubated with IL-6 (12 hours) before stimulation with IL-4 (30 minutes) (Representative immunoblot of 3 independent experiments shown). (b) qRT-PCR analyses of Ctrl and Il6ra−/− BMDM that were stimulated with IL-6 (12 hours) and subsequently exposed to IL-4 or IL-4 + IL-6 for 24 hours (n=6 vs 6; *p≤0.01 vs Ctrl; **p≤0.001 vs NT; Data are expressed as % of NT Ctrl). (c) FACS analysis of CD206/MRC1 and ARG1 expression in Ctrl BMDM that were left untreated or stimulated with IL-6, IL-4 or IL-4 + IL-6 (n=5; *p≤0.05 vs IL-6 + IL-4; **p≤0.001). (d) FACS analysis of CD206/MRC1 expression in WAT and BAT (n=5; *p≤0.05 vs IL-6+IL-4; **p≤0.01 ***p≤0.001) or (e) blood and peritoneum of Ctrl mice that were treated with IL-6, IL-4 or IL-4 + IL-6 (n=5; *p≤0.05 vs IL-6+IL-4; **p≤0.01). (f) GTT area under the curve (AUC) of HFD Ctrl and Il6raΔmyel mice before (NT) and after a 4-week treatment period with IL-4 (n=15 vs 17; *p≤0.05 vs Ctrl; **p≤0.01). (g) qRT-PCR analyses of HFD Ctrl and Il6raΔmyel mice treated with IL-4 for 4 weeks (n=7–8 vs 7; *p≤0.05; **p≤0.01 ***p≤0.001; Data are expressed as % of NT Ctrl). (h) Immunohistochemical staining of CD206/MRC1 in livers from HFD Ctrl and Il6raΔmyel mice that were left untreated (NT) or treated with IL-4 for 1 week (Representative images of 3 per group shown; scale bars represent 50μm).
Because IL-4-STAT6 regulates macrophage polarization by inducing M2-associated genes such as mannose receptor 1 (Mrc1), arginase 1 (Arg1), Il10 and resistin-like alpha (Retnla)22, we asked whether IL-6 modulates the expression of M2 genes in macrophages. IL-6 induced expression of Mrc1 and Arg1 in control but not Il6ra−/− BMDM, whereas the expression of suppressor of cytokine signaling 1 (Socs1) or Retnla was not modulated by IL-6 (Supplementary Fig. 6a). Treatment with IL-4 alone induced similar expression of Socs1, Mrc1, Arg1, Il10 and Retnla in control and Il6ra−/− BMDM treated (Fig. 5b), while IL-6 + IL-4 synergistically enhanced expression of M2-associated markers in control but not in Il6ra−/− BMDM (Fig. 5b). IL-4 and IL-6 synergized to induce expression of CD206 (MRC1) and arginase 1 protein expression as assessed by flow cytometry in control BMDM (Fig. 5c and Supplementary Fig. 6b). Moreover, in control but not Il6ra−/− macrophages, IL-6 synergized with IL-4 to inhibit the gene expression of Nr3c2 and Tnf, which both represent classical M1-macrophage markers (Supplementary Fig. 6c).
We next assessed whether IL-6 synergizes with IL-4 to induce M2-macrophage polarization in vivo. We treated control mice with either IL-6, IL-4 or IL-4 + IL-6 and assessed expression of the M2 marker CD206 via flow cytometry in different macrophage compartments. In line with what we observed in BMDM, IL-6 enhanced IL-4-induced M2 polarization in cells from WAT, BAT, blood and peritoneum in vivo (Fig. 5d, e and Supplementary Fig. 6d, e).
IL-4 regulates the onset of diet-induced obesity and insulin resistance23. Four weeks of exposure to IL-4 significantly improved glucose tolerance in HFD control but not HFD Il6raΔmyel mice (Fig. 5f and Supplementary Fig. 7a). In addition, IL-4 treatment reduced the HOMA-IR indices in HFD control mice to a greater extent than in HFD Il6raΔmyel mice (Supplementary Fig. 7b). qRT-PCR analyses revealed a significant reduction of IL-4-induced expression of Mrc1, Retnla and Arg1, as classical IL-4 targets and M2 macrophage markers, in WAT, BAT and liver of HFD Il6raΔmyel mice compared to HFD control mice in vivo (Fig. 5g). Furthermore, in line with the mRNA expression data, CD206 (MRC1) immunoreactivity was reduced in livers of IL-4-treated HFD Il6raΔmyel mice compared to HFD controls (Fig. 5h). Thus, myeloid cell-restricted Il6ra-deficiency leads to resistance against the beneficial effects of IL-4 on glucose homeostasis and insulin sensitivity in obesity. Furthermore, IL-6-action in macrophages is a necessary prerequisite for IL-4-induced alternative macrophage activation in vivo.
IL-6 signaling in myeloid cells limits LPS-induced endotoxemia
Next, we aimed to determine whether IL-6, in addition to augmenting the IL-4-STAT6 axis, also exhibits anti-inflammatory properties during conditions that promote both classical M1- and alternative M2-macrophage activation independent of metabolic inflammation. Therefore, we stimulated control and Il6ra−/− BMDM with bacterial lipopolysaccharide (LPS) and determined gene expression of typical M1 markers and inflammatory cytokines. LPS alone induced expression of Nos2, Tnf, Il1b and Il12a to the same extent in both genotypes in vitro (Fig. 6a). However, co-stimulation with IL-6 blunted LPS-induced inflammatory gene expression by approximately 50% in control but not in Il6ra−/− macrophages (Fig. 6a). Thus, the transcription of classical LPS-induced cytokines was blunted by IL-6 and this response was not present in macrophages derived from Il6raΔmyel mice.
Figure 6. Myeloid cell IL-6 signaling limits LPS-endotoxemia.

(a) qRT-PCR analyses of control (Ctrl) and Il6ra−/− BMDM that were left untreated or stimulated with IL-6 (12 hours) and subsequently exposed to bacterial lipopolysaccharides (LPS) alone or LPS + IL-6 for an additional 24 hours (n=6 vs 6; *p≤0.05 vs LPS + IL-6 stimulated Ctrl; **p≤0.05, ***p≤0.01; Data are expressed as % of LPS stimulated Ctrl). (b) Body weight loss of Ctrl and Il6raΔmyel mice 24 hours after exposure to 1mg/kg LPS (n=7 vs 8; *p≤0.05). (c) Food intake of Ctrl and Il6raΔmyel mice before (basal) and 24 hours after exposure to LPS (n=7 vs 8; *p≤0.01). (d) Respiratory exchange ratio (RER) of Ctrl and Il6raΔmyel mice before (basal) and 24 hours after exposure 1mg/kg LPS (n=7 vs 8; *p≤0.01). (e) Cytokine concentration in circulation of Ctrl and Il6raΔmyel mice 6 hours after exposure to 1mg/kg LPS (n=7 vs 7; **p≤0.01, p=0.06 for IL-4). (f) qRT-PCR analyses of liver and WAT from Ctrl and Il6raΔmyel mice 48 hours after exposure to 1mg/kg LPS (n=7 vs 8; *p≤0.05; Data are expressed as % of Ctrl). (Values are expressed as mean ± sem).
To assess whether IL-6 signaling in macrophages also regulates the inflammatory response to LPS in vivo, we treated control and Il6raΔmyel mice with a sublethal dose of LPS (1mg/kgBW). While LPS treatment significantly reduced body weight, food intake and respiratory exchange ratio (RER) in both genotypes, these responses were significantly stronger in Il6raΔmyel mice (Fig. 6b–d). This was paralleled by a significant increase of pro-inflammatory cytokines TNF, IL-1β and IL-12 in circulation after LPS injection in Il6raΔmyel mice compared to control mice (Fig. 6e). Additionally, IL-4 was increased in the circulation of LPS-treated Il6raΔmyel mice compared to control mice, indicating a possible compensatory upregulation due to IL-4 resistance in these animals (Fig. 6e). Analysis of gene expression in liver and WAT 48 hours following LPS injection revealed a significant increase in pro-inflammatory gene expression in Il6raΔmyel mice compared to control mice, while expression of Il4ra and Retnla was significantly reduced in WAT of Il6raΔmyel mice (Fig. 6f). Collectively, these experiments revealed that IL-6 signaling in macrophages not only limits diet-induced inflammation and insulin resistance but also LPS-induced inflammatory responses (Supplementary Fig. 7c).
Discussion
In the current study we demonstrate, through disruption of the IL-6 receptor in myeloid cells, that IL-6 is a novel determinant of macrophage polarization both upon high fat feeding and during endotoxemia. HFD mice with a myeloid cell-specific Il6ra deficiency have an increased propensity to disruption of glucose homeostasis and show increased M1 and reduced M2 macrophage marker expression concomitant with enhanced JNK activation in WAT, BAT and liver. Activation of pro-inflammatory gene expression induces activation of JNK, which is a well-characterized effector of obesity-associated insulin resistance18,24. Pro-inflammatory M1 macrophages are central mediators of obesity-induced inflammation and insulin resistance25 and specific ablation of these cells normalizes insulin sensitivity in obese, insulin resistant mice26. Conversely, genetic deletion of transcription factors that promote alternative M2 macrophage activation, such as peroxisome proliferator-activated receptor γ (PPARγ), predisposes lean mice to the development of glucose intolerance and insulin resistance27. Hence, M2 macrophages are critical for metabolic control and, upon the development of obesity, increased abundance of M1 macrophages leads to the disruption of glucose homeostasis.
Our results identify IL-6 as a critical instigator of M2 macrophage polarization and assign it an unexpected beneficial role in the prevention of obesity-associated insulin resistance. This finding may appear surprising in light of the general assumption that IL-6 acts as a pro-inflammatory cytokine28. In fact, blocking of IL-6 signaling has proven a valuable clinical approach for the treatment of rheumatoid arthritis29. However, the role of IL-6 in obesity-associated inflammation until now had remained controversial. This stems predominantly from clamp experiments indicating that IL-6 infusion causes insulin resistance12 and from the positive correlation between obesity and the amounts of IL-6 in the circulation11. Nevertheless, studies that have taken alternative approaches, particularly more recently, indicate that IL-6 may rather exert beneficial metabolic effects since conventional IL-6 deficient mice develop late onset obesity and insulin resistance13. HFD-induced inflammation and deterioration of glucose metabolism is enhanced in IL-6 knockout mice17, hepatic inactivation of IL-6 signaling promotes insulin resistance14, transgenic overexpression of human IL-6 in mice improves energy and glucose homeostasis30 and IL-6 enhances insulin secretion via GLP-1 production31.
On a cell-autonomous level, we reveal that IL-6 directly induces expression of the IL-4 receptor and therefore primes macrophages for IL-4-dependent activation of STAT6. Importantly, the IL-4-STAT6 axis is the most potent inducer of M2 macrophage polarization and genetic ablation of either IL-4Rα or STAT6 strongly inhibits alternative macrophage activation in vitro and in vivo32,33. A recent study demonstrated that during inflammatory conditions, upregulation of IL-4Rα was observed specifically in myeloid effector cells34. This upregulation is dependent on a soluble protein34, which in light of our findings is likely to be IL-6. In addition, evidence exists from the tumor immunology field that IL-6 signaling is involved in polarization of tumor-associated macrophages35, macrophage subtypes that are similar to obesity-associated M2 macrophages. However, these studies have not demonstrated the potent IL-4-sensitizing effect of IL-6 that we document here.
Our observations that IL-6 acts not only to prime myeloid cells for IL-4 signaling during obesity but also to limit LPS-endotoxemia demonstrate that IL-6 generally functions as an anti-inflammatory cytokine. This role has been assigned to IL-6 more than a decade ago36, however our results further extend this notion and demonstrate that these anti-inflammatory properties depend on direct IL-6 signaling in myeloid cells. In further support of our findings, it was recently demonstrated that myeloid cell-specific disruption of SOCS3, the negative regulator of the IL-6-STAT3 axis, skews macrophages towards an M2 phenotype and renders mice more resistant to LPS-induced endotoxemia37, underlining the importance of intact IL-6 signaling in this context.
Following these observations, we propose that during inflammatory conditions, i.e. in the presence of elevated free fatty acids and/or bacterial LPS, IL-6 limits pro-inflammatory gene expression and augments IL-4 responsiveness in macrophages. It thus acts to partially counterbalance the typical shift of macrophage populations towards a pro-inflammatory M1 phenotype, ultimately reducing inflammation and associated insulin resistance. Taken together, our data demonstrate that IL-6 has a homeostatic role in limiting obesity-associated insulin resistance and inflammation and define a novel mechanism in the control of macrophage polarization. This may set the basis to investigate the role of IL-6 signaling in macrophages in conditions where alternative macrophages activation is also of critical importance, such as fibrosis, wound healing and carcinogenesis.
Methods
Animals
All animal procedures were conducted in compliance with protocols approved by local government authorities (Bezirksregierung, Cologne, Germany; Alfred Medical Research and Education Precinct (AMREP) Animal Ethics Committee, Melbourne, Australia; Institutional Animal Care and Use Committee (IACUC), San Francisco, USA) and were in accordance with NIH guidelines. Mice were housed in groups of 3–5 at 22–24°C in a 12:12 h light/dark cycle with lights on at 6 a.m. Animals were either fed normal chow diet (Teklad Global Rodent #T.2018.R12; Harlan, Germany) containing 53.5% of carbohydrates, 18.5% of protein, and 5.5% of fat (12% of calories from fat) or from week 4 of age a high fat containing diet (# C1057; Altromin, Germany) containing 32.7%, 20% and 35.5% of carbohydrates, protein and fat (55.2% of calories from fat), respectively. Water was available ad libitum and food was only withdrawn if required for an experiment. Body weight was measured once a week. Experiments on mice were performed at 10–24 weeks of age.
Generation of Il6raΔmyel mice
Mice bearing the Il6rafl/fl allele were generated as previously described14. LysM-Cre15 mice were mated with Il6rafl/fl mice, and a breeding colony was maintained by mating Il6rafl/fl with LysM-CreTg/wt Il6rafl/fl mice (Il6raΔmyel) as previously described39. These lines had been backcrossed to C57Bl/6J mice for at least 10 generations. LysM-Cre mice were genotyped by PCR as previously described40, Il6rafl/fl were genotyped by PCR with primers crossing the loxP site as previously described14. LysM-CreTg/wt mice and wildtype controls were sacrificed at the age of 104 weeks and tissues were isolated for RNA extraction. Tissues of HFD conventional IL-6 knockout mice were collected as previously described17.
Analysis of body composition
Body composition was determined using nuclear magnetic resonance (NMR) in vivo via the NMR Analyzer minispec mq7.5 (Bruker Optik).
Indirect calorimetry
A PhenoMaster System (TSE Systems) was used for calorimetry measurements. Mice were maintained at 22°C to 24°C in 7.1-l chambers of the PhenoMaster open circuit calorimetry. Prior to starting a 48h measurement period, mice were allowed to adapt to the chambers for 24h. Food and water were provided ad libitum and measured by the integrated automatic instruments.
Glucose and insulin tolerance tests, IL-4 injection and HOMA-IR calculation
Glucose tolerance tests were performed with 20–24-week-old animals after a 16h fasting period. After determination of fasted blood glucose levels, each animal received an i.p. injection of 20% glucose (10ml/kg) (DeltaSelect, Germany). Blood glucose levels were detected after 15, 30, 60 and 120 minutes.
Insulin tolerance tests were performed with 10-week-old animals fed ad libitum. After determination of basal blood glucose levels, each animal obtained an i.p. injection of Insulin, 0.75U/kg (Actrapid; Novo Nordisk A/S, Denmark), and blood glucose was measured 15, 30 and 60 minutes after insulin injection.
IL-4 treatment was performed as previously described23. Briefly, 24-week-old HFD control and Il6raΔmyel mice were i.p.-injected twice per week with 2 μg IL-4 (PeproTech) complexed with an IL-4 specific antibody (BD Biosciences). HOMA-IR was calculated as previously described41. To promote alternative activation of macrophages in vivo, mice were given a single injection of IL4 in sterile PBS (50μg/kg). IL-6 (R&D) was used at 25μg/kg for in vivo studies.
Hyperinsulinemic-euglycemic clamp studies in awake mice
Surgical implantation of catheters in the jugular vein was performed as previously described42. After 5–6 days of recovery, only mice that had lost less than 10% of their preoperative weight were included. Each animal was fasted for 6 hours in the morning of the experiment and then placed in a restrainer for the duration of the clamp experiment. All infusates used in the experiment were prepared using a 3% plasma solution obtained from fasted donor animals. A primed-continuous tracer D-[3-3H]-Glucose infusion started 50 minutes prior to the clamp (PerkinElmer, 5μCi prime, 0.05 μCi/min). After a 50-minute basal period, a blood sample (40μl) was collected from the tail tip for determination of basal parameters. To minimize blood loss, red blood cells were collected by centrifugation, resuspended in saline, and reinfused. The clamp started with a primed-continuous insulin infusion (INSUMAN rapid, Sanofi-Aventis; 60μU/g prime, 4μU/g/min) and blood glucose levels were measured every 10 minutes (B-Glucose Analyzer; Hemocue). Physiological blood glucose levels (between 120 and 140 mg/dl) were maintained by adjusting a 20% glucose infusion (DeltaSelect). Approximately 120 minutes before the end of the experiment 2-[1-14C]-Deoxy-D-glucose (American Radiolabeled Chemicals, Inc., 10μCi) was infused and blood samples (10μl) were collected until reaching the steady state. Steady state was ascertained when a fixed glucose infusion rate maintained blood glucose constant for 30 minutes. During the steady state, blood samples (40μl) were collected for the measurement of steady state parameters.
At the end of the experiment, mice were sacrificed by cervical dislocation, and liver, adipose tissue and skeletal muscle were dissected and stored at −80°C. Plasma [3-3H]Glucose radioactivity of basal and steady state was measured as previously described42. Plasma 2-[1-14C]-Deoxy-D-glucose radioactivity was directly measured in liquid scintillation counter. Adipose tissue and skeletal muscle lysates were processed through ion exchange chromatography columns (Poly-PrepR Prefilled Chromatography Columns, AGR1-X8 formate resin, 200–400 mesh dry; Bio Rad Laboratories) to separate 2-[1-14C]-Deoxy-D-glucose (2DG) from 2-[1-14C]-Deoxy-D-glucose-6-phosphate, 2-[1-14C] (2DG6P).
Glucose turnover rate (mg × kg−1× min−1) was calculated as previously described42. In vivo glucose uptake for adipose tissue and skeletal muscle (nmol × g−1× min−1) was calculated based on the accumulation of 2DG6P in the respective tissue and the disappearance rate of 2DG from plasma as described previously43.
LPS-endotoxemia
9–10 weeks old control and Il6raΔmyel mice were aclimatized to the PhenoMaster System cages for five days. Subsequently, all mice were challenged by an i.p. injection of 1mg/kg body weight LPS (E. coli-derived, serotype O55:B5, # L5418, Sigma-Aldrich) in 0.9 % saline, and measurements of food intake (NCD) and energy expenditure were resumed. Whole blood was collected into heparinized tubes 6 hours post injection, and spun at 7000 g (4° C) for 7 minutes to obtain plasma. LPS serotype and preparation as well as administration route, were chosen based on previous publications44,45. Levels of cytokines were quantified in plasma (diluted 1:2) using Multiplex Bead Immunoassays (Life Technologies, CA) and a Bio-Plex 200 system (Bio-Rad, UK) according to the manufacturer’s instructions.
Analytical procedures
Blood glucose levels were determined from whole venous blood using an automatic glucose monitor (Bayer Contour, Bayer, Germany). Leptin and insulin levels in serum were measured by ELISA using mouse standards according to manufacturer’s guidelines (Mouse Leptin ELISA; Crystal Chem, IL, USA/Mouse Insulin ELISA; Crystal Chem, IL, USA). Serum non-esterified free fatty acids (NEFA) were quantified with according to the manufacturer’s instructions (WAKO Chemicals GmbH, Germany). Liver and serum triglyceride concentrations were determined in 6 hour fasted animals. Liver triglycerides were extracted as previously described46. Liver and serum triglyceride content was quantified according to the manufacturer’s instructions (Sigma, USA).
Generation of bone marrow-derived macrophages
Mice were sacrificed by CO2 anesthesia, rinsed in 70% (v/v) ethanol and bone marrow was isolated from femurs and tibias. Bone marrow cells were plated at a concentration of 1–2*106 cells/ml in RPMI 1640 (supplemented with 10% FCS, 1% glutamine, 1% penicillin-streptomycin and 10ng/ml M-CSF (PeproTech, Hamburg, Germany)) on 15 cm petridishes and differentiated for 8–10 days. 24 hours before all the experiments, M-CSF was removed and cells were washed two times with sterile PBS.
Primary macrophage culture
Bone marrow-derived macrophages were stimulated with recombinant IL-6 (R&D), IL-4, IL-10 (both from PeproTech) or E. coli-derived lipopolysaccharides (LPS O55:B5; Sigma). For pre-incubation experiments, cells were incubated with 50 ng/ml IL-6 for 12 or 24 hours and subsequently exposed to either IL-4 or LPS alone or in combination with IL-6 for the indicated time points. For antibody-mediated neutralization of IL-10, cells were preincubated for 1 hour with either isotype control (10 μg/ml Purified NA/LE Rat IgG1, #554682 BD Pharmingen) or anti-IL10 antibody (10 μg/ml Purified NA/LE Rat Anti-Mouse IL-10, #554421) and subsequently exposed to IL-6 (50ng/ml) or IL-10 (10ng/ml) in the presence of either isotype control or anti-IL10 antibody for 12 hours. BMDM were transfected with 600 pmol of siRNA oligos against Stat3, Il6ra or a nontargeting control (all LifeTechnologies) as previously described40.
Flow cytometry
Adipose tissue was homogenized for 30 seconds with a tissue lyser (Miltenyi) and digested with collagenase I (2 mg/ml, Worthington) for 20 min at 37 °C in a shaker (250 rpm) as previously described47. The digested cell suspension was centrifuged at 1,600 rpm for 5 min to separate stromal-vascular fraction from adipocytes. Pelleted cells were resuspended in FACS buffer (PBS containing 5% FBS and 1% L-glutamine) and passed through a 40 μm strainer (BD Biosciences). Antibodies directed against mouse CD11b, CD45, CD115, CD206 and F4/80 (Biolegend, 1:200 dilution); arginase-1 (Santa Cruz Biotech, 1:200 dilution); and anti-goat IgG (Invitrogen, 1:1000 dilution) were used. Samples were stored in FACS buffer with 1% paraformaldehyde at 4 °C prior to analysis. Data were acquired on FACSVerse (BD Biosciences) and data analysis was performed using FlowJo (Treestar).
Western blot analysis
Protein isolation was performed as previously described40. Immunoblot analysis was performed as previously described40, with antibodies raised against STAT3 (#9132, Cell Signaling), phospho-STAT3 (#9145, Cell Signaling), STAT6 (#9362, Cell Signalling), phospho-STAT6 (#54461, Abcam), Interleukin 4 receptor α subunit (#00884; Novus Biologicals) and SAPK/JNK (#9252, Cell Signaling) as well as α-Tubulin (#T6047; Sigma), calnexin (#208880; Calbiochem) or Hsc70 (#sc-7298, Santa-Cruz) as loading controls. JNK MAPKinase assay (#8794, Cell Signaling) was carried out according to the manufacturer’s instructions. Briefly, immunoprecipitation of active JNK from tissue lysates was performed with an antibody directed against phospho-JNK (Thr183/Tyr185) Rabbit mAb coupled to sepharose beads. Upon addition of kinase buffer, c-Jun fusion protein and ATP, JNK phosphorylates the c-Jun substrate. Phospho-c-Jun (Ser63) Antibody was then used to measure JNK activity by immunoblotting. Straight blots were carried out with the input lysates to adjust for differences in protein concentration. Quantification of immunoblot signal intensity was performed with ImageJ6448.
Analysis of gene expression
Isolated total RNA from adipose tissue, liver and primary macrophages was analyzed using qRT-PCR. RNA was isolated from tissue and cells using the Qiagen RNeasy Kit (Qiagen, Germany). The RNA was reversely transcribed with High Capacity cDNA RT Kit and amplified using TaqMan® Universal PCR-Master Mix, NO AmpErase UNG with TaqMan® Assay-on-demand kits (Applied Biosystems, CA, USA). Relative expression of target mRNAs was adjusted for total RNA content by hypoxanthine guanine phosphoribosyl transferase 1 (Hprt1) mRNA qRT-PCR. Calculations were performed by a comparative method (2−ΔΔCT). qRT-PCR was performed on an ABI-PRISM 7900 HT Sequence Detection system (Applied Biosystems, Germany).
Microarrays
RNA for microarray expression analysis was isolated from bone marrow-derived macrophages. Quality of RNA was assessed using the Experion™ RNA StdSens Analysis Kit (BIO-RAD). Biotin-labeled cDNA was synthesized using GeneChip Whole Transcript Sense Target Labeling Assay (Affymetrix) according to the manufacturer’s guidelines. Following fragmentation, cDNAs were subsequently hybridized for 17 h at 45°C to GeneChip Mouse Gene 1.0 ST Arrays (Affymetrix) (n=4 per genotype). Arrays were washed and stained in the GeneChip Fluidics Station 450 (Affymetrix) and scanned on a GeneChip Scanner 3000 7G (Affymetrix). Affymetrix Powertools and robust multi-array average (RMA) were used for background correction, quantile normalization and summarization of the raw intensity values. RMA, statistical analyses (Student’s t-test), calculation of the fold change of intensity values and graphical illustrations were performed using R software (version 2.13.1) along with the Bioconductor software package. Genes were considered significantly deregulated if p-values ≤ 0.05.
Chromatin immunoprecipitation
Cells (5 million) were cross-linked with 1% formaldehyde for 10 min, followed by quenching with 12.5 mM glycine for 5 min. After cytoplasmic lysis (500 μl, 15 min 4°C, 50 mM Hepes-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0,25% TritonX, Pierce protease and phosphatase inhibitors) and nuclear lysis (200 μl, 15 min 4°C, 50 mM Tris-HCl pH 8, 10 mM EDTA, 1% SDS, Pierce protease and phosphatase inhibitors), chromatin was sheared with a Bioruptor Pico sonicator (10x 20On/30Off, Diagenode) to yield fragment sizes of 100–500bp. After saving 10% as input control, chromatin was diluted with IP buffer (20 mM Tris-HCl pH 8, 2 mM EDTA, 150 mM NaCl, 1% TritonX, Pierce protease and phosphatase inhibitors) and immunoprecipitation with phospho-STAT3 (Cell Signaling, #9145) and normal rabbit IgG (Cell Signaling, #2729) antibodies was carried out with sheared chromatin of ~2.5 million cells over night, followed by 1 hour incubation at 4°C with Protein A/G magnetic beads (Millipore). Washing and reverse crosslinking was performed as previously described49. DNA was further purified using ChIP DNA Clean and Concentrator Kit (DCC, Zymoresearch). Real-time PCR was performed using SYBR Select PCR master mix (Lifetechnologies) on an ABI PRISM 7900 HT Sequence Detection system (Lifetechnologies). Primer pairs designed for qRT-PCR analysis are depicted in (Supplementary Table 1). Commercially available IGX1A primer pairs were used as negative control (Qiagen, #GPM100001C(-)01A). The relative promoter occupancy was calculated by IgG background subtraction.
Dual luciferase reporter assay
For reporter construct generation, different segments of the murine Il4ra-promoter were inserted upstream of the Firefly-luciferase open reading frame using specific primers. Immortalized murine macrophages21 (2×105 cells per well) were plated on 24-well plates and, on the next day, transfected with 500ng DNA using FuGENE HD transfection reagent (Promega) at a ratio of 4.5:1. The Renilla-luciferase reporter gene (50 ng) was used as an internal control. Dual luciferase reporter assays were carried out 48h after transfection using a Luciferase Assay System (Promega) according to the manufacturer’s instructions. In all cases, the data were normalized for transfection efficiency by dividing the Firefly-luciferase signal by that of the Renilla luciferase.
Immunohistochemistry
Immunohistochemistry was performed as previously described40. Quantification of F4/80 (#ab6640, Abcam) and Mac-2 (#CL8942B, Cedarlane) -positive crown-like structures (CLS) was performed with AxioVision 4.2 (Carl Zeiss MicroImaging GmbH, Oberkochen, Germany).
Statistics
In all figures, data are presented as mean ± sem. P-values were calculated with two-tailed unpaired student’s t-test or, if more than two conditions were compared, with 1- or 2-Way ANOVA followed by Bonferroni’s or Tukey’s post-test. P ≤ 0.05 was considered significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We wish to thank G. Schmall and T. Rayle for excellent secretarial assistance as well as B. Hampel and D. Kutyniok for outstanding technical assistance. This work was supported by the DFG (SFB 612 and SFB 670 to J.C.B.), the Leibniz Preis (BR 1492/7-1 to J.C.B.), the Cologne Excellence Cluster on Cellular Stress Responses in Aging Associated Diseases (CECAD; funded by the DFG within the Excellence Initiative by German Federal and State Governments), NIH Director’s Pioneer Award (DP1AR064158 to A.C.), and NIH (HL076746, DK094641 both to A.C.). M.A.F is supported by NHMRC Australia (SPRF APP1021168, Project Grants APP1041760, APP1042465).
Footnotes
Database accession
The microarray data presented in this publication have been deposited in NCBI’s Gene Expression Omnibus38 and are accessible through GEO Series accession number GSE47426 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE47426)
Contributions
J.M., J.R., A.C., F.T.W. and J.C.B designed the experiments. J.M., B.C., J.G., M.C.V., J.R. and K.D.N. performed the experiments. S.T. helped with the flow cytometry. A.C.H. performed the clamp operations. J.S. assisted during the ChIP analyses. H.S.B. performed the indirect calorimetry. E.E., T.L.A., A.M., L.P. and M.A.F. supplied reagents. J.M. and J.C.B. wrote the manuscript.
References
- 1.Swinburn BA, et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet. 2011;378:804–814. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- 2.Xu H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellen KE. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arkan MC, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 6.Cai D, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinridders A, et al. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 9.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen BK, Febbraio MA. Point: Interleukin-6 does have a beneficial role in insulin sensitivity and glucose homeostasis. J Appl Physiol. 2007;102:814–816. doi: 10.1152/japplphysiol.01208.2006. [DOI] [PubMed] [Google Scholar]
- 11.Weiss R, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350:2362–2374. doi: 10.1056/NEJMoa031049. [DOI] [PubMed] [Google Scholar]
- 12.Kim HJ, et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes. 2004;53:1060–1067. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- 13.Wallenius V, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–79. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 14.Wunderlich FT, et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010;12:237–249. doi: 10.1016/j.cmet.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 16.Vogt MC, Brüning JC. CNS insulin signaling in the control of energy homeostasis and glucose metabolism – from embryo to old age. Trends Endocrinol Metab. 2013;24:76–84. doi: 10.1016/j.tem.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Matthews VB, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53:2431–2441. doi: 10.1007/s00125-010-1865-y. [DOI] [PubMed] [Google Scholar]
- 18.Belgardt BF, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proceedings of the National Academy of Sciences. 2010;107:6028–6033. doi: 10.1073/pnas.1001796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119:e110–e119. doi: 10.1182/blood-2011-09-381483. [DOI] [PubMed] [Google Scholar]
- 20.Portales-Casamar E, et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–10. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baumgartl J, et al. Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab. 2006;3:247–256. doi: 10.1016/j.cmet.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricardo-Gonzalez RR, et al. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci USA. 2010;107:22617–22622. doi: 10.1073/pnas.1009152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 25.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patsouris D, et al. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–309. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odegaard JI, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 30.Sadagurski M, et al. Human IL6 enhances leptin action in mice. Diabetologia. 2009;53:525–535. doi: 10.1007/s00125-009-1580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellingsgaard H, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med. 2011;17:1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herbert DR, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 33.Vats D, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wermeling F, Anthony RM, Brombacher F, Ravetch JV. Acute inflammation primes myeloid effector cells for anti-inflammatory STAT6 signaling. Proceedings of the National Academy of Sciences. 2013 doi: 10.1073/pnas.1312525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duluc D, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–4330. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 36.Xing Z, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spence S, et al. Suppressors of Cytokine Signaling 2 and 3 Diametrically Control Macrophage Polarization. Immunity. 2013;38:66–78. doi: 10.1016/j.immuni.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 38.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wunderlich CM, et al. Cutting edge: Inhibition of IL-6 trans-signaling protects from malaria-induced lethality in mice. The Journal of Immunology. 2012;188:4141–4144. doi: 10.4049/jimmunol.1102137. [DOI] [PubMed] [Google Scholar]
- 40.Mauer J, et al. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010;6:e1000938. doi: 10.1371/journal.pgen.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan SD, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol. 2011;13:434–446. doi: 10.1038/ncb2211. [DOI] [PubMed] [Google Scholar]
- 42.Könner AC, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Ferre P, Leturque A, Burnol AF, Pénicaud L, Girard J. A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaesthetized rat. Biochem J. 1985;228:103–110. doi: 10.1042/bj2280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaurasia B, et al. Phosphoinositide-dependent kinase 1 provides negative feedback inhibition to Toll-like receptor-mediated NF-kappaB activation in macrophages. Mol Cell Biol. 2010;30:4354–4366. doi: 10.1128/MCB.00069-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruud J, et al. Inflammation- and tumor-induced anorexia and weight loss require MyD88 in hematopoietic/myeloid cells but not in brain endothelial or neural cells. The FASEB Journal. 2013;27:1973–1980. doi: 10.1096/fj.12-225433. [DOI] [PubMed] [Google Scholar]
- 46.Norris AW, et al. Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest. 2003;112:608–618. doi: 10.1172/JCI17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen KD, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480:104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Günschmann C, et al. Insulin/IGF-1 controls epidermal morphogenesis via regulation of FoxO-mediated p63 inhibition. Dev Cell. 2013;26:176–187. doi: 10.1016/j.devcel.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.