

Abstract
Background
The mariner family of transposable elements is one of the most widespread in the Metazoa. It is subdivided into several subfamilies that do not mirror the phylogeny of these species, suggesting an ancient diversification. Previous hybridization and PCR studies allowed a partial survey of mariner diversity in the Metazoa. In this work, we used a comparative genomics approach to access the genus-wide diversity and evolution of mariner transposable elements in twenty Drosophila sequenced genomes.
Results
We identified 36 different mariner lineages belonging to six distinct subfamilies, including a subfamily not described previously. Wide variation in lineage abundance and copy number were observed among species and among mariner lineages, suggesting continuous turn-over. Most mariner lineages are inactive and contain a high proportion of damaged copies. We showed that, in addition to substitutions that rapidly inactivate copies, internal deletion is a major mechanism contributing to element decay and the generation of non-autonomous sublineages. Hence, 23% of copies correspond to several Miniature Inverted-repeat Transposable Elements (MITE) sublineages, the first ever described in Drosophila for mariner. In the most successful MITEs, internal deletion is often associated with internal rearrangement, which sheds light on the process of MITE origin. The estimation of the transposition rates over time revealed that all lineages followed a similar progression consisting of a rapid amplification burst followed by a rapid decrease in transposition. We detected some instances of multiple or ongoing transposition bursts. Different amplification times were observed for mariner lineages shared by different species, a finding best explained by either horizontal transmission or a reactivation process. Different lineages within one species have also amplified at different times, corresponding to successive invasions. Finally, we detected a preference for insertion into short TA-rich regions, which appears to be specific to some subfamilies.
Conclusions
This analysis is the first comprehensive survey of this family of transposable elements at a genus scale. It provides precise measures of the different evolutionary processes that were hypothesized previously for this family based on PCR data analysis. mariner lineages were observed at almost all “life cycle” stages: recent amplification, subsequent decay and potential (re)-invasion or invasion of genomes.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-727) contains supplementary material, which is available to authorized users.
Keywords: Drosophila, Comparative genomics, Tc1-mariner, Transposable elements, MITEs, Deletion rate
Background
Mariner is a Class II transposon (i.e., transposing through a cut-and-paste mechanism) belonging to the large Tc1-mariner-IS630 superfamily of transposable elements present in almost all groups of living organisms. It was first discovered in Drosophila mauritiana [1], a species from the melanogaster subgroup. Since then, mariner-like elements (MLEs) have been identified in a wide range of species, primarily metazoans. MLEs form a particularly homogeneous group of transposable elements (TEs), as all full-length copies share a size of approximately 1.3 kb, terminal inverted repeats (TIRs) of 28–30 bp and a unique open reading frame (ORF) encoding a transposase of approximately 345 amino acids. As with other members of the Tc1-mariner superfamily, transposases of the mariner family contain the typical catalytic DDE motif, which is necessary for transposition [2, 3]. However, in the mariner family, the DD(35)E signature is replaced by DD(34)D. Other conserved motifs with known (DNA-binding domain) or unknown functions have also been identified [4].
Based on the transposase phylogeny, the numerous MLEs can be grouped into several distinct subfamilies. Typically, MLEs within one subfamily share at least 40% identity at the amino acid level and between 40 and 56% at the nucleotide level [5]. Historically, the term MLE was used to designate any sequence showing sequence similarities with the original mariner elements (peach and Mos1 copies), including consensus sequences derived from independent, closely related clones identified in the same species [6, 7]. The ability to derive a consensus sequence illustrates that the copies share a recent common ancestral copy (presumably similar to the consensus sequence) and constitute a (monophyletic) phylogenetic clade. It is assumed that within such a clade, any one mobilizable copy can be cross-mobilized by the active transposase of another copy, such as the inactive but full-length peach copy in D. mauritiana, which excises using the transposase of the Mos1 copy [8]. These two copies are 99% identical at the nucleotide level [9] and belong to the same functional clade (usually referred to as Dmmar1, from the mauritiana subfamily of MLEs, and hereafter called Dromar1). Mariner clades are sometimes referred to as ‘types’ [10], ‘tribes’ [11], or ‘lineages’ [12]; this latter terminology will be used hereafter. Hence, the mariner family is composed of several subfamilies, each of which comprises several lineages.
While cross-mobilization is the rule within a lineage, copies from two different subfamilies are generally not expected to cross-mobilize because their nucleotide sequences will differ sufficiently such that the transposase from one lineage cannot recognize the TIRs of the other. In vitro experiments revealed that a 16% or more difference in the TIRs can preclude the binding of the transposase [13]. However, if the TIRs are similar enough, cross-mobilization may occur between two lineages from the same subfamily. Therefore, if two mariner lineages are sufficiently different, they may evolve and coexist independently within the same genome. Indeed, the coexistence of different lineages/subfamilies within the same genome is not uncommon [14, 15].
MLEs in animals are characterized by a patchy distribution, a high proportion of inactive copies and several suspected cases of horizontal transfer (HT). These properties led Lohe et al. [16] to propose a mariner lifecycle with HT as the starting point of the cycle. Amplification within the genome and the population is followed by diversification (vertical inactivation) and ultimately stochastic loss [16, 17]. In fact, this lifecycle may be a general rule for all transposable elements, as suggested by the recent theoretical studies of Le Rouzic et al. [18, 19]. The horizontal transfer step is the step under selection for the obvious reason that full activity is required for the transferred element to invade the new genome [20, 21]. However, once installed in a genome, TEs are subject to little or no selective pressure, as observed by Witherspoon and Robertson [14] in the Caenorhabditis elegans and C. briggsae genomes. In the absence of selection, mutations can accumulate, and this may explain the vertical inactivation.
Although mariner elements were first identified in Drosophila, a few different lineages have been found in this genus. Apart from Dmmar1, only two other lineages from the mellifera and irritans subfamilies have been found in D. erecta and D. ananassae, respectively, in studies aiming to identify MLEs in a wide range of insects and arthropods [5, 16]. One relic copy has also been identified in D. melanogaster [22]. In the present study, these elements are referred to as Dromar6 (D. erecta), Dromar5 (D. ananassae) and Dromar14 (D. melanogaster). In the Drosophilidae, the Dmmar1 (Mos1) lineage is the most thoroughly investigated lineage [23–26]. A wide range of species displays some hybridization signal (Southern blot or dot blot), which has been confirmed by sequencing in only some species, potentially reflecting the existence of other lineages from the mauritiana subfamily. A very patchy distribution has been found in the Drosophilidae, resembling the pattern observed at the larger scales of arthropods or metazoans. However, more recently, Wallau et al. [27] detected various MLEs from the mellifera, mauritiana and irritans subfamilies in 23 neotropical drosophilid species. Hence, MLEs are also diversified in the Drosophilidae.
The recently sequenced Drosophila genomes by the Drosophila 12 Genomes Consortium [28] and the Drosophila modENCODE project (Piano and Cherbas [29], http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/modENCODE_ComparativeGenomics_WhitePaper.pdf) offer a unique opportunity to investigate i) the diversity of the mariner family in a group of both closely and distantly related species of the Drosophila genus, and ii) the evolutionary history of related but independent elements (see Figure 1B for a phylogeny of these species).
Figure 1.
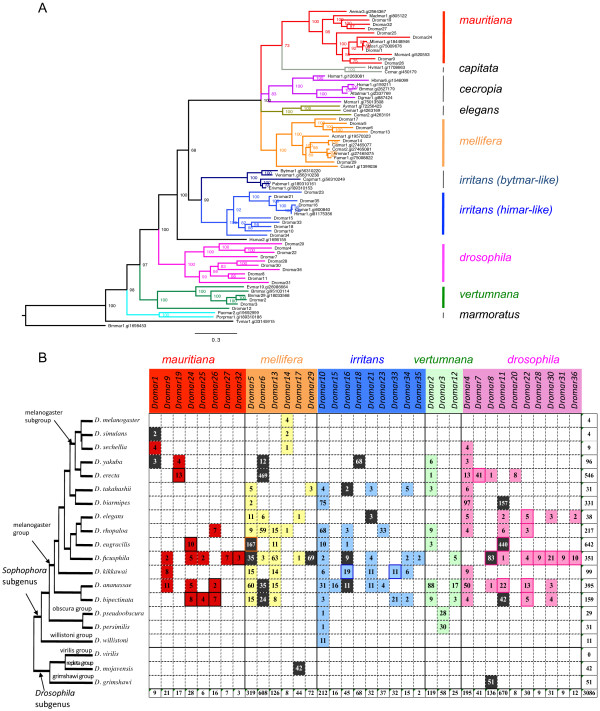
mariner lineages identified in this study. (A) Phylogeny of mariner-like transposases generated by a Bayesian analysis using the WAG + G amino acid substitution model. Posterior probability of each node is indicated. Clade colors denote the different subfamilies of MLEs (indicated to the right of the tree). The NCBI accession numbers of the transposases found in the database are indicated after the element names. (B) Distribution of MLE lineages in Drosophila genomes, indicating the number of independent copies, not truncated by assembly. Subfamilies are colored as in A. Dark colors indicate lineages with potentially coding copies. Framed boxes indicate lineages with MITEs.
The Drosophila genus is separated into two subgenera: Sophophora, which contains D. melanogaster, and Drosophila. They diverged approximately 47 Mya ago, according to TimeTree2 [30]. Each subgenus is composed of multiple groups and subgroups, but Sophophora is most represented among the 20 sequenced genomes, with 17 species versus 3 for the Drosophila subgenus.
An exhaustive homology-based search in the 20 genomes recovered 36 lineages of MLEs, most of them not described previously. Phylogenetic analysis suggested that the 36 lineages belong to 5 different subfamilies, including one not identified previously. Although the genome dataset is highly biased toward species from the melanogaster group (Sophophora subgenus), comparison of these different mariner lineages within the different species provides a view of this TE family within one genus and offers clues to the evolution of MLEs by revealing mariner elements at each stage of their life cycle.
Results
MLE distribution and diversity within the genus Drosophila
A panel of 18 transposase sequences spanning eight of the main known mariner subfamilies (Table 1) were used in a TBLASTN search against the 20 Drosophila sequenced genomes (Table 2). The results were analyzed using an automatic procedure to separate the elements into different lineages and recover full sequences, including TIRs (see Methods and Figure 2). We identified 3685 copies corresponding to 36 different MLE lineages that we named Dromar1-Dromar36 (Additional file 1 and Additional file 2).
Table 1.
mariner transposases used in the first TBLASTN search
| GI prot | UNIPROT | Subfamily (clade) | Species | |
|---|---|---|---|---|
| Mos1 | 75009676 | Q7JQ07_DROMA | mauritiana | Drosophila mauritiana |
| Madmar1 | 805122 | Q25436_MAYDE | mauritiana | Mayetiola destructor |
| Mcmar1-1 | 75013508 | Q869A8_MELCH | Meloidogyne chitwoodi | |
| Hsmar1 | 1263081 | Q13579_HUMAN | cecropia | Homo sapiens |
| Dtmar1 | 887424 | Q24693_DUGTI | cecropia | Dugesia tigrina |
| Avmar1 | 72256423 | Q45FI1_9BILA | elegans | Adineta vaga |
| Cemar2 | 7331821 | Q9N523_CAEEL | elegans | Caenorhabditis elegans |
| Cemar1 | 7331903 | Q9N4X9_CAEEL | elegans | Caenorhabditis elegans |
| Avmar1 | 72256423 | Q45FH8_9BILA | elegans | Adineta vaga |
| Ccmar1 | 1399036 | Q17312_CERCA | mellifera | Ceratitis capitata |
| Acmar1 | 19570323 | Q8T0Y0_APICE | mellifera | Apis cerana |
| Camar1 | 27465077 | Q8I8E8_CHYAM | mellifera | Chymomyza amoena |
| Ccmar2 | 27465081 | Q8I8E6_CERCA | mellifera | Ceratitis capitata |
| Famar1 | 75008822 | Q6XLA0_FORAU | mellifera | Forficula auricularia |
| Hsmar2 | 1698455 | Q13539_HUMAN | irritans (Hsmar-like) | Homo sapiens |
| Cpmar1 | 600840 | Q04514_CHRPL | irritans (Himar-like) | Chrysoperla plorabunda |
| Bytmar1 | 56310220 | Q5QT23_9EUCA | irritans (Bytmar-like) | Bythogtaea thermydron |
| Vesmar1 | 56310238 | Q5QT20_9CRUS | irritans (Bytmar-like) | Ventiella sulfuris |
The historical irritans subfamily is divided into three clades following Bui et al. [31].
Table 2.
Characteristics of the genome assemblies used in this study
| Scaffold number | Size (Mb) | WGS accession | Assembly | |
|---|---|---|---|---|
| D. melanogaster | 15 | 139 | AABU01 | Dmel_caf1 |
| D. simulans | 10,601 | 137 | AAGH01 | Dsim_caf1 |
| D. sechellia | 14,730 | 166 | AAKO01 | Dsec_caf1 |
| D. yakuba | 8,122 | 165 | AAEU02 | Dyak_caf1 |
| D. erecta | 5,124 | 152 | AAPQ01 | Dere_caf1 |
| D. takahashii | 1,792 | 182 | AFFI02 | Dtak_2.0 |
| D. biarmipes | 1,136 | 169 | AFFD02 | Dbia_2.0 |
| D. elegans | 1,063 | 171 | AFFF02 | Dele_2.0 |
| D. rhopaloa | 4,435 | 197 | AFPP02 | Drho_2.0 |
| D. eugracilis | 955 | 156 | AFPQ02 | Deug_2.0 |
| D ficusphila | 1,284 | 152 | AFFG02 | Dfic_2.0 |
| D. kikkawai | 1,284 | 164 | AFFH02 | Dkik_2.0 |
| D. ananassae | 13,749 | 231 | AAPP01 | Dana_caf1 |
| D. bipectinata | 1,552 | 167 | AFFE02 | Dbip_2.0 |
| D. pseudoobscura | 2,661 | 152 | AAFS01 | Dpse_caf1 |
| D. persimilis | 12,838 | 188 | AAIZ01 | Dper_caf1 |
| D. willistoni | 14,838 | 235 | AAQB01 | Dwil_caf1 |
| D. virilis | 13,530 | 206 | AANI01 | Dvir_caf1 |
| D. mojavensis | 6,841 | 193 | AAPU01 | Dmoj_caf1 |
| D. grimshawi | 17,440 | 200 | AAPT01 | Dgr0_caf1 |
Figure 2.
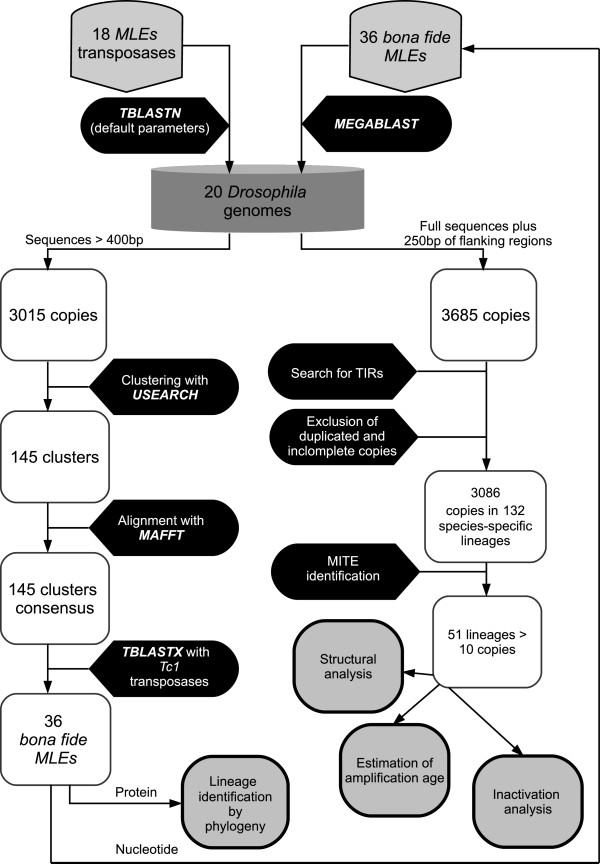
Experimental design procedure showing all steps of the analysis. We first searched by TBLASTN all 20 Drosophila using a set of 18 MLEs transposases representative of all described subfamilies of the mariner family. All sequences longer than 400 bp were clustered with a threshold of 80% identity. A consensus of each cluster was blasted against a transposase database composed of Tc1 family elements to exclude sequences from this family. For the remaining 36 clusters (bona fide MLEs), consensus conceptual translations were used in a phylogenetic analysis together with the MLEs transposases, and the nucleotide consensus were used as queries in a MEGABLAST search in the same 20 genomes to correctly identify all copies. All hits plus 250 bp of flanking regions were retrieved. Structural and evolutionary analyses were performed on a clean dataset from which duplicated copies (segmental duplications) and incomplete copies (ends of contigs) were excluded.
The protein sequences translated from the 36 nucleotide consensus sequences were aligned with 36 other mariner transposases and subjected to phylogenetic analysis (Figure 1A). This analysis revealed that 26 MLE lineages belonged to 4 known subfamilies (mauritiana, mellifera, irritans and vertumnana). The 10 remaining lineages grouped together into a highly supported clade that appears to correspond to a new subfamily, which we designated drosophila. The clade structure derived from the transposase analysis was supported by the comparison of TIRs, which showed that some blocks of nucleotides are clade (subfamily)-specific (see Additional file 3).
For each lineage, we analyzed flanking regions to filter out duplicated copies (segmental duplication) and retain only independent copies resulting from transposition. All truncated copies located at contig ends were also discarded. This resulted in a clean dataset of 3085 copies, corresponding to 132 different MLE/species lineage combinations that were used in all further analyses (Figure 1B). The copy number varies from 1 to 469 (Dromar6 in D. erecta) per lineage per species. In the mauritiana subfamily, all lineages presented a low copy number ranging from 1 to 13, in agreement with what was observed previously for the historical Dmmar1 (Dromar1) lineage in D. mauritiana [1], which does not exceed 20 copies. In contrast, higher copy numbers were observed in other subfamilies. For instance, with 1142 independent copies and 10 different lineages, the Drosophila subfamily appears to be the most successful in Drosophila. This large variation may reflect some subfamily-specific properties.
On average, six MLE lineages coexist in the same genome. However, large differences are observed among species. Globally, the subgenus Drosophila, composed of D. grimshawi, D. virilis and D. mojavensis, appears to be poor in MLEs (only two lineages identified). Indeed, in D. virilis, no mariner was detected in a BLASTN search using the 36 mariner sequences with default parameters. The search with blastn-short option detected one fragment < 500 bp, which was distantly related to Dromar18. Therefore, this genome appears to be prone to eradication of such elements. Only one lineage was present in each of D. grimshawi and D. mojavensis. However, in both cases, these elements presented a substantial copy number (51 and 42 independent copies, respectively, totaling 90 and 52, respectively; see Additional file 1), most of them very similar. In addition, they included potential autonomous elements, which suggests a recent origin (see below). In the subgenus Sophophora, all species contained at least traces of MLEs and, with the exception of D. melanogaster and D. willistoni, contained at least two different MLE lineages. In D. erecta and D. yakuba, 8 and 6 lineages, respectively, were found, representing 4 and 5 different subfamilies, respectively. This diversity is particularly prevalent in D. ananassae and D. ficusphila, where the 5 subfamilies are represented by 18 and 23 lineages, respectively.
Nine MLEs lineages are restricted to only one species. Among the 33 lineages detected in the melanogaster group (Sophophora subgenus), twelve are subgroup-specific, and twenty-one are shared by two or more subgroups of species. Only two lineages are also present in other groups of the same subgenus (Dromar10 in D. melanogaster, D. obscura and D. willistoni) or in species from the Drosophila subgenus (Dromar8) (Figure 1B). Hence, large differences are observed among species in copy number and diversity of MLEs, which may reflect specific properties of the genome or merely the independent evolutionary history of these lineages.
MITEs arising from internal deletion and/or rearrangement
Short non-autonomous copies are often found among Class II elements. The amplification of such short copies can ultimately lead to the emergence of a MITE (Miniature Inverted repeat Transposable Element). MITEs are present in numerous species and can be very abundant, but to date, few MITEs have been identified in Drosophila species or within the mariner family. Non-autonomous short copies that have transposed at least once (i.e., are present in at least two independent copies) were identified in fourteen MLE lineages, primarily from the mauritiana and drosophila subfamilies. A total of 27 independent sublineages could be distinguished, as one MLE lineage can derive more than one MITE sublineage in different species (e.g., Dromar22 and Dromar11) or within the same species (e.g., Dromar7 and Dromar11). Each sublineage was characterized by a specific independent deletion/rearrangement pattern, illustrating its origin from a single copy (see Figure 3).
Figure 3.
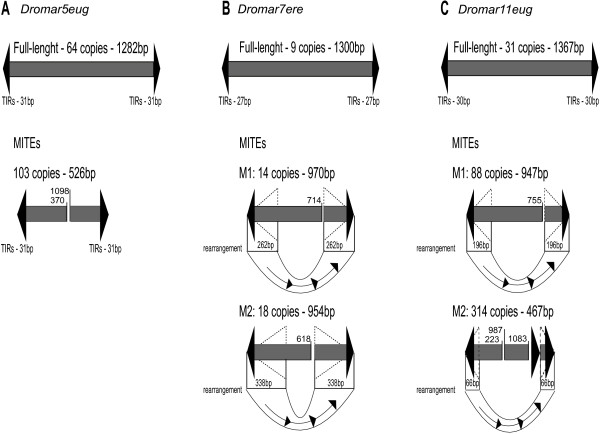
Schematic representation of different MITEs and their progenitor full-length elements. (A) Dromar5eug identified in D. eugracilis (B) Dromar7ere from D. erecta. (C) Dromar11eug from D. eugracilis. The rearrangements, which consistently involved the replacement of the 3′ region of the element with the 5′ region, are depicted below the elements. The sizes of the rearranged fragments are indicated and correspond to the size of the new inverted terminal repeats, shown as dashed triangles. Numbers above elements correspond to the position of breakpoints relative to the full-length element.
In total, MITEs represent 724 copies, corresponding to 23% of all mariner copies. The copy number of full-length mariner was generally low except in a few lineages in which hundreds of copies are found. Ten of 27 MITE sublineages contained 10 or more (up to ca. 300) amplified short copies (see Additional file 4). One third of the lineages, mainly characterized by a low copy number, had no larger relatives in the genome, and most were species specific (except for a few copies in Dromar22 that are shared by more than one species). They likely represent aborted MITE amplifications due to the loss of the autonomous partner.
The typical size of MITEs varied between 900 and 1000 bp; however, in four lineages, copies were approximately 460–560 bp (Additional file 4). The two most successful MITEs are the ones with the shortest sizes.
For all MITE families, internal sequences and TIRs were fully homologous to the sequences of the full-length copies, with no traces of long, non-homologous sequences. Indeed, most of the MITE lineages corresponded to internally deleted elements; for example, Dromar5eug contains 112 MITE copies of 526 bp and 63 full-length copies (Figure 3A). This lineage is widely distributed across the melanogaster group, with a high copy number not only in D. eugracilis but also D. ananassae (77 copies). However, the MITE sublineage was specific to D. eugracilis.
Seven MITE sublineages from the drosophila subfamily clearly originated from rearrangements: two for Dromar7 in D. erecta, three for Dromar11 in D. eugracilis and one each for Dromar28 and Dromar36. All resulted from the replacement of the 3′ portion by the 5′ sequence (Figure 3B, C). These rearrangements cause these copies to have unusually long TIRs. Hence, internal rearrangements appear to be a major process involved in the generation of MITEs.
MITEs have been associated with genes in plant species [32, 33]. We investigated whether this association was present in the high-copy-number Dromar11 and Dromar5 MITEs in D. eugracilis. Because this genome is not yet annotated, we first identified potentially coding regions using a TBLASTN search with the protein database of D. melanogaster and then computed the distance of Dromar11eug and Dromar5eug copies to these potential CDSs (Additional file 5). For both lineages, the proportion of copies present in the same supercontig as a putative CDS was higher for MITEs than for non-MITE copies. However, for copies present in a CDS-carrying contig, we could not detect significant differences in the distance-to-CDS distribution of MITE and non–MITE copies (KS test). Nevertheless, a significant difference was observed between Dromar5Meug and Dromar11M1eug, as well as between Dromar5 and Dromar11 lineages overall. These two lineages differ in age, with Dromar11 being more recent (see below); its copies were also globally closer to CDS, which suggests that older lineages are farther from genes. This finding equally supports two hypotheses: long-term selection against insertion near genes versus the preferential insertion of new copies near genes pushing away old insertions.
These MITE families were separated from their associated full-length elements for subsequent analyses.
Mobility and coding potential in each lineage
In each species, for each lineage with at least ten copies (52 species-mariner lineages representing a total of 2673 copies), we counted the number of i) potentially mobilizable copies (characterized by two full-length TIRs), and ii) potentially coding copies (i.e., presenting an uninterrupted ORF of the expected size of between 330 – 363 aa). These various (non-mutually exclusive) types of copies may provide information on how MLEs decay over time; i.e., through loss of transposition ability or loss of activity. However, this crude analysis is likely to yield an overestimation of the number of active copies, particularly in old lineages, as substitutions in the sequence may lead to inactivation of the transposase without affecting the size of the ORF. In addition, we characterized the divergence of these lineages relative to their respective consensus sequence. Although it may be biased by the structuration of the lineage, the divergence from the majority-rule consensus is often used as an estimate of lineage age (Figure 4A, B).
Figure 4.
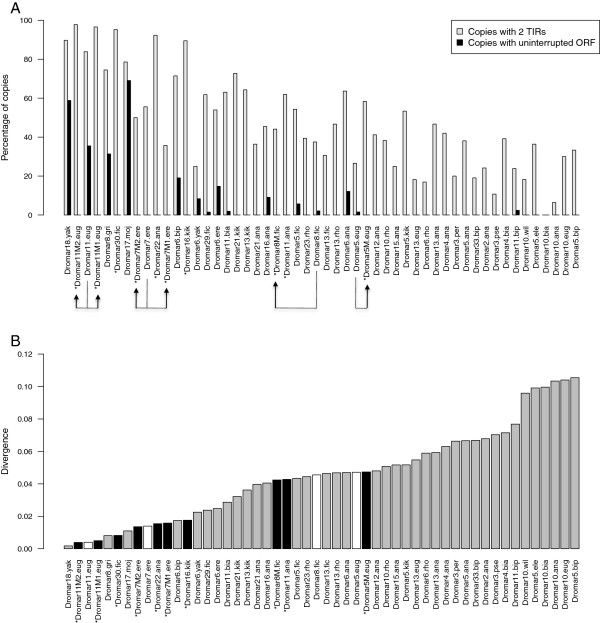
Structural characteristics of mariner lineages copies. A - Proportion of copies at two different conservation stages: with 2 TIRs and with an uninterrupted ORF between 330 and 363 AA in all lineages with at least 10 independent copies. An asterisk before the name denotes MITE sublineages. Arrows indicates full-length lineages and the derived MITEs sublineages. B - Mean divergence of the same lineages relative to the consensus sequence. Black bars are MITE sublineages, whites bars are their full-length partners.
First, we observed that only 15 lineages contained copies with an uninterrupted ORF. Four lineages containing between 20 and 50% of potentially active copies also corresponded to the less diverged lineages (less than 2%). In less conserved lineages, the proportion of copies with an uninterrupted ORF was typically very low, below 20%. Second, more than half of all copies (54%) still harbor both TIRs and may likely be mobilizable. Again, less diverged lineages approximately corresponded to those with more potentially mobilizable copies. MLE copies have previously been found to be frequently inactive due to frameshifts and stop codons [16, 26], which our analyses confirm. The loss of TIRs by deletion also appears to be important in explaining the decay of a lineage. mariner lineages appear to inactivate rapidly through mechanisms that impair either the coding ability of copies (modification of the ORF) or its ability to move (modification of the TIRs).
The few lineages with a high proportion of potentially active copies and a high proportion of 2-TIRs copies also contain a high number of copies, which likely reflects recent amplification activity. However, all other combinations were also observed, and no correlation could be established among copy number, mean divergence time and the presence of functional ORF or TIRs. Hence, some lineages are recent and active but with few copies (e.g., Dromar1, for which activity is biologically demonstrated); others are recent but inactive with few copies (e.g., Dromar9 and Dromar7). Others are ancient with many relic copies (e.g., Dromar2 and Dromar10). This analysis illustrates that each lineage has its own evolutionary history and that no one rule applies, even within the same genome.
Finally, among recent (less diverged) lineages, we found seven of the ten MITE sublineages included in our analysis. However, only two (Dromar11M1eug and Dromar11M2eug) were associated with a full-length lineage with potentially active copies in the same genome. Two others (Dromar5Meug and Dromar8Mfic) also have potentially active partners in the genome, but belong to older lineages. For the four “orphan” MITE lineages with no full-length partners (Dromar30fic, Dromar11ana, Dromar22ana, and Dromar16kik), we could not identify close active lineages that might have provided the transposase needed for transposition. However, we cannot exclude the possibility that active copies still exist in other individuals or populations of the species.
Mechanisms of inactivation
To determine whether some processes of inactivation are more prevalent than others, , we calculated the number of nucleotides lost, the number of nucleotides gained by insertion and the number of substitutions for each of the 52 species-mariner lineages. For indels, we distinguished cases in which deletion encompassed TE sequences only (internal deletions, potentially impairing ORF) from those extending into flanking regions, in which one end of the TE is lacking (truncation, affecting at least the mobility of the copy). A large insertion, alone or followed by other rearrangements, may also lead to the apparent truncation of the copy.
The first graph (Figure 5A) plots the average size of copies against lineage divergence (both relative to the consensus, excluding insertion). In agreement with the results shown in Figure 4, the observed negative correlation indicates that size reduction is a continuous process that, along with substitutions, contributes to the aging, inactivation and potentially, the disappearance of the lineages. Indeed, up to half of the original nucleotides can be lost in old lineages, ignoring the too-short copies (<400 bp) discarded during the initial search. Counting the insertions only reduces the coefficient of correlation (−0.7656), indicating that nucleotide gain by insertion cannot offset the number of nucleotides lost by internal deletion or truncation. Indeed, the number of nucleotides lost by internal deletion almost always exceeded the number gained by insertion, regardless of lineage age (mean divergence) (Figure 5B). The same pattern was revealed when computing the number of events (not shown, see Additional file 6). However, most internal deletion events are of very small size. The average size of deletions did not exceed 102 nt, and the maximum median deletion size was 22 nt (Additional file 6).
Figure 5.
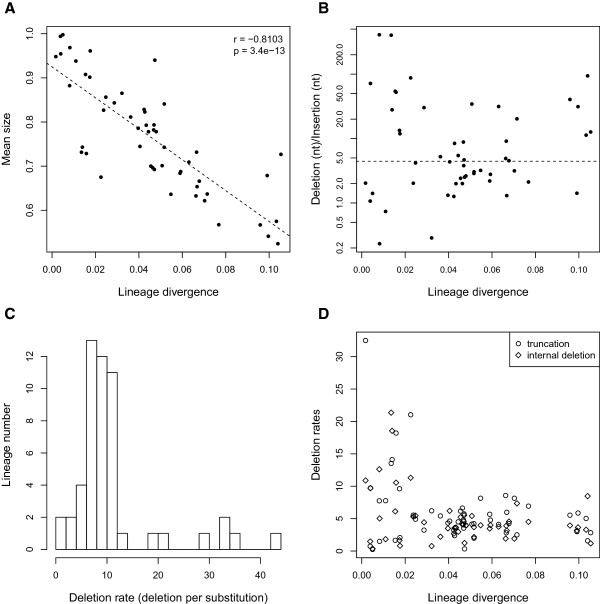
Degeneration patterns observed for lineages with at least 10 independent copies. (A) Mean size of copies in each lineage as a function of lineage divergence. The dotted line indicates the linear regression. (B) Ratio of nucleotides lost (by truncation or internal deletion) to nucleotides gained by insertion (log scale) as a function of lineage divergence. The dotted line corresponds to the median ratio. (C) Histogram of the deletion rate (number of nucleotides lost per substitution per lineage) distribution. (D) Deletion rates (internal deletion rate and truncation rate are dissociated) as a function of lineage divergence.
The ratio of deleted nucleotides to substitutions (Figure 5C) ranged from 1.5 to 45, but was between 4 and 12 for most lineages. This indicates that deletion is consistently more prevalent than substitution and is thus a major process leading to family inactivation. Plotting this ratio against nucleotide divergence (as a measure of lineage age) suggests that this may be particularly true for old lineages, whereas young lineages display more variable, sometimes very high, ratios (Figure 5D). A likely explanation for this pattern is that the high variability in young lineages is due to stochastic effects (occurrence of rare large deletions or truncations), whereas in old families, the effect of large deletions is offset by a higher number of substitutions. Alternatively, multiple large deletions in an old lineage will lead to the disappearance of copies and subsequently the lineage itself.
Amplification age and evolutionary history
In all our analyses, deletions affect copy functionality in an easily detectable way (ORF shortening and frameshifts), whereas substitutions leading to inactivation are only visible when they create a stop codon. Hence, among lineages that have apparent coding potential, some may actually be inactive. The divergence from consensus measure provides indications of the age of the lineages and the time of expansion, but it does not reveal whether the lineage remains active. Furthermore, this measure is only accurate when the consensus sequence reflects the ancestral active sequence. To estimate the global dynamics and activity of mariner lineages, we used the methodology developed by Le Rouzic, Payen and Hua-Van [34], based on phylogeny. In the tree, each node corresponds to a duplicative transposition and can be computed in a Lineage-Through-Time (LTT) plot depicting the rate of duplicative transposition events through time (Figure 6A). By this method, the variation in transposition rate per copy over time can be readily evidenced and amplification burst(s) within a species can be dated (in divergence units), allowing comparisons among species or MLE lineages. More simply, the evolutionary pattern can be inferred from the shape of the curve. Three schematic patterns are shown in Figure 6A that reflect the major patterns that arise from the analysis of the main mariner lineages. The first pattern is a past amplification burst followed by a stabilization in copy number, suggesting a loss of activity. The second pattern reveals successive amplification bursts. The third pattern involves an increase in transposition rate with time, compatible with an ongoing burst.
Figure 6.
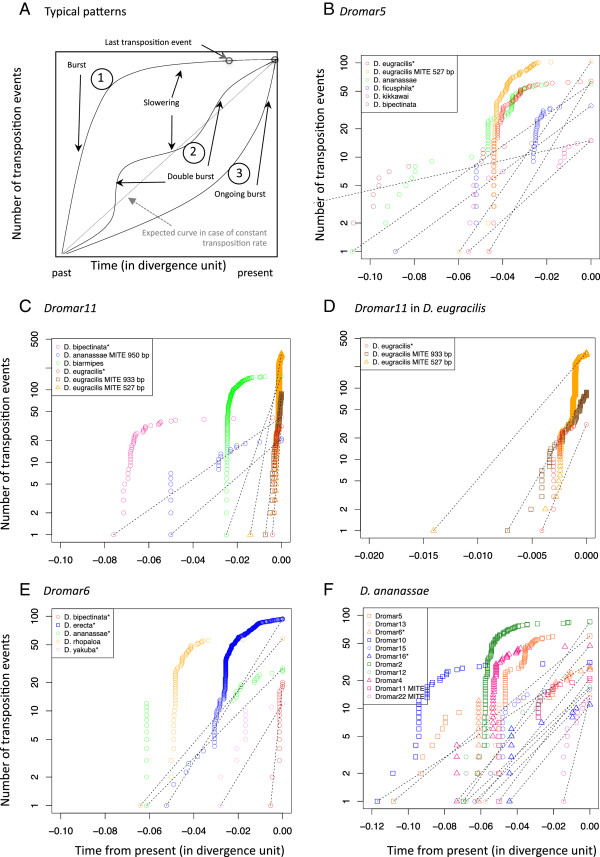
Dynamics analysis of different lineages in different species. (A) Interpretation of curve shape in theoretical LTT (Lineage-Through-Time) plot (cumulative number of transposition events over time, measured in genetic divergence units). (B) LTT plot of Dromar5 in 5 species. The dotted lines represent the theoretical curves assuming a constant transposition rate per copy over time (exponential transposition). (C) Same analysis as in B for Dromar11. (D) - Magnification of C for the three recent D. eugracilis Dromar11 sublineages. (E) Same analysis as in B for Dromar6. (F) LTT plot of different lineages in D. ananassae. Colors reflect the subfamily of each lineage following the color code in Figure 1.
We used this methodology to analyze (i) the same lineages in different species and (ii) different mariner lineages within the same species.
We first focused on some lineages exhibiting a large distribution, a high copy number and some potentially active copies (in Dromar5, Dromar6 and Dromar11). Dromar5 is present in all species from the melanogaster group with the exception of the melanogaster subgroup. This suggests that it was present in the common ancestor of this group, an inference supported by the presence of ancient copies and ancient bursts detected in some species (Figure 6B). However, in each species, the lineage amplified at different time (assuming a comparable molecular clock among all species). This lineage has been particularly successful in D. eugracilis, with numerous copies including a MITE family, and in D. ananassae and D. ficusphila. In D. eugracilis, MITE and non-MITE copies amplified at the same time. However, the non-MITE lineage has continued amplifying more recently than the MITE lineage. In D. ananassae and D. bipectinata, the lineage appears to be extinct; it also appears to be extinct in D. ficusphila, despite the presence of a potentially active copy in this species. In contrast, the lineage may not be totally extinct in D. kikkawai, although no potentially active copy has been detected in the sequenced genome.
Dromar11 is present in several species from the melanogaster group, and MITE lineages have been detected in D. eugracilis and D. ananassae. Again, amplification appears to have occurred at different times in each species (Figure 6C). An ancient transpositional burst followed by a decrease in activity is apparent in three species: D. biarmipes, D. bipectinata, and D. ananassae. The lineages may not be completely inactivated, as recent transpositions were also detected, compatible with the presence of potentially active copies in D. biarmipes and D. bipectinata. However, this is not expected for D. ananassae, in which only a MITE sublineage persists. In D. eugracilis, the lineage amplified more recently. For the two MITE sublineages, amplification is clearly ongoing (Figure 6D). However, it appears to have slowed down for the non-MITE partners. This situation illustrates the competition that may occur between autonomous and non-autonomous elements [35].
Dromar6 contains potentially active copies in four species. The analysis shown in Figure 6E revealed that the lineage is still transposing in these four species and that it is inactive in D. rhopaloa, which contains no potential active copies. In D. bipectinata, a species closely related to D. ananassae, the lineage appears recent and in an ongoing burst; this may be explained by recent acquisition through horizontal transfer.
Figure 6F illustrates the dynamics of different mariner lineages within D. ananassae, which contains numerous lineages. Among the 11 lineages analyzed, two old ones (Dromar10 and Dromar5) appeared to be completely extinct, following one or more transposition periods (double burst). Three displayed recent transposition: the MITE lineage Dromar22, which is the most recent in the genome but has no autonomous partner, and the two lineages with potentially active copies that have suffered several transposition bursts, suggesting reinvasion or reactivation of lineages. Indeed, these lineages do not appear to be more recent than several other lineages that are now extinct, and they first amplified during the same period (0.4 to 0.6 divergence units from present). Hence, amplification of MLE lineages occurs regularly, with each transposition burst eventually replaced by another one from another lineage.
TA insertion sites are not random
Mariner elements, like all elements of the Tc1-mariner- IS630 superfamily, insert into TA dinucleotides that are duplicated upon insertion. Few studies have searched for preferences other than this strict target site. To determine if there are differences in the immediate environment between insertion and non-insertion sites, we analyzed the region spanning 5 nt on either side of the insertion sites of some lineages from the five subfamilies. The percentage of T and A at each position was compared to the average percentage across all TAs of the genome (Figure 7). Some biases were observed within the 3 nt on either side of the insertion. A strong bias was observed for the mellifera subfamily, which appears to insert in very TA-rich regions. A similar tendency was observed for mauritiana and drosophila copies. For irritans, a bias was observed at the −3 and +3 positions. An analysis of the preference of the mellifera and drosophila subfamilies for trinucleotides revealed that not all TA-rich sequences are used. For example, for mellifera, the sequence ATA is highly preferred over the sequence ATT in the 5′ end of the insertion site, although in this case, we did not compute the frequency of these trinucleotides across the entire genome (Additional file 7). A WebLogo analysis confirmed the existence of a consensus sequence composed of an AT stretch for the mellifera, drosophila and mauritiana lineages (Additional file 8). In contrast, in the vertumnana and the irritans subfamilies, no consensus could be detected. The presence of the consensus was independent of copy number, active status, or species, but may be affected by the age of the lineage in a species (e.g., Dromar11 and Dromar8, which both have different amplification times between species). In this case, the apparent weakness of the consensus in old lineages may be explained by mutations that appeared after insertion. This suggests that the stringency of the TSD choice depends mainly on the peculiarity of each transposase, in terms of its binding affinity to or cutting efficiency of a particular sequence and is conserved within subfamilies.
Figure 7.
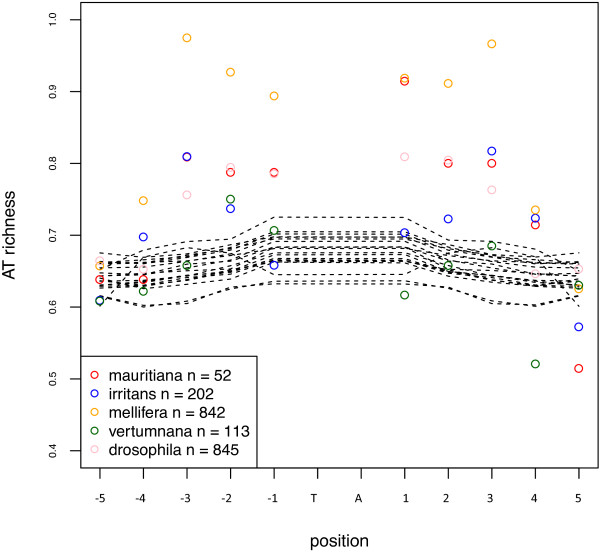
Percentage of TA at each position around the insertion TA. Results for each subfamily are shown as colored open circles (n: number of analyzed TA) and are compared to that obtained for all TAs in the 20 genomes (dotted black lines).
Discussion
Methodology
The use of both TBLASTN with 18 query transposases and MEGABLAST allowed us to identify more than 3685 copies representing 36 different mariner lineages. TBLASTN provided several hits that were identified as Tc1-like sequences, which indicates that the search was likely to be exhaustive. However, non-autonomous lineages could have been missed because we did not recover too-short sequences (<400 bp) or sequences with no conserved transposase domain, generated by internal deletions. Copies interrupted by insertions of less than 1000 nt could be reassociated. However, if an insertion is longer than 1000 nt, one copy could appear as two independent truncated copies. However, this imprecision is not expected to strongly bias the results. Hence, the panel of retrieved copies can be considered as representative of the mariner panorama in the Drosophila sequenced genomes.
Distribution, diversity and copy number across the genus
The 20 Drosophila genomes evaluated here belong mainly to the Sophophora subgenus and melanogaster group. Species from the same subgroup can have very different mariner content (e.g., D. melanogaster and D. yakuba). However, mariner lineages in the genomes of sister species appear more similar (e.g., D. simulans and D. sechellia, and D. pseudoobscura and D. persimillis). In this latter case, the majority of the copies lie at orthologous sites, allowing us to date the expansion of this lineage prior to speciation.
In the subgenus Drosophila, only 2 mariner lineages could be detected after the filtration process. This finding suggests that this subgenus is poor in mariner lineages. However, PCR searches for mariner elements in several species from the Drosophila subgenus have revealed the presence of at least three different subfamilies (mellifera, irritans and mauritina) in several neotropical species [27]. The low representation of MLEs in this subgenus may be due to poor sequencing sampling and the fact that those species are distantly related to those tested by Wallau et al. [27].
The 36 lineages identified across the Drosophila genus belong to 5 different subfamilies. One subfamily appeared as a new uncharacterized subfamily, and we named it the drosophila subfamily. However, representatives of this subfamily have been detected in a very distant genome (the cnidarian Hydra magnipapillata), which is described as a genome rich in Tc1-mariner in which several horizontal transfers have occurred [36].
Several of the 36 lineages are restricted to one or a few closely related species. Most of the lineages are in very low copy number and can therefore be easily lost. These findings suggest that the total number of different lineages within the Drosophila genus is large and that analyzing new species will uncover new lineages.
Marineractivity and inactivation
In addition to the wide diversity of mariner lineages, most of them correspond to inactive lineages. The absence of potentially active copies does not mean that the lineage is extinct, as the data come from highly inbred lines and represent little of the variability in natural populations. Nevertheless, this result supports previous findings that mariner elements are easily vertically inactivated [16]. In the inactivation process, the loss of nucleotides appears to play an important role. In old lineages, almost half of the nucleotides are lost. Therefore, the fate of MLEs in a genome appears to be elimination. Complete removal of a lineage can occur through a persistently low level of reinsertion or vertical inactivation followed by rapid elimination by the genome. The large variety of mariner lineages in the Drosophila genomes, along with the deep phylogenetic relationships, high deletion rate (Figure 4) and very recent amplifications of these elements (Figures 4 and 6), suggest that this diversity is ancestral and that these genomes might be regularly fed with the old lineages through horizontal transfer events. The deletion rate data are in agreement with the high rate of deletion found for the helena non-LTR retroelement in the D. melanogaster and D. virilis genomes [37, 38]. Some MLEs presented a higher deletion rate than other mariner lineages, as is shown in Figure 4D. However, these lineages with high deletion rates are among the more recently amplified ones and were found in different species; therefore, it is not clear whether some deletion mechanisms are responsible or if this pattern is due to stochastic effects.
MITEs
Originally, MITEs were described as high copy number, short elements in plants [39], reaching thousands of copies. In Drosophila, few MITEs have been characterized to date, and the copy number has not exceeded 100 [40–42]. Furthermore, few mariner MITEs have been described, the most notable case being the MiHsmar1 family in humans [43]. Almost a quarter of the copies corresponded to 27 different MITE sublineages from 14 MLE lineages, 17 from the drosophila subfamily and 6 from the mauritiana subfamily, suggesting that these subfamilies were particularly likely to generate such shorter elements. The copy number was typically very low among both the MITE sequences and the longer elements. The term “MITEs” might therefore appear inappropriate, although these copies transposed at least once, as indicated by their flanking sequences. However, it cannot be excluded that these low copy number MITEs reached a high copy number, as is theoretically possible for the few lineages with high copy number (from 100 to more than 300), which has never before been observed in Drosophila.
The approximate size uniformity (23 sub-lineages between 900 and 970 bp, and 4 between 467 and 560 bp) was particularly unexpected and suggests that MITE transposition ability may be strongly constrained by either size or structure. This scenario is supported by the observation that in other internally deleted elements (that have not amplified), the size distribution does not exhibit any strong bias (not shown). Size constraints have been reported for some mariner elements [44], although there are also examples of successful transposition events of very short elements among MLEs [43, 45].
Although knowledge of the dynamics and the transposition process of MITEs continues to grow, the origins of these elements remain unclear. Indeed, sometimes the sequence similarities to the putative autonomous partner are restricted to the TIRs. In other cases, large TIRs are present, but similarities are only visible at the tip of the TIRs [46]. The de novo formation hypothesis (that similar solo-TIR-like sequences close to each other may generate these short non-autonomous TEs) has not found empirical support to date. The alternative hypothesis assumes an internal deletion of the autonomous element, with degeneration or substitution of internal sequence [40, 47]. Amplification could result from the recognition of TIRs by the transposase of active copies from the same lineage or from distantly related lineages [48]. In our study, all MITE lineages could be traced back to a putative mariner full-length lineage due to homologous parts in the internal region, as expected from the homology-based strategy used. No instance of severe degeneration was detected. Our analyses indicate that internal deletion appears to be a major process in the generation of non-autonomous copies and MITEs. However, we detected seven cases where internal deletion was accompanied (or caused) by rearrangements systematically consisting of the replacement of the 3′ part of the element by a 5′ portion. In one case, corresponding to the most abundant MITE, two TIRs were present in the 3′ region. The absence of MITE copies with only one 3′ terminal TIR indicates that the most external TIR is strongly favored for transposition, perhaps providing the element with optimal size. Optimal size may add further to the potential absence of a repressive motif in the MITEs, explaining MITE success [49]. A side effect of the rearrangement process is the large increase in the size of the inverted repetition, which may increase transposition ability by stabilizing the synaptic complex. Although experimental evidence is lacking, successful MITEs, such as the first ones identified in plants, exhibit a high potential to form stable secondary structures [50]. Finally, whereas the origin of internal deletions can easily be explained by abortive gap repair [51], the origin of more complex rearrangement remains elusive.
Dynamics of transposition and the horizontal transfer hypothesis
The dynamics analysis revealed several patterns that conflict with the hypothesis of a constant transposition rate per copy. The temporal variation in transposition rate per copy appears to consistently follow the same progression: an initial high transposition rate corresponding to a burst of amplification, followed by progressive attenuation. This pattern may be due to two non-exclusive processes. First, copy inactivation acts rapidly, diminishing the proportion of active copies (encoding an active transposase). Therefore, if transposition is limited by transposase availability, the transposition rate per copy will decrease. Second, the decrease in transposition rate per copy over time may reflect the initiation of regulatory processes. The relative contributions of these processes can likely be estimated from the dynamics by considering active (encoding) and inactive (but trans-mobilizable) copies over time, which we aim to investigate in the future.
The similarities in the amplification dynamics suggests a general pattern, in accordance with theoretical models [52] and models derived from experimental observations [16]. However, the dynamics observed in other species and other Class II elements may be very different, such as those observed for pogo-like elements in the fungus Fusarium oxysporum [34]. In this latter case, transposition rates per copy do not vary widely over time.
Mariner is known to undergo frequent horizontal transfer (HT) [53]. Posited factors supporting HT are a patchy distribution, a TE phylogeny incongruent with that of the host and the presence of highly similar sequences in distantly related species [54]; this latter hypothesis is difficult to test definitely, as the true donor may not be part of the sample. Another possible factor that may reflect HT are differences in amplification times or patterns of the same lineage observed between different species.
Horizontal transfer followed by rapid amplification burst may explain why lineages appear as recent and active in some species, while old and inactive in others. Dromar8 displays a very patchy distribution, and it is present in distantly related species in which it displays varying amplification times. This lineage is very young in D. grimshawi, with a high proportion of potentially coding copies relative to the apparently older lineage present in the distantly related D. ficusphila. This lineage may represent a good candidate for horizontal transfer. Dromar18 and Dromar17 are two other recent lineages with a restricted species-specific distribution, and may also have arisen by HT. In particular, Dromar18, which is not present in closely related species, may have arisen by HT. Dromar17 contains some old copies and may have resulted from a re-activation process.
However, several mariner lineages are widely distributed within the melanogaster group. Such a distribution suggests an ancestral presence and vertical transmission. Another hypothesis is that amplification of the lineage predated speciation within the group. However, the Dromar5, Dromar6 and Dromar11 lineages exhibited some amplification time differences among species, as did Dromar4 (not shown), which suggests that the ancestral presence of the elements may not fully explain the observed pattern. Horizontal transfer with replacement of old copies or reactivation may have occurred. The detailed analysis of the amplification dynamics of Dromar6 provides another example of a probable horizontal transfer. This lineage is present in the closely related species D. ananassae and D. bipectinata, and although this suggests its potential presence in the ancestor of the two species, the lineage in D. bipectinata latter species is much more recent and is therefore better explained by horizontal transfer.
As stressed previously [24], only a combination of these arguments constitutes a convincing basis for invoking HT. The results of the analysis of amplification time provide a new, additional argument. However, these arguments are insufficient to demonstrate HT, and further analyses are needed to test the HT hypothesis. One approach may include a new methodology for HT detection, which compares the synonymous substitution rates in elements and genes [20]; however, the results should also be interpreted with care [55]. We are currently developing a new method that automatically compares TEs and genes and is based on substitution rates and other parameters. This method may help detect HT at a comparative genomic scale.
Competition at different levels
Transposable elements are pieces of selfish DNA; i.e., DNA that promotes its own perpetuation in the genome without participating in the survival of the host cell or organism [56, 57]. Nevertheless, the exponential or infinite multiplication of TEs may be limited by their elimination through natural selection, once such multiplication becomes too harmful to the host [58, 59]. The establishment of regulatory mechanisms also explains why TE copy number may remain relatively low. Regulation by silencing through epigenetic processes is a universal strategy used by genomes to minimize the harmfulness of invading sequences [60]. The genome wide epigenetic silencing of TEs appears to result from the combination of processes targeted toward particular TE families and that rely on sequence homology. Hence, this host defense system is highly dynamic and illustrates the arms race between the genome and TEs [61]. Some authors have proposed an ecological view of the genome in which TE copies and families are viewed analogous to individuals and species that compete for the same ecological niches [19, 62, 63]. Competition between copies of the same lineage can occur when transposase resources become limited due to the progressive inactivation of TEs. At the extreme, some copies becomes parasitic on others, and MITEs constitute the most convincing example. The dynamics of Dromar11 in D. eugracilis illustrates this process, as the full-length sub-lineage shows a marked lower transposition rate than the associated MITEs, at a recent time. As a counter example, the Dromar5 full-length lineage appears to be more efficient in promoting its own transposition than was the associated MITE in the recent past. This finding may reflect that this older lineage is involved in a different stage of the cyclical interaction predicted by theoretical studies [35]. Further analysis of the mutational pattern may be useful in testing this hypothesis.
Parasitism and competition might even occur between copies from closely related lineages (same subfamilies) when they are able to cross-mobilize [49]. In genomes with numerous mariner lineages, there are several examples of concomitant amplification, suggesting that competition between lineages is limited. Nevertheless, we noticed that concomitant amplification never involved lineages from the same subfamilies. At the same time, several non-autonomous elements lack autonomous partners, but are recently amplified. No close partners could be identified, possibly because the sequenced genome of one individual, while potentially representative of the average genome of a population, does not contain every copy present in the population.
Conclusions
With 36 different mariner lineages identified, this analysis sheds light on the powerful ability of mariner to diversify. We detected some subfamily specificity (low copy number for mauritiana, ability to form MITE sublineages for mauritiana and drosophila, strong insertion site bias for mauritiana and mellifera), although the causes remain unclear. The dynamics analysis revealed that amplification is the result of a short transposition burst followed by stagnation (and ultimately decay), most likely due to rapid inactivation or regulation. Horizontal transmission or reactivation may compensate for the continuous inactivation of copies that leads to the death of the lineage. This scheme is in accordance with the mariner life cycle suggested by Lohe et al. [16] from their analysis of MLE PCR fragments in various animals. The dynamics analysis also provides a way to access the competition process, particularly between non-autonomous and autonomous partners, previously evidenced in a theoretical study [19]. The different examples of MITEs detected here provide insights into the creation and amplification of these elements. This genus-scale analysis illustrates the power of comparative genomics to decipher the evolution of transposable elements. In the near future, accumulating population genomics data should permit deeper analysis and improved understanding of TE evolution.
Methods
Detection of marinersequences
The strategy used to recover MLEs is depicted in Figure 2. We used several programs from the BLAST + suite [64]. Based on their availability in the protein database and their diversity, we selected 18 transposases (Table 1), covering at least 8 subfamilies, and used them as queries in a batch TBLASTN search performed on the WGS sequences of 20 Drosophila genomes [28] and Piano, Cherbas [29], using the default cut-off value. We recovered 14977 different hits (High Scoring Pairs) and reassembled them into 3694 copies according to the following criteria. Two hits from the same scaffold are grouped together if the distance between the center of each hit is less than 1000 bp and if they have the same orientation. Only copies longer than 400 pb were retained. Clustering with USEARCH v 6.0 [65] using a threshold of 80% identity provided 145 Clusters. A BLASTX search against transposases from the Tc1 family was conducted with the consensus sequences of the 145 clusters as queries. Forty-six clusters were found to be more related to Tc1 elements and were excluded from analysis. The remaining clusters were inspected manually. At the same time, a global alignment was performed with MAFFT v.7 [66] on the sequences and their flanking regions (250 bp each side). From these analyses, 36 clusters were considered bona fide MLEs, and consensus sequences were derived and translated into protein for phylogenetic purposes. Consensus sequences were also used as queries in a MEGABLAST search against the 20 Drosophila genomes to precisely determine copy numbers and copy ends (including truncated or deleted copies lacking recognizable conserved protein motifs). This resulted in 3685 sequences. The full sequence presenting homology along with 250 of each flanking sequences were then extracted. Duplicated sequences (same flanking regions) as well as truncated copies located at the end of contigs and supercontigs (ends of the sequence, presence of ‘N’ nucleotides) were further eliminated to obtain a clean dataset of 3084 sequences.
Alignment and consensus
Nucleotides and proteins were aligned with MAFFT v.7 [66] or MUSCLE3.8 [67], and evaluated and refined by hand. Consensus sequences were derived using the relative majority rule. Gaps were distinguished between internal gaps and truncations. When the majority was an internal gap, a gap was included in the consensus. Gaps at the end of the sequences were not counted. Consensus sequences were evaluated and corrected as needed to obtain full-length sequences.
Distance to genes
The distance of elements to genes was calculated for Dromar5 and Dromar11 in D. eugracilis. The D. melanogaster protein database was used for TBLASTN queries on the D. eugracilis genome to identify homologous gene sequences positions. The distance to the closest putative gene was then computed for copies present in the same contig as a gene.
Inactivation analysis
Copies were assumed to contain 2 TIRs if they were not truncated at the end. Hence, mismatches or substitutions in the TIRs, which may affect mobility, were not accounted for. Copies were considered to possess an uninterrupted ORF if the ORF comprised between 330 and 363 codons. It is likely that this relaxed constraint leads to the overestimation of the number of such copies. Furthermore, the ability to encode an active transposase also depends on the protein sequence. The lineage divergence is based on the divergence to the consensus, recalculated for each lineage in each genome. It takes into account the total number of substitutions and the total numbers of insertion and deletion events. The sum is then divided by the total number of nucleotides aligning with the consensus sequence (large insertions are not counted). For indels, the numbers of nucleotides inserted and deleted were calculated relative to the consensus sequence. For deletions, two cases were considered: internal deletions or end truncations. Deletions events are sometimes shared by several sequences because they occurred before transposition. All these redundant deletions were counted only once.
Shorter copies (less than 1000 bp) with evidence of transposition (at least 2 copies with TIRs, bordered by different flanking regions) were considered as MITEs. MITE classification relied on homology with longer MLEs (in the TIRs and internal sequences) and on the breakpoints of deletion/rearrangements. In this analysis, MITEs with no internal homology could not be retrieved.
Analysis of amplification dynamics
Species-specific amplification dynamics of single lineages were inferred using a new method based on the phylogenetic tree node distributions over time. This method relies on the topology of the tree and offers a visualization of the variation in transposition rate over time. More details are available in Le Rouzic, Payen, Hua-Van [34]. The trees were rooted with an outgroup corresponding to the consensus sequence of the closest MLE lineage. All trees were reconstructed using FastTree 2.1 [68]. All insertions were removed from the sequence dataset.
Phylogenetic analyses
The phylogenetic analysis of transposases was performed with MrBayes 3.2.1 [69] using the transposase derived from the consensus of each lineage along with the MLE proteins used in the BLASTP searches. For this analysis, we used the amino acid substitution model WAG + G, suggested as the best model by ProtTest 3 [70]. Two million generations were evaluated, sampling the most probable tree every 100 generations and burning 25% of those. Sequences used as outgroups were Bmmar1 from Bombyx mori, a Tc1-mariner element from the maT (DD37D) family and Tvmar1 from Trichomonas vaginalis. The resulting tree can be acessed at TreeBASE repository [71].
Insertion sites
For each subfamily, the percentage of T or A at the 10 positions surrounding the TA insertion site was computed and compared to the percentage found for all TAs in the 20 genomes. For each mariner lineage, a consensus derived from the immediate flanking sequence was calculated with WebLogo [72].
Availability of supporting data
The genome sequences of the Drosophila species can be accessed at FlyBase (http://flybase.org/) or at NCBI (http://www.ncbi.nlm.nih.gov/). All mariner copies can be retrieved using their positions given in Additional file 2: Table S2.
Electronic supplementary material
Additional file 1: Table S1: Description of all lineages identified in the 20 species. (PDF 80 KB)
Additional file 2: Table S2: Supercontig locations of all mariner copies identified in this study. Positions refer to start and end of regions reported as similar to the consensus sequence after MEGABLAST analysis. They may sometimes slightly differ from positions after manual curation. (XLSX 163 KB)
Additional file 3: Figure S1: Open Reading Frames and TIRs. For each lineage, (+ +) indicated the presence of uninterrupted ORF in both copies and consensus sequence, (− +) indicated the presence of uninterrupted ORF in the consensus sequence only. TIR consensus are reported on the right. (PDF 310 KB)
Additional file 4: Table S3: Description of MITE sublineages. (PDF 57 KB)
Additional file 5: Figure S2: Violin pots depicting the distribution of copies relative to genes for Dromar5 and Dromar11 in D. Eugracilis. Significant differences (Kolgomorov-Smirnov test) are indicated. M: MITE lineage, FL: full-length lineage. n: copy number (percentage of copies located on genes-containing contigs). (PDF 101 KB)
Additional file 6: Table S4: Sequence evolution characteristics in mariner lineages. (XLSX 54 KB)
Additional file 7: Figure S3: Analysis of the trinucleotide preference in 5′ (A) and 3′ (B) of the insertion TA site for the mellifera and drosophila subfamilies. Only trinucleotides found more than 5 times in the data are presented. (PDF 151 KB)
Additional file 8: Figure S4: Consensus analysis of the region surrounding the insertion sites of different lineages, using WebLogo [71]. The duplicated target TA is present on each site (central TAs). The elements have been removed. The numbers into parentheses indicated the number of 5′ and 3′ flanking regions analysed. (PDF 291 KB)
Acknowledgments
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenção de Aperfeiíoamento de Pessoal de Nível Superior (CAPES) and the Centre National de la Recherche Scientifique (CNRS). The first author of this manuscript was a “Bolsista da CAPES – Proc. N° 6145116”. We thank Arnaud Le Rouzic for his critique of the manuscript and valuable comments. We thank AJE for English editing.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GLW carried out the bioinformatics analyses of the study, participated in the study design and drafted the manuscript. PC participated in the design of the study and helped draft the manuscript. EL participated in the design of the study and helped draft the manuscript. AH conceived of the study, participated in its design and coordination and helped draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Gabriel Luz Wallau, Email: gabriel.wallau@gmail.com.
Pierre Capy, Email: pierre.capy@legs.cnrs-gif.fr.
Elgion Loreto, Email: elgionl@gmail.com.
Aurélie Hua-Van, Email: Aurelie.Hua-Van@legs.cnrs-gif.fr.
References
- 1.Jacobson JW, Medhora MM, Hartl DL. Molecular structure of a somatically unstable transposable element in Drosophila. Proc Natl Acad Sci U S A. 1986;83:8684–8688. doi: 10.1073/pnas.83.22.8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson HM. The mariner transposable element is widespread in insects. Nature. 1993;362:241–245. doi: 10.1038/362241a0. [DOI] [PubMed] [Google Scholar]
- 3.Doak TG, Doerder FP, Jahn CL, Herrick G. A proposed superfamily of transposase genes: transposon-like elements in ciliated protozoa and a common “D35E” motif. Proc Natl Acad Sci U S A. 1994;91:942–946. doi: 10.1073/pnas.91.3.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brillet B, Yves B, Corinne A-G. Assembly of the Tc1 and mariner transposition initiation complexes depends on the origins of their transposase DNA binding domains. Genetica. 2007;130(2):105–120. doi: 10.1007/s10709-006-0025-2. [DOI] [PubMed] [Google Scholar]
- 5.Robertson HM, MacLeod EG. Five major subfamilies of mariner transposable elements in insects, including the Mediterranean fruit fly, and related arthropods. Insect Mol Biol. 1993;2:125–139. doi: 10.1111/j.1365-2583.1993.tb00132.x. [DOI] [PubMed] [Google Scholar]
- 6.Bigot Y, Hamelin MH, Capy P, Periquet G. Mariner-like elements in hymenopteran species: insertion site and distribution. Proc Natl Acad Sci U S A. 1994;91:3408–3412. doi: 10.1073/pnas.91.8.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robertson HM. The Tc1-mariner superfamily of transposons in animals. J Insect Physiol. 1995;41:99–105. doi: 10.1016/0022-1910(94)00082-R. [DOI] [Google Scholar]
- 8.Garza D, Medhora M, Koga A, Hartl DL. Introduction of the transposable element mariner into the germline of Drosophila melanogaster. Genetics. 1991;128:303–310. doi: 10.1093/genetics/128.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medhora M, Maruyama K, Hartl DL. Molecular and functional analysis of the mariner mutator element Mos1 in Drosophila. Genetics. 1991;128:311–318. doi: 10.1093/genetics/128.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casse N, Bui QT, Nicolas V, Renault S, Bigot Y, Laulier M. Species sympatry and horizontal transfers of Mariner transposons in marine crustacean genomes. Mol Phylogenet Evol. 2006;40:609–619. doi: 10.1016/j.ympev.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Rouault JD, Casse N, Chenais B, Hua-Van A, Filee J, Capy P. Automatic classification within families of transposable elements: application to the mariner Family. Gene. 2009;448:227–232. doi: 10.1016/j.gene.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Robertson HM, Asplund ML. Bmmar1: a basal lineage of the mariner family of transposable elements in the silkworm moth, Bombyx mori. Insect Biochem Mol Biol. 1996;26:945–954. doi: 10.1016/S0965-1748(96)00061-6. [DOI] [PubMed] [Google Scholar]
- 13.Lampe DJ, Walden KK, Robertson HM. Loss of transposase-DNA interaction may underlie the divergence of mariner family transposable elements and the ability of more than one mariner to occupy the same genome. Mol Biol Evol. 2001;18:954–961. doi: 10.1093/oxfordjournals.molbev.a003896. [DOI] [PubMed] [Google Scholar]
- 14.Witherspoon DJ, Robertson HM. Neutral evolution of ten types of mariner transposons in the genomes of Caenorhabditis elegans and Caenorhabditis briggsae. J Mol Evol. 2003;56:751–769. doi: 10.1007/s00239-002-2450-x. [DOI] [PubMed] [Google Scholar]
- 15.Lorite P, Maside X, Sanllorente O, Torres MI, Periquet G, Palomeque T. The ant genomes have been invaded by several types of mariner transposable elements. Naturwissenschaften. 2012;99:1007–1020. doi: 10.1007/s00114-012-0982-5. [DOI] [PubMed] [Google Scholar]
- 16.Lohe AR, Moriyama EN, Lidholm DA, Hartl DL. Horizontal transmission, vertical inactivation, and stochastic loss of mariner-like transposable elements. Mol Biol Evol. 1995;12:62–72. doi: 10.1093/oxfordjournals.molbev.a040191. [DOI] [PubMed] [Google Scholar]
- 17.Hartl DL, Lohe AR, Lozovskaya ER. Modern thoughts on an ancyent marinere: function, evolution, regulation. Annu Rev Genet. 1997;31:337–358. doi: 10.1146/annurev.genet.31.1.337. [DOI] [PubMed] [Google Scholar]
- 18.Le Rouzic A, Capy P. The first steps of transposable elements invasion: parasitic strategy vs. genetic drift. Genetics. 2005;169:1033–1043. doi: 10.1534/genetics.104.031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Rouzic A, Boutin TS, Capy P. Long-term evolution of transposable elements. Proc Natl Acad Sci U S A. 2007;104:19375–19380. doi: 10.1073/pnas.0705238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva JC, Kidwell MG. Horizontal transfer and selection in the evolution of P elements. Mol Biol Evol. 2000;17:1542–1557. doi: 10.1093/oxfordjournals.molbev.a026253. [DOI] [PubMed] [Google Scholar]
- 21.Lampe DJ, Witherspoon DJ, Soto-Adames FN, Robertson HM. Recent horizontal transfer of mellifera subfamily mariner transposons into insect lineages representing four different orders shows that selection acts only during horizontal transfer. Mol Biol Evol. 2003;20:554–562. doi: 10.1093/molbev/msg069. [DOI] [PubMed] [Google Scholar]
- 22.Augé-Gouillou C, Bigot Y, Periquet G. Mariner-like sequences are present in the genome of the fruitfly, Drosophila melanogaster. J Evol Biol. 1999;12:742–745. doi: 10.1046/j.1420-9101.1999.00068.x. [DOI] [Google Scholar]
- 23.Brunet F, Godin F, David JR, Capy P. The mariner transposable element in the Drosophilidae family. Heredity. 1994;73(Pt 4):377–385. doi: 10.1038/hdy.1994.185. [DOI] [PubMed] [Google Scholar]
- 24.Capy P, Langin T, Bigot Y, Brunet F, Daboussi MJ, Periquet G, David JR, Hartl DL. Horizontal transmission versus ancient origin: mariner in the witness box. Genetica. 1994;93:161–170. doi: 10.1007/BF01435248. [DOI] [PubMed] [Google Scholar]
- 25.Brunet F, Godin F, Bazin C, Capy P. Phylogenetic analysis of Mos1-like transposable elements in the Drosophilidae. J Mol Evol. 1999;49:760–768. doi: 10.1007/PL00006598. [DOI] [PubMed] [Google Scholar]
- 26.Brunet F, Giraud T, Godin F, Capy P. Do deletions of mos1-like elements occur randomly in the drosophilidae family? J Mol Evol. 2002;54:227–234. doi: 10.1007/s0023901-0004-2. [DOI] [PubMed] [Google Scholar]
- 27.Wallau GL, Hua-Van A, Capy P, Loreto EL. The evolutionary history of mariner-like elements in Neotropical drosophilids. Genetica. 2011;139:327–338. doi: 10.1007/s10709-011-9552-6. [DOI] [PubMed] [Google Scholar]
- 28.Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 29.Piano , Cherbas 2008.http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/modENCODE_ComparativeGenomics_WhitePaper.pdf
- 30.Kumar S, Hedges SB. TimeTree2: species divergence times on the iPhone. Bioinformatics. 2011;27:2023–2024. doi: 10.1093/bioinformatics/btr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bui QT, Casse N, Leignel V, Nicolas V, Chenais B. Widespread occurence of mariner transposons in coastal crabs. Mol Phylogenet Evol. 2008;47:1181–1189. doi: 10.1016/j.ympev.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 32.Kuang H, Padmanabha C, Li F, Kamei A, Bhaskar PB, Ouyang S, Jiang J, Buell CR, Baker B. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res. 2009;19(1):42–56. doi: 10.1101/gr.078196.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wessler S. Transposable elements and the evolution of gene expression. Symp Soc Exp Biol. 1998;51:115–122. [PubMed] [Google Scholar]
- 34.Le Rouzic A, Payen T, Hua-Van A. Reconstructing the evolutionary history of transposable elements. Genome Biol Evol. 2013;5:77–86. doi: 10.1093/gbe/evs130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Rouzic A, Capy P. Population genetics models of competition between transposable element subfamilies. Genetics. 2006;174:785–793. doi: 10.1534/genetics.105.052241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chapman JA, Kirkness EF, Simakov O, Hampson SE, Mitros T, Weinmaier T, Rattei T, Balasubramanian PG, Borman J, Busam D, Disbennett K, Pfannkoch C, Sumin N, Sutton GG, Viswanathan LD, Walenz B, Goodstein DM, Hellsten U, Kawashima T, Prochnik SE, Putnam NH, Shu S, Blumberg B, Dana CE, Gee L, Kibler DF, Law L, Lindgens D, Martinez DE, Peng J, et al. The dynamic genome of Hydra. Nature. 2010;2010(464):592–596. doi: 10.1038/nature08830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrov DA, Hartl DL. High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol Biol Evol. 1998;15:293–302. doi: 10.1093/oxfordjournals.molbev.a025926. [DOI] [PubMed] [Google Scholar]
- 38.Petrov DA. DNA loss and evolution of genome size in Drosophila. Genetica. 2002;115:81–91. doi: 10.1023/A:1016076215168. [DOI] [PubMed] [Google Scholar]
- 39.Wessler SR, Bureau TE, White SE. LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995;5:814–821. doi: 10.1016/0959-437X(95)80016-X. [DOI] [PubMed] [Google Scholar]
- 40.De Freitas OM, Silva LE. The hobo-related elements in the melanogaster species group. Genet Res. 2008;90:243–252. doi: 10.1017/S0016672308009312. [DOI] [PubMed] [Google Scholar]
- 41.Dias ES, Carareto CM. msechBari, a new MITE-like element in Drosophila sechellia related to the Bari transposon. Genet Res (Camb) 2011;93:381–385. doi: 10.1017/S0016672311000371. [DOI] [PubMed] [Google Scholar]
- 42.Depra M, Ludwig A, Valente VL, Loreto EL. Mar, a MITE family of hAT transposons in Drosophila. Mob DNA. 2012;3:13. doi: 10.1186/1759-8753-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z. The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol. 2007;27:4589–4600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lohe AR, Hartl DL. Reduced germline mobility of a mariner vector containing exogenous DNA: effect of size or site? Genetics. 1996;143:1299–1306. doi: 10.1093/genetics/143.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinzelle L, Jegot G, Brillet B, Rouleux-Bonnin F, Bigot Y, Auge-Gouillou C. Factors acting on Mos1 transposition efficiency. BMC Mol Biol. 2008;9:106. doi: 10.1186/1471-2199-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rezsohazy R, van Luenen HG, Durbin RM, Plasterk RH. Tc7, a Tc1-hitch hiking transposon in Caenorhabditis elegans. Nucleic Acids Res. 1997;1997(25):4048–4054. doi: 10.1093/nar/25.20.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feschotte C, Zhang X, Wessler SR. Miniature inverted-repeat transposable elements and their relationship to established DNA transposons. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington DC.: ASM Press; 2002. pp. 1147–1158. [Google Scholar]
- 48.Jiang N, Feschotte C, Zhang X, Wessler SR. Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs) Curr Opin Plant Biol. 2004;7:115–119. doi: 10.1016/j.pbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR. Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science. 2009;325:1391–1394. doi: 10.1126/science.1175688. [DOI] [PubMed] [Google Scholar]
- 50.Tu Z. Three novel families of miniature inverted-repeat transposable elements are associated with genes of the yellow fever mosquito, Aedes aegypti. Proc Natl Acad Sci U S A. 1997;94:7475–7480. doi: 10.1073/pnas.94.14.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubin E, Levy AA. Abortive gap repair: underlying mechanism for Ds element formation. Mol Cell Biol. 1997;17:6294–6302. doi: 10.1128/mcb.17.11.6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Rouzic A, Dupas S, Capy P. Genome ecosystem and transposable elements species. Gene. 2007;2007(390):214–220. doi: 10.1016/j.gene.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 53.Robertson HM, Soto-Adames FN, Walden KK, Avancini RMP, Lampe DJ. The mariner Transposons of Animals: Horizontally Jumping Genes. In: Syvanen M, Kado CI, editors. Horizontal Gene Transfer. 2002. pp. 173–185. [Google Scholar]
- 54.Loreto EL, Carareto CM, Capy P. Revisiting horizontal transfer of transposable elements in Drosophila. Heredity (Edinb) 2008;100:545–554. doi: 10.1038/sj.hdy.6801094. [DOI] [PubMed] [Google Scholar]
- 55.Wallau GL, Ortiz MF, Loreto EL. Horizontal transposon transfer in eukarya: detection, bias, and perspectives. Genome Biol Evol. 2012;4:689–699. doi: 10.1093/gbe/evs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doolittle WF, Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980;284:601–603. doi: 10.1038/284601a0. [DOI] [PubMed] [Google Scholar]
- 57.Orgel LE, Crick FH. Selfish DNA: the ultimate parasite. Nature. 1980;284:604–607. doi: 10.1038/284604a0. [DOI] [PubMed] [Google Scholar]
- 58.Charlesworth B, Sniegowski P, Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature. 1994;371:215–220. doi: 10.1038/371215a0. [DOI] [PubMed] [Google Scholar]
- 59.Nuzhdin SV. Sure facts, speculations, and open questions about the evolution of transposable element copy number. Genetica. 1999;107:129–137. doi: 10.1023/A:1003957323876. [DOI] [PubMed] [Google Scholar]
- 60.Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 61.Obbard DJ, Gordon KH, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc Lond B Biol Sci. 2009;364:99–115. doi: 10.1098/rstb.2008.0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brookfield JF. The ecology of the genome - mobile DNA elements and their hosts. Nat Rev Genet. 2005;6:128–136. doi: 10.1038/nrg1524. [DOI] [PubMed] [Google Scholar]
- 63.Venner S, Feschotte C, Biemont C. Dynamics of transposable elements: towards a community ecology of the genome. Trends Genet. 2009;25:317–323. doi: 10.1016/j.tig.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 66.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 70.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Data set available at the TreeBASE repositoryhttp://purl.org/phylo/treebase/phylows/study/TB2:S16144
- 72.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Description of all lineages identified in the 20 species. (PDF 80 KB)
Additional file 2: Table S2: Supercontig locations of all mariner copies identified in this study. Positions refer to start and end of regions reported as similar to the consensus sequence after MEGABLAST analysis. They may sometimes slightly differ from positions after manual curation. (XLSX 163 KB)
Additional file 3: Figure S1: Open Reading Frames and TIRs. For each lineage, (+ +) indicated the presence of uninterrupted ORF in both copies and consensus sequence, (− +) indicated the presence of uninterrupted ORF in the consensus sequence only. TIR consensus are reported on the right. (PDF 310 KB)
Additional file 4: Table S3: Description of MITE sublineages. (PDF 57 KB)
Additional file 5: Figure S2: Violin pots depicting the distribution of copies relative to genes for Dromar5 and Dromar11 in D. Eugracilis. Significant differences (Kolgomorov-Smirnov test) are indicated. M: MITE lineage, FL: full-length lineage. n: copy number (percentage of copies located on genes-containing contigs). (PDF 101 KB)
Additional file 6: Table S4: Sequence evolution characteristics in mariner lineages. (XLSX 54 KB)
Additional file 7: Figure S3: Analysis of the trinucleotide preference in 5′ (A) and 3′ (B) of the insertion TA site for the mellifera and drosophila subfamilies. Only trinucleotides found more than 5 times in the data are presented. (PDF 151 KB)
Additional file 8: Figure S4: Consensus analysis of the region surrounding the insertion sites of different lineages, using WebLogo [71]. The duplicated target TA is present on each site (central TAs). The elements have been removed. The numbers into parentheses indicated the number of 5′ and 3′ flanking regions analysed. (PDF 291 KB)