

Background: AT1R and BK channels are key in defining vascular tone.
Results: AT1R closely interacts with BK (its voltage-sensing-conduction cassette suffices) and is responsible for inhibiting BK via constitutive and agonist-mediated G-protein-independent mechanisms in arterial myocytes and expressing cells.
Conclusion: AT1R and BK channels form a tight complex facilitating their functional coupling.
Significance: AT1R-BK association is a new mechanism behind vascular tone regulation.
Keywords: Angiotensin II, Cell Surface Receptor, Patch Clamp, Potassium Channel, Protein Complex, Vascular Smooth Muscle Cells, Angiotensin II Type 1A Receptor, Large Conductance Voltage- and Ca2+-activated K+ Channel
Abstract
Angiotensin II (ANG-II) and BK channels play important roles in the regulation of blood pressure. In arterial smooth muscle, ANG-II inhibits BK channels, but the underlying molecular mechanisms are unknown. Here, we first investigated whether ANG-II utilizes its type 1 receptor (AT1R) to modulate BK activity. Pharmacological, biochemical, and molecular evidence supports a role for AT1R. In renal arterial myocytes, the AT1R antagonist losartan (10 μm) abolished the ANG-II (1 μm)-induced reduction of whole cell BK currents, and BK channels and ANG-II receptors were found to co-localize at the cell periphery. We also found that BK inhibition via ANG-II-activated AT1R was independent of G-protein activation (assessed with 500 μm GDPβS). In BK-expressing HEK293T cells, ANG-II (1 μm) also induced a reduction of BK currents, which was contingent on AT1R expression. The molecular mechanisms of AT1R and BK channel coupling were investigated in co-transfected cells. Co-immunoprecipitation showed formation of a macromolecular complex, and live immunolabeling demonstrated that both proteins co-localized at the plasma membrane with high proximity indexes as in arterial myocytes. Consistent with a close association, we discovered that the sole AT1R expression could decrease BK channel voltage sensitivity. Truncated BK proteins revealed that the voltage-sensing conduction cassette is sufficient for BK-AT1R association. Finally, C-terminal yellow and cyan fluorescent fusion proteins, AT1R-YFP and BK-CFP, displayed robust co-localized Förster resonance energy transfer, demonstrating intermolecular interactions at their C termini. Overall, our results strongly suggest that AT1R regulates BK channels through a close protein-protein interaction involving multiple BK regions and independent of G-protein activation.
Introduction
The seven transmembrane angiotensin II type 1 receptor (AT1R)4 and the large conductance voltage- and Ca2+-activated K+ (BK, BKCa, MaxiK, Slo1) channel are abundantly expressed in vascular smooth muscle cells and play significant roles in various physiological functions (1, 2). As two major tone determinants in arteries (3, 4), activation of AT1R by angiotensin II (ANG-II) leads to powerful vasoconstrictions (3, 5), whereas the BK channel, depending on its activity, can promote vasorelaxation or vasoconstriction (1, 6).
ANG-II activates AT1R and a series of G-protein-dependent or -independent signaling cascades in vascular smooth muscle cells (7, 8) that in turn regulate various physiological effects, such as cell contraction (9, 10), proliferation (11, 12), apoptosis (13), inflammatory response (14), and vascular senescence (15). Our laboratory (16) and Lu et al. (17) have shown that ANG-II inhibits the activity of BK channels in coronary smooth muscle. However, the molecular mechanisms underlying ANG-II and BK functional coupling and whether this regulation extends to other arterial beds are unanswered questions. Because renal arteries are involved in salt and fluid balance (18) and increased renal vascular tone has been demonstrated in multiple studies of hypertension (19, 20), investigating ANG-II-BK coupling in renal arteries is of particular interest.
We demonstrate that in renal arterial smooth muscle cells, ANG-II—through AT1R—inhibits BK currents using a mechanism that is independent of G-protein activation. We also provide evidence indicating that the receptor is in close proximity to BK channels and modifies the channel voltage dependence even in the absence of ANG-II stimulation. Molecular analysis revealed that BK channel N-terminal regions are sufficient for its association with AT1R, and Förster resonance energy transfer demonstrated intermolecular interactions between both proteins.
EXPERIMENTAL PROCEDURES
Animals
Sprague-Dawley male rats (3 months old) were euthanized with an overdose of inhaled isoflurane. Protocols received institutional approval.
Antibodies
Monoclonal anti-FLAG and anti-c-Myc polyclonal antibodies were from Sigma. Anti-pERK1/2 and anti-ERK1/2 polyclonal antibodies (pAbs) were from Cell Signaling. Anti-BK mAb was from NeuroMab. Anti-AT1R pAb was custom-made by AnaSpec and raised against N-terminal residues 10GIKRIQDDCPKAGRH24 of AT1R.
Constructs
Human AT1R and BK α-subunit clones were used (accession numbers NM_001006939 and U11058, respectively). For biochemistry and immunocytochemistry we used C-terminal c-Myc-tagged AT1R (AT1R-c-Myc) in pCMV6-Entry (Origene), N-terminal FLAG-tagged AT1R (FLAG-AT1R) in pcDNA3 (that includes the signal peptide, KTIIALSYIFCLVFA, to promote membrane expression), and BK α-subunit without tag (BK) or with an N-terminal c-Myc tag (c-Myc-BK) (21) in pcDNA3. For electrophysiology, we used AT1R-IRES-c-Myc-BK construct in pIRES vector to ensure concurrent expression of AT1R and BK channels.
Transient Transfection
HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. For electrophysiology, cells were transfected at 30% confluency with 0.1 μg of AT1R-IRES-BK construct per 35-mm dish. For biochemistry and immunocytochemistry, cells were transfected at 70–80% confluency with 2 μg of BK and/or 5 μg of AT1R plasmid DNAs per 60-mm dish. Plasmid DNAs were incubated with Lipofectamine 2000 in OPTI-MEM (Invitrogen) for 20 min at room temperature. Plasmid-Lipofectamine mixtures were incubated with cells at 37 °C in a CO2 incubator. Culture medium with 20% FBS was added to the cells after 5–7 h. Cells were used 24–48 h after transfection.
Co-immunoprecipitation (co-IP)
HEK293T cells expressing target proteins were harvested and solubilized with cell lysis buffer (50 mm Tris, 150 mm NaCl, 5 mm EDTA, 0.1% nonylphenyl polyethylene glycol (Nonidet P-40 alternative), 0.25% sodium deoxycholate, pH 7.4) supplemented with complete protease inhibitor mixture (Roche, 1 tablet/25 ml buffer), and 1 μm PMSF was added immediately before use. Protein G beads (GE Healthcare) were saturated with antibodies (8 μg of anti-c-Myc pAb/30 μl of protein G) and incubated with cell lysates (0.8–1 mg of protein). Beads were washed five times with cell lysis buffer and eluted at 37 °C for 1 h with 3× SDS sample buffer (Biolabs). Lysates and immunoprecipitated proteins were analyzed by SDS-PAGE and immunoblotting.
Immunocytochemistry
Cells were plated on coverslips covered with poly-d-lysine (1 mg/ml) and collagen (0.1 mg/ml) in PBS. Two protocols were used: (i) With live labeling (see Fig. 3, A–C), living cells that had been transfected with c-Myc-BK and FLAG-AT1R were labeled with anti-c-Myc pAb and anti-FLAG mAb for 1 h on ice in a 95% air, 5% CO2 incubator at 37 °C. Cells were then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and labeled with secondary antibodies in PBS containing 1% normal goat serum for 1 h at room temperature. Cells were mounted with Prolong. (ii) With labeling of permeabilized cells (see Figs. 3, D–F, and 6), cells were first fixed with 4% paraformaldehyde in PBS for 20 min. Cells were washed three times (5 min each) with permeabilization buffer (0.2% Triton X-100 in PBS) and incubated with 10% normal goat serum in permeabilization buffer for 30 min at room temperature. The cells were then incubated overnight at 4 °C with primary antibody in permeabilization buffer containing 1% normal goat serum. Labeling with secondary antibodies and mounting were as for live labeling. Images were taken using an Olympus confocal microscope.
FIGURE 3.

BK and AT1R co-localization in HEK293T cells and in arterial myocytes. A–C, live labeling of BK (green) and AT1R (red) in transiently expressing HEK293T cells displaying co-localization at the plasma membrane. HEK293T cells were co-transfected with N-terminally tagged FLAG-AT1R and c-Myc-BK. Anti-FLAG mAb and anti-c-Myc pAb were used for labeling. D–F, labeling of BK (red) and AT1R (green) after permeabilization showing total expression and subplasmalemma co-localization in rat renal arterial smooth muscle cells. Anti-BK mAb and anti-AT1R pAb were used for labeling. G, a representative three-dimensional cross-correlation plot of BK → AT1R as a function of pixel shift in the x and y axes (arrows in A and B indicate the cell used for the plot). The cross-correlation surface has a peak at zero pixel shift that decays abruptly by shifting the image few pixels indicative of specific co-localization. H, quantification of co-localization in HEK293T cells and smooth muscle cells (SMCs) by PPI analysis (see “Experimental Procedures”). In HEK293T cells, PPI is 0.68 ± 0.08 for BK → AT1R and 0.87 ± 0.08 for AT1R → BK (n = 16 cells, three independent transfections). In rat renal arterial SMCs, PPI is 0.62 ± 0.08 for AT1R → BK and 0.71 ± 0.08 for BK → AT1R (n = 18 cells, two independent cell isolations). Error bars indicate S.D. values.
FIGURE 6.

Molecular analysis of BK-AT1R association. A, BK topology (left) and truncated BK constructs that were used for immunocytochemistry (right). RCK, regulator of conductance for K+. B, overlaid confocal images of cells co-expressing AT1R (red) and full-length or truncated BK (green) molecules: BK, BK1–711, BK1–441, BK1–343, and BK323–1113. Note that BK323–1113 appears intracellular. Larger yellow squares show 5× magnification of the regions in the smaller yellow squares. C, specific co-localization analysis by PPI (25) demonstrating that the first 1–343 amino acids of BK are sufficient for its association with AT1R. PPI values were 0.68 ± 0.1 for BK to AT1R and 0.87 ± 0.08 for AT1R to BK (n = 16 cells, four independent experiments); 0.63 ± 0.06 for BK1–711 to AT1R and 0.83 ± 0.12 for AT1R to BK1–711 (n = 9 cells, two independent experiments); 0.75 ± 0.1 for BK1–441 to AT1R and 0.88 ± 0.12 for AT1R to BK1–441 (n = 13 cells, two independent experiments); 0.83 ± 0.11 for BK1–343 to AT1R and 0.91 ± 0.07 for AT1R to BK1–343 (n = 15 cells, three independent experiments); and 0.19 ± 0.08 for BK322–1113 to AT1R and 0.26 ± 0.09 for AT1R to BK322–1113 (n = 17 cells, three independent experiments). Error bars indicate S.D. values. *, p < 0.05.
Co-localization Analysis
Images were median filtered. A protein proximity index algorithm was used to quantify co-localization of AT1R and BK channels (22).
Isolation of Rat Renal Arterial Smooth Muscle Cells
Freshly isolated left and right renal arteries from rats were cut into small pieces in ice-cold oxygenated Tyrode solution: 130 mm NaCl, 5.4 mm KCl, 0.6 mm NaH2PO4, 1.02 mm MgCl2, 10 mm HEPES, 5 mm glucose, and 20 mm taurine, pH 7.4. Pieces were incubated for 1 h on ice in the same Tyrode solution that contained 40 units/ml papain (Sigma) and 0.2% bovine serum albumin followed by digestion in a 37 °C water bath for 15 min. The arterial vessels were further digested in collagenase type 2 (Worthington)/Tyrode solution at 37 °C for ∼35 min until the tissue was observed loose under the microscope. Then the tissue pieces were washed with Tyrode solution and gently triturated with a fire-polished glass pipette until the cells were completely dissociated. Smooth muscle cells were plated onto small coverslips that were coated with 0.1 mg/ml poly-d-lysine hydrobromide in PBS: 2.67 mm KCl, 137.93 mm NaCl, 1.47 mm KH2PO4, and 8.06 mm Na2HPO4, pH 7.4; stored at 4 °C; and used within 8 h.
Inside-out Patch Clamp
Pipette resistances were ∼3 MΩ when filled with pipette solution (bath solution is the same): 105 mm KOH, 5 mm KCl, 10 mm HEPES, 5 mm N-(2-hydroxyethyl)ethylenediamine-N,N′,N′-triacetic acid (HEDTA), 0.01 mm CaCl2 (6.7 μm free Ca2+), pH 7.4 (adjusted with methanesulfonic acid). The holding potential was 0 mV; test pulses were from −180 to 160 mV; repolarizing pulses were set to −70 mV. All experiments were performed at room temperature. Voltage-dependent activation curves were obtained by measuring the instantaneous tail currents (I) (at the beginning of repolarizing pulses) and fitting the data to a Boltzmann distribution of the form: FPo = G/Gmax = I/Imax = 1/(1 + exp[(V½ − V) zδF/RT]), where FPo is the fractional open probability, G is the macroscopic conductance [G = I/(V − EK), where I is the instantaneous tail current, V is the voltage of the constant repolarizing pulse, in this case −70 mV, and EK is the reversal potential for K+, in this case 0 mV)], Gmax is the maximal macroscopic conductance [(Gmax = Imax/(V − EK), where Imax is the maximal instantaneous tail current], V is the voltage of the test pulse, V½ is the half-activation potential, zδ is the effective valence, and F, R, and T have their usual thermodynamic meanings.
Whole Cell BK Current Recording
Pipette resistances were ∼1 MΩ when filled with the pipette solution: 143 mm KCl, 1 mm MgCl2, 3 mm HEDTA, 0.0193 mm CaCl2 (free [Ca2+] = 2.3 μm), and 10 mm HEPES, with pH adjusted to 7.2 with KOH. Free Ca2+ was measured with a Ca2+ electrode. Bath solution was 135 mm NaCl, 4 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm HEPES, and 10 mm glucose, with pH adjusted to 7.4 with NaOH. Whole cell BK currents were evoked by voltage steps delivered from a holding potential of 0 mV to test potentials ranging from −90 to +160 mV in 10-mV increments. The time course of drug action on BK currents was always recorded in the same cell with continuous test pulses at 100 mV from a holding potential of 0 mV for ∼3 min. All current traces were leak-subtracted, and experiments were performed at room temperature.
GDPβS trilithium salt (Sigma) and losartan (Sigma) stock solutions (500 and 10 mm, respectively) were prepared in DMSO and diluted to working concentrations in bath solution. ANG-II (Sigma) stock solution (1 mm) was made in HEPES buffer: 5 mm HEPES, and 0.01% BSA, pH 7.4, with NaOH.
The effectiveness of losartan and ANG-II were evaluated biochemically. Twenty-four hours after transfection, HEK293T cells expressing FLAG-AT1R and c-Myc-BK were starved for 16 h, incubated with 10 μm losartan for 20 min or left untreated, and then stimulated with 1 μm ANG-II for 5 min or left untreated. Cells were harvested, and ERK1/2 phosphorylation was examined by immunoblotting with pERK1/2 Ab.
Co-localized FRET
CFP and YFP were selected as donor and acceptor fluorophores, respectively, in FRET experiments. To measure FRET between each donor/acceptor pair, in each confocal microscope acquisition session, three types of samples were prepared: (i) cells expressing only donor to evaluate donor bleed-through coefficient, (ii) cells expressing only acceptor to evaluate acceptor bleed-through coefficient, and (iii) cells co-expressing both. For each type of sample, three sets of images were acquired using the following confocal acquisition settings: for the CFP channel, the excitation wavelength was set at 403 nm, and the emission collecting window was between 470 and 500 nm with PMT1; for the YFP channel, the excitation was set at 488 nm, and the emission collecting window was between 550 and 600 nm with PMT2; for the CFP to YFP channel (FRET channel), the excitation was at 403 nm, and the emission collecting window was between 550 and 600 nm with PMT2. Images were acquired with a Nikon microscope and subjected to pixel by pixel analysis using ImageJ (FRET and co-localization analyzer) for co-localized FRET. Co-localization of two fluorophores was correlated with FRET to eliminate false FRET signals (23).
Statistics
Unless otherwise stated in figure legends, data are expressed as means ± S.D., n is the number of cells examined, and at least three independent experiments were performed. Statistical comparisons were analyzed with Student's t test, and p < 0.05 was considered statistically significant.
RESULTS
AT1R Mediates ANG-II-induced BK Macroscopic Current Reduction in Renal Arterial Myocytes via a G-protein-independent Mechanism
The whole cell patch configuration was used to examine the effect of ANG-II on BK currents in freshly isolated renal arterial myocytes. The pipette solution contained 2.3 μm free Ca2+, which mimics the concentration reached during spontaneous Ca2+ sparks in arterial myocytes that activate BK channels (24). To inactivate other voltage-dependent potassium channels, the holding potential was set to 0 mV. Confirming the identity of the recorded currents as originating from BK activity, its specific blocker iberiotoxin (100 nm) reduced K+ currents (measured at the end of 20-ms test pulses to +100 mV) by 73 ± 13% (n = 7 cells). The degree of blockade in native cells was within the range attained in an experiment in which BK channels and AT1R were simultaneously expressed in HEK293T cells (∼75%) using the same recording protocol (not shown). Thus, it is reasonable to refer to the recorded currents as BK currents.
To evaluate the effect of ANG-II on BK currents, continuous recordings were obtained from the same cells before and after application of ANG-II. Representative current traces of voltage-dependent activation before (control) and ∼3 min after ANG-II application are shown in Fig. 1 (A and B), demonstrating that ANG-II (1 μm) caused a clear reduction in current amplitude. Mean current values were measured at the end of 100 mV continuous pulses in similar paired experiments and normalized to control levels (Fig. 1C), demonstrating that 1 μm ANG-II suppressed whole cell BK currents by 38 ± 7% (n = 5 cells).
FIGURE 1.
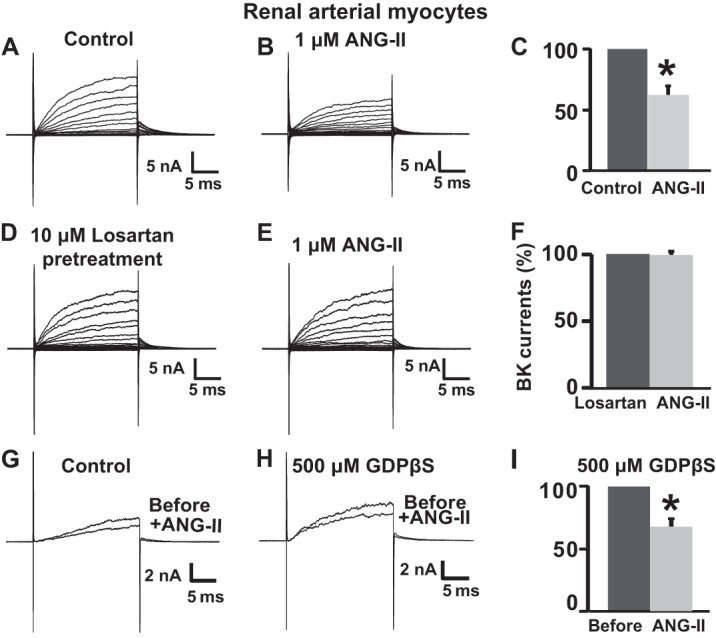
ANG-II inhibits whole cell BK currents in freshly dissociated rat renal arterial smooth muscle cells via losartan-sensitive AT1R and independent of G-protein activation. A and B, whole cell BK currents at baseline (control) and after 1 μm ANG-II treatment in the same arterial myocyte. Whole cell currents were elicited by 20-ms pulses from −90 to 160 mV from a holding potential of 0 mV. C, mean percentage values normalized to control. The currents were reduced to 62 ± 7% (n = 5 cells, four independent experiments) of the original current after ANG-II treatment. D and E, ANG-II failed to inhibit BK whole cell currents when the same myocyte was pretreated with 10 μm losartan (20 min), an AT1R inhibitor. F, mean percentage BK current values were the same in cells pretreated with losartan and after stimulation with ANG-II (n = 9 cells, three independent experiments). G and H, superimposed traces with test pulses of 100 mV before and after ANG-II (1 μm) treatment in a control cell and in a cell intracellularly perfused with 500 μm GDPβS, an inhibitor of G-protein activation. I, mean values of paired experiments as in H showing that in the presence of GDPβS, ANG-II reduced currents to 68 ± 5% (n = 5 cells, two independent cell isolations) of its original value. Measurements were at the end of test pulses to 100 mV. In this and the following figures, the current traces are just prior to drug application and after the effect of drug treatment reached steady state. Error bars indicate S.D. values. *, p < 0.05.
To determine whether ANG-II inhibitory effect on BK channels occurs via activation of AT1R, we used losartan, a biphenyltetrazol derivative that binds selectively to AT1R and competes with ANG-II for binding to the receptor preventing its effects. Fig. 1 (D and E) illustrates currents from a typical experiment in which a cell pretreated with 10 μm losartan for 20 min (Fig. 1D) failed to respond to 1 μm ANG-II perfusion (Fig. 1E). Mean percentage values of similar paired experiments (at +100 mV) clearly show that losartan pretreatment completely prevented the ANG-II effect (n = 9 cells; Fig. 1F). To control the effectiveness of 10 μm losartan in preventing ANG-II induced AT1R activity, we analyzed the levels of pERK½, a well established downstream signaling marker in cells co-expressing AT1R and BK channels (n = 3 experiments; Fig. 2H).
FIGURE 2.

AT1R expression is essential for ANG-II inhibitory effect on BK channels. A and C, representative traces before and after application of 1 μm ANG-II in BK and AT1R-IRES-BK transfected cells, respectively. B and D, corresponding mean % values ± S.D. (n = 6 cells, four independent transfections for BK alone, and n = 3 cells, one transfection for AT1R + BK). Only when AT1R is expressed, ANG-II reduced BK currents to 66 ± 10% of its original value. E, example of time course of ANG-II action on BK currents. A similar time course was collected for other drugs. F, current traces before and after 1 μm ANG-II in AT1R-IRES-BK transfected cells pretreated with 10 μm losartan. G, corresponding mean % values (n = 5 cells, one transfection). H, example of immunoblot analysis of pERK1/2 in lysates from cells expressing AT1R and BK that were untreated (lane 1), treated with ANG-II alone (lane 2), treated with losartan alone (lane 3), and pretreated with losartan and stimulated with ANG-II (lane 4). pERK1/2 levels were higher in ANG-II treated cells (lane 2) and returned to near baseline in cells preincubated with losartan (lane 4) (n = 3 experiments). Loading and expression controls were ERK1/2, AT1R, and BK blots. All lanes were loaded with 30 μg of protein. Antibody concentrations were 186 ng/ml pERK1/2 pAb, 2.85 ng/ml ERK1/2 pAb, 250 ng/ml c-Myc pAb, and 525 ng/ml BK mAb. Error bars indicate S.D. values. *, p < 0.05. Ctrl, control; Los, losartan.
To gain insight into the mechanism of AT1R-BK coupling, we next examined whether inhibition of BK currents entailed a G-protein-dependent mechanism. To this end, we used GDPβS, a hydrolysis-resistant GDP analog that binds to Gα protein preventing its activation. Intracellular perfusion of 500 μm GDPβS via the pipette failed to prevent ANG-II-induced inhibition of BK currents (Fig. 1, G–I), yielding an inhibition of 32 ± 5% (n = 5 cells) similar to the one obtained when GDPβS was absent (Fig. 1C). Overall, the results indicate that AT1R via a G-protein-independent mechanism mediates the ANG-II-induced inhibition of BK channels from renal arterial myocytes and suggest that this regulatory system may be present in various arterial beds.
AT1R Expression Is Required for ANG-II-mediated Inhibition of BK Channel
To further confirm the role of AT1R and rule out the remote possibility of a direct effect of ANG-II on the channel protein, we investigated ANG-II effects in HEK293T cells transfected with BK alone or BK + AT1R (in pIRES vector). Whole cell BK currents were continuously recorded at a test potential of 100 mV before and during the application of drugs until their effects reached steady state. Holding potential was 0 mV.
ANG-II (1–2 μm) had no effect on BK current amplitude in the absence of AT1R co-expression (n = 13 cells, five independent transfections) as shown for 1 μm ANG-II (n = 6 cells) in Fig. 2 (A and B). In contrast, when AT1R was co-expressed with BK, extracellular application of 1 μm ANG-II suppressed the whole cell BK currents by 34 ± 10% (n = 3 cells) (Fig. 2, C and D) similar to that observed in native cells. ANG-II inhibitory effect on BK + AT1R currents reached steady state within 1 min as illustrated in Fig. 2E. This time frame was similar in native cells as well.
As expected, pretreatment with 10 μm losartan to inhibit AT1R prevented the BK-induced inhibition by ANG-II (n = 5 cells) (Fig. 2, F and G). At the concentration used, losartan (10 μm) effectively prevented the ANG-II (1 μm)-induced activation of AT1R as measured by the levels of phosphorylated ERK1/2 (pERK1/2) (Fig. 2H, lane 4 versus lane 2) in cells co-expressing AT1R + BK (n = 3 experiments). These results demonstrate that AT1R and its activation are required for the ANG-II-induced inhibition of BK channels.
AT1R Co-localizes with BK Channels
We next examined whether BK channels and AT1R share similar subcellular localization that would facilitate their functional coupling. To this end, we immunolabeled HEK293T cells expressing N-terminally tagged FLAG-AT1R and c-Myc-BK channel, as well as native AT1R and BK proteins in freshly dissociated rat renal arterial smooth muscle cells. In HEK293T cells, live labeling revealed a substantial co-localization of AT1R and BK at the plasma membrane (Fig. 3, A–C). Similarly, permeabilized renal arterial myocytes showed a remarkable AT1R and BK co-localization at the cell periphery (Fig. 3, D–F). Quantification of co-localization was performed evaluating their protein proximity index (PPI) as described earlier (25, 26). This method is user-independent and can differentiate specific protein proximity or “co-localization” from nonspecific co-localization signals that may arise from coincidental expression or antibody background signal. Fig. 3G is a three-dimensional graph of the cross-correlation of BK and AT1R signals versus pixel shift in the x and y axes of the marked cell (arrows) in panels A and B of Fig. 3. The graph shows a sharp peak that corresponds to the specific correlation (co-localization), which was used to calculate PPI. PPI was at the cell surface 0.87 ± 0.08 for AT1R → BK and 0.68 ± 0.10 for BK → AT1R in HEK293T cells (n = 16 cells). These results can be interpreted as if ∼87% of the expressed AT1R is proximal to BK and ∼68% of the expressed BK is proximal to AT1R. Notoriously, rat renal arterial smooth muscle cells also showed robust proximity indexes. PPI was 0.62 ± 0.08 for AT1R → BK and 0.71 ± 0.08 for BK → AT1R (n = 18 cells) (Fig. 3H). Given the resolution of confocal microscopy, these data indicate that BK and AT1R are within 250–500-nm distance, supporting the hypothesis that they may coexist in a macromolecular complex.
AT1R Expression Constitutively Modulates the Voltage Sensitivity of the BK Channel
Because AT1R and BK channels co-localize in the absence of agonist, we wondered whether this proximal localization could constitutively affect BK channel voltage activation properties. To investigate this point, a study using the inside-out patch clamp configuration was performed in transfected HEK293T cells expressing BK alone or BK + AT1R. To assure co-expression of BK and AT1R, we used AT1R-IRES-c-Myc-BK construct. The recording protocol consisted of a holding potential of 0 mV and 20-ms test pulses from −180 to 160 mV, followed by a 15-ms repolarizing pulse to −70 mV (Fig. 4A). With this protocol and isotonic K+, tail currents are observed at the beginning of negative test pulses and during the repolarizing pulse to −70 mV (Fig. 4, B and C). The latter were used to analyze changes in voltage sensitivity by constructing voltage activation curves (FPo = G/Gmax = I/Imax versus voltage) and derive the half-activation potential, V½, from the fit to a Boltzmann distribution (see “Experimental Procedures”). Note that this parameter reflects the voltage sensitivity of BK channels and is independent of the number of channels in the patch that can vary from patch to patch.
FIGURE 4.

Constitutive inhibition of BK channel activity by AT1R expression. A, voltage stimulation protocol. B and C, representative inside-out patch recordings of type 1 and 3 currents in HEK293T cells co-transfected with BK and AT1R. Fitted V½ values were 0.4 and 36.8 mV, respectively. V½ values were calculated using instantaneous tail currents, I, to obtain FPo (G/Gmax = I/Imax) as a function of the preceding test pulse voltage and fitting the data to a Boltzmann distribution as in F (see “Experimental Procedures”). D, examples of normalized BK tail currents (I/Imax) in cells expressing BK alone or BK + AT1R. The scheme (top trace) shows the pulse protocol. Current traces demonstrate that peak I/Imax (FPo) magnitude followed the trend BK ≈ type 1 AT1R + BK > type 2 AT1R + BK ≈ type 3 AT1R + BK. E, BK activation kinetics was slowed down by AT1R co-expression in the order type 3 > type 2 > type 1 (test pulse = 60 mV) consistent with a larger decrease in Po in type 3 AT1R + BK channels. F, average voltage activation curves (FPo versus voltage) of BK expressed alone or in combination with AT1R. The error bars are within symbols and indicate S.E. Continuous lines are the means of the fitted curves of each experiment. Average V½ values were −0.4 ± 3.2 for BK (n = 24 cells), −0.3 ± 2 for AT1R + BK type 1 (n = 22 cells), 13 ± 2 for AT1R + BK type 2 (n = 17 cells), and AT1R + BK type 3 = 35 ± 2 (n = 12 cells). Type 3 channels displayed the highest inhibition by AT1R co-expression (FPo is lower at a given potential when compared with BK expressed alone). G, V½ distribution in all patches (n = 79) from cells co-expressing BK and AT1R. Ca2+ concentration facing the intracellular side of the channels was 6.7 μm.
In the presence of AT1R, BK currents with diverse voltage sensitivities were observed that could be classified in three main types. Fig. 4 (B and C) shows representative traces of AT1R + BK currents classified as “type 1” and “type 3,” respectively. Clear-cut differences were manifested between these two groups (e.g. notice the difference in activation kinetics, already denoting distinct channel Po). In type 3 currents, voltage activation analysis demonstrated that AT1R expression caused the largest decrease in the channel response to membrane depolarization, yielding the most positive V½ values, which in this specific example was ∼37 mV. In contrast, type 1 currents responded more efficiently to voltage as voltages ∼0 mV already elicited an FPo of 0.5 (i.e. V½ is ∼0 mV). As shown later, this behavior resembles that of BK alone.
To better illustrate the change in voltage sensitivity of the three types of AT1R + BK currents with respect to BK expressed alone, Fig. 4D shows individual traces of tail currents, I, normalized to the maximum peak tail current, Imax, in each patch elicited by changing the voltage from a single test pulse, in this case 0 mV, to −70 mV. Note that normalized tail currents of BK expressed alone and type 1 AT1R + BK were similar in peak amplitude, type 2 currents showed a modest reduction, and type 3 AT1R + BK currents showed the largest decrease in peak tail current or FPo (FPo = I/Imax). Mean FPo ± S.E. values at 0 mV were 0.52 ± 0.08 for BK (n = 24 cells), 0.49 ± 0.02 for type 1 (n = 22 cells), 0.33 ± 0.07 for type 2 (n = 17 cells), and 0.12 ± 0.04 for type 3 (n = 12 cells) AT1R + BK currents (see Fig. 4F for other potentials). The robust reduction in FPo in response to voltage caused by AT1R expression in type 3 currents is also clearly observed, as mentioned before, as a decrease in activation kinetics (Fig. 4, C and E). Fig. 4E illustrates this change for currents at a single test potential (+60 mV) at which BK and type 1 AT1R + BK currents show similar activation, whereas type 3 currents show the slowest activation kinetics.
Average voltage activation curves of the three AT1R + BK current types (open symbols) and from currents from patches expressing BK channel alone (closed symbols) are displayed in Fig. 4F demonstrating that the sole expression of AT1R can shift the voltage activation curves toward more positive potentials. V½ shifts of the three AT1R + BK types compared with patches from BK alone (V½ = −0.4 ± 3.2, n = 24 cells) are practically none for type 1 (V½ = −0.3 ± 2, n = 22 cells), ∼13 mV for type 2 (V½ = 13 ± 2, n = 17 cells), and ∼35 mV for type 3 (V½ = 35 ± 2, n = 12 cells) AT1R + BK currents (Fig. 4F).
A distribution histogram of a total of 78 patches from AT1R + BK-expressing cells confirmed that V½ values (binned every 7 mV) distributed in three types (Fig. 4G). Experimental peaks were at V½ = −0.3 ± 2 (n = 22 cells) for type 1, at V½ = 13 ± 1.8 mV (n = 17 cells) for type 2, and at V½ = 34 ± 2.1 (n = 12 cells) for type 3. In agreement, the histogram could be well fitted to three Gaussian distributions with means (μ) at 1 ± 0.43 mV (type 1), 16 ± 0.35 mV (type 2), and 34 ± 0.8 mV (type 3); the areas were 249 ± 25, 123 ± 20, and 100 ± 24, respectively. These results indicate that ∼50% of the recorded currents showed an inhibition by AT1R. In summary, the results demonstrate that in the absence of agonist, AT1R can constitutively regulate BK channels by decreasing its activity in response to voltage and support the idea that AT1R and BK channels are proximal to each other at the plasma membrane.
AT1R Is in Complex with the BK Channel in HEK293T Cells
To examine whether AT1R and BK (Fig. 5A) form a macromolecular complex, co-immunoprecipitation experiments were performed using HEK293T cells expressing both proteins (n = 5 independent experiments). Fig. 5B is an example showing that AT1R tagged with c-Myc at the C terminus (AT1R-c-Myc) is able to co-immunoprecipitate BK protein (lane 4) (n = 3). Negative controls included: (i) mock transfected cells (lane 1), (ii) cells expressing either AT1R-c-Myc (lane 2) or BK (lane 3) alone, (iii) immunoprecipitation using rabbit IgG (lane 5) or beads alone (lane 6), and (iv) immunoprecipitation using a mixture of lysates from cells independently expressing either AT1R-c-Myc or BK (lane 7). The fact that in the latter experiment we were unable to co-immunoprecipitate BK (Fig. 5B, lane 7), even when AT1R was readily immunoprecipitated (Fig. 5C, lane 7), argues in favor of a specific interaction between AT1R and BK in co-transfected cells and rules out the possibility that co-immunoprecipitation only occurs because of overexpression of the proteins as expected from the mass law. Fig. 5C confirms that AT1R-c-Myc was efficiently immunoprecipitated by c-Myc pAb in all instances in which the receptor was transfected (lanes 2, 4, and 7). Immunoblots of the input lysates showed similar expression of BK or AT1R under the different conditions under which the respective proteins were expressed (Fig. 5, B and C, lower panels). Reverse IP in another two experiments showed similar results. In this case, c-Myc-BK, which was effectively immunoprecipitated (Fig. 5E, lanes 2 and 3), pulled down Flag-AT1R only when both proteins were co-expressed (Fig. 5D, lane 3). Immunoblots of input lysates (Fig. 5, D and E, bottom panels) confirm appropriate expression of both proteins. Overall, these data demonstrate that AT1R can form a complex with BK channels.
FIGURE 5.

AT1R forms a complex with BK in HEK293T cells. A, cartoons of seven-transmembrane domain BK α-subunit (left) and AT1R (right). B, AT1R pulls down BK (n = 3 independent experiments). Lanes 1–4, IP using anti-c-Myc pAb recognizing AT1R-c-Myc in lysates from transfected (+) or untransfected (−) cells with AT1R-c-Myc and/or BK; lanes 5 and 6, IP with rabbit IgG or protein G beads using lysates from cells co-transfected with AT1R-c-Myc and BK; lane 7, IP using a mixture of lysates from cells independently expressing AT1R-c-Myc or BK. Loading was equal in all lanes (25 μl). Lower panel, control immunoblot of BK expression in input cell lysates (30 μg of protein/lane). Immunoblots were with 525 ng/ml anti-BK mAb. C, control showing effective IP of AT1R in same cell lysates or mixture of lysates. Loading was equal in all lanes (5 μl). Lower panel, AT1R expression in input cell lysates (30 μg of protein/lane) probed with 250 ng/ml anti-c-Myc pAb. D, BK also pulls down AT1R (n = 2 independent experiments). Lanes 1–3, IP of lysates from cells expressing only AT1R, expressing only BK or co-expressing both proteins. IP was with anti-c-Myc pAb recognizing c-Myc-BK, and immunoblot was with 495 ng/ml anti-FLAG mAb recognizing AT1R. Lane 4, negative control using IgG to IP lysates from cells expressing both AT1R and BK. Lower panel, immunoblot of input lysates. E, control of effective IP of BK. Lower panel, expression of BK in corresponding input cell lysates. Immunoblot was with 525 ng/ml anti-BK mAb. IB, immunoblot; IE, independently expressing.
The N-terminal Transmembrane Segment of BK Channels Is Sufficient for AT1R Association
We next investigated which regions in BK channel are relevant for its association with AT1R. To this end, we used a series of constructs encoding truncated BK molecules (Fig. 6A) and measured their degree of co-localization in HEK cells co-transfected with FLAG-AT1R. Labeling was done under permeabilized conditions. BK1–711, BK1–441, and BK1–343 were c-Myc tagged at the N terminus for visualization. BK322–1113 encompasses the whole cytosolic region and was recognized by anti-BK pAb. Visual inspection revealed that BK, as well as C-terminal truncations BK1–711, BK1–441, and BK1–343, expressed at the cell periphery and could also be observed intracellularly likely because of their normal traffic to the membrane.
Overlays of BK constructs (green) and FLAG-AT1R (red) signals are illustrated in Fig. 6B. BK1–711, BK1–441, and BK1–343 showed co-localization to AT1R similar to BK full-length proteins. On the contrary, BK C terminus (BK323–1113) did not co-localize with AT1R, most likely because of its cytosolic localization; thus, the possibility exists that the C terminus when in the context of the whole protein can participate in the complex formation. The results show, however, that the first 343 amino acids of BK are sufficient for AT1R+ BK association.
Single cells expressing both AT1R and the truncated BK molecule were selected to quantify the specific co-localization using the protein proximity index method. The results presented in Fig. 6C confirmed the visual inspection, indicating that the N-terminal region including the voltage sensing cassette of BK channels is sufficient for its association with AT1R (values are given in the figure legend).
AT1R Is Physically Associated with BK Channels
To further understand the interaction between AT1R and BK channels, we performed FRET experiments in HEK293T cells. BK-CFP, AT1R-YFP C-terminal fusion proteins, and control constructs (see below) were expressed as follows: (i) BK-CFP + AT1R-YFP; (ii) CFP-linker-YFP (positive control), a construct encoding a fusion protein of CFP and YFP with a 6-amino acid (DPPVAT) linker in between assuring FRET signals; (iii) CFP + YFP (negative control); (iv) BK-CFP + YFP (negative control); and (v) CFP + AT1R-YFP (negative control).
Strong co-localized FRET signals were found in cells co-transfected with BK-CFP + AT1R-YFP or with CFP-Linker-YFP but not in negative controls. Fig. 7A (top panels) shows BK-CFP and AT1R-YFP expression mainly at the cell periphery and striking co-localized FRET signals. Note that in these and following panels, YFP signals were pseudocolored in green. Images in the middle panels correspond to fluorescent signals of the CFP-linker-YFP in the CFP and YFP channels, respectively. As expected, the degree of co-localized FRET is also high. In contrast, if CFP and YFP are expressed independently, no co-localized FRET is observed (bottom panels) validating the specificity of FRET signals observed between BK-CFP and AT1R-YFP. Supporting this view, Fig. 7B shows a good correlation of donor-acceptor co-localization and FRET in cells co-expressing BK-CFP + AT1R-YFP, as well as in cells expressing CFP-linker-YFP (red dots) but not in cells expressing CFP + YFP (blue dots). The mean values of normalized co-localized FRET demonstrated that FRET was present in BK-CFP + AT1R-YFP and CFP-linker-YFP groups and was negligible in control groups (Fig. 7C; values are in the figure legend). These results support the conclusion that AT1R and BK channels are not only in the same cell compartment but are also physically associated with each other.
FIGURE 7.

AT1R and BK channels co-localized FRET. A, co-localized FRET in cells co-expressing AT1R-YFP and BK-CFP fusion proteins, expressing CFP-linker-YFP (positive control), and co-expressing CFP and YFP independently (negative control). Bleed-through was calculated using cells individually expressing each molecule (data not shown). B, correlation of CFP-YFP co-localization and FRET signal. Red dots indicate high correlation, and blue dots indicate no correlation. C, mean values of normalized co-localized FRET were as follows for cells expressing: (i) CFP-linker-YFP, 46 ± 10.3 (n = 14); (ii) CFP + YFP 0.5 ± 0.8 (n = 16, two independent experiments); (iii) BK-CFP + AT1R-YFP, 28 ± 7.5 (n = 20); (iv) CFP + AT1R-YFP, 2.4 ± 3 (n = 10); and (v) BK-CFP + YFP, 1.1 ± 1.6 (n = 11). Co-localized FRET was analyzed with a pixel by pixel method using a FRET analyzer from ImageJ. *, p < 0.05 with respect to controls. coFRET, co-localized FRET.
DISCUSSION
The present study addresses for the first time the molecular mechanisms underlying ANG-II-BK channel crosstalk. We have identified a close physical and functional interaction between AT1R and BK channels and discovered that AT1R by itself can inhibit BK channels. This close partnership likely facilitates the inhibition of BK channels by ANG-II via AT1R in arterial myocytes.
Co-localized FRET experiments (Fig. 7) demonstrate that AT1R and BK channels are within an intermolecular distance of ∼1–10 nanometers, strongly supporting a direct protein-protein interaction. In addition to FRET, other lines of evidence that in conjunction agree with a close interaction between the receptor and the channel proteins are: (i) co-immunoprecipitation and specific co-localization of BK and AT1R (Figs. 3 and 5), (ii) modification of BK channel voltage sensitivity by the sole AT1R expression (Fig. 4), and (iii) ANG-II-AT1R-mediated inhibition of BK currents independent of G-protein activation (Fig. 1).
The FRET findings together with the constitutive modulation of BK by AT1R (in the absence of ANG-II) raise the possibility that AT1R association might directly cause conformational changes on BK channels and subsequent alterations in channel activity. This conjecture is also supported by the identification of AT1R-associating regions on the BK molecule (Fig. 6).
The deletion mutant studies demonstrate that the first 343 amino acids of BK containing the voltage sensing and conduction cassettes are sufficient for the proteins to associate (Fig. 6). However, the participation of BK C terminus cannot be excluded and is supported by FRET experiments, because the fluorophores were fused at the C terminus of each protein (Fig. 7). Thus, it is apparent that the interaction between BK and AT1R most likely involves several regions throughout BK channels. At present, the precise residues in BK or AT1R facilitating their interaction remain to be pinpointed.
The association between AT1R and BK channels in heterologously expressing cells is specific as demonstrated by: (i) a lack of co-immunoprecipitation when lysates of independently expressed AT1R and BK were mixed (Fig. 5) and (ii) the fact that the Drosophila homolog of BK, DSlo, did not co-immunoprecipitate with BK (n = 3, not shown). In addition, the degree of co-localization that occurs in heterologously expressing cells mimics the one observed in freshly isolated arterial cells (Fig. 3).
Functionally, a role of AT1R in mediating ANG-II inhibition of BK channels in arterial smooth muscle has been suggested based on correlated segregation of total membrane proteins in lipid rafts (17); however, direct evidence for the AT1R-BK interaction was lacking. Our electrophysiological and biochemical studies in native arterial myocytes and in heterologously expressing cells demonstrate that AT1R expression is needed for the inhibitory action of ANG-II on BK channels (Figs. 1 and 2) and that both proteins form a macromolecular complex (Figs. 3, 5, and 7). Conclusive evidence for AT1R requirement was given by heterologous expression, because receptor expression was a sine qua non condition for the ANG-II effect. Additional evidence was the observation that the inhibitory effect of ANG-II is prevented by losartan, a specific inhibitor of AT1R both in heterologous expression experiments and in freshly isolated renal arterial smooth muscle cells. Interestingly, a role of AT1R in mediating BK channel inhibition, as measured by losartan sensitivity, has also been found in endothelial cells (27).
Increasing evidence indicates that positive or negative regulation of BK activity by G-protein-coupled receptors can occur via two basic mechanisms: one that is G-protein-dependent like in the case of activatory β-adrenergic (28–30) and inhibitory muscarinic receptors (31) and another that is G-protein-independent as is the case of activatory μ-opioid (32) and inhibitory thromboxane A2 receptors (22). We now show that this is also the case for AT1R in smooth muscle because GDPβS pretreatment did not block ANG-II effects on BK in renal arterial smooth muscle cells (Fig. 1). These results are consistent with our previous results in lipid bilayers, where ANG-II-mediated reduction of coronary arterial BK channel open probability did not require putative G-protein activation as it occurred in the absence of GTP and Mg2+ (16). Based on the GTP dispensability, in reconstituted lipid bilayers, we proposed earlier that ANG-II may directly inhibit BK channels (16); however, this mechanism is ruled out by our present studies, in which ANG-II failed to inhibit BK currents in HEK293T cells expressing only BK channels (Fig. 2).
Given the fact that BK channels and vasoconstrictor AT1R are key regulators of arterial tone, inhibition of BK channels by ANG-II-induced AT1R activation might contribute to increased contractility and the progression of diseased states. In line with this view, inhibiting AT1R with losartan enhanced the relaxation induced by NS1619, a BK channel opener, in mesenteric arteries from a type 2 diabetic rat model (33). In other words, this experiment indicates that in mesenteric arteries of type 2 diabetic animals, a proportion of BK channels are originally inhibited by AT1R activity. Interestingly, in a rat model of type-1 diabetes, ANG-II-mediated channel inhibition in coronary arteries was lacking with respect to control (17), indicating that ANG-II to BK channel regulation is disease-dependent. The mechanisms we unveiled in this study might also apply to other functionally important systems with concurrent expression of AT1R and BK channels, for example in adrenal glomerulosa cells (34, 35) and neurons (36–38).
This work was supported, in whole or in part, by National Institutes of Health Grants HL107418 and HL096740 (to L. T. and E. S.) and HL088640 (to E. S.).
- AT1R
- angiotensin II type 1 receptor
- BK
- large conductance voltage- and Ca2+-activated K+ channel
- ANG-II
- angiotensin II
- pAb
- polyclonal antibody
- CFP
- cyan fluorescent protein
- IP
- immunoprecipitation
- GDPβS
- guanosine 5′-[β-thio]diphosphate
- PPI
- protein proximity index.
REFERENCES
- 1. Hu X. Q., Zhang L. (2012) Function and regulation of large conductance Ca2+-activated K+ channel in vascular smooth muscle cells. Drug Discov. Today 17, 974–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Touyz R. M., Schiffrin E. L. (2000) Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 52, 639–672 [PubMed] [Google Scholar]
- 3. Nguyen Dinh C. A., Touyz R. M. (2011) Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr. Hypertens. Rep. 13, 122–128 [DOI] [PubMed] [Google Scholar]
- 4. Grimm P. R., Sansom S. C. (2010) BK channels and a new form of hypertension. Kidney Int. 78, 956–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayabuchi Y., Nakaya Y., Yasui S., Mawatari K., Mori K., Suzuki M., Kagami S. (2006) Angiotensin II activates intermediate-conductance Ca2+-activated K+ channels in arterial smooth muscle cells. J. Mol. Cell Cardiol. 41, 972–979 [DOI] [PubMed] [Google Scholar]
- 6. Alioua A., Mahajan A., Nishimaru K., Zarei M. M., Stefani E., Toro L. (2002) Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc. Natl. Acad. Sci. U.S.A. 99, 14560–14565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goette A., Lendeckel U. (2008) Electrophysiological effects of angiotensin II. Part I: signal transduction and basic electrophysiological mechanisms. Europace 10, 238–241 [DOI] [PubMed] [Google Scholar]
- 8. Mehta P. K., Griendling K. K. (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292, C82–C97 [DOI] [PubMed] [Google Scholar]
- 9. Wynne B. M., Chiao C. W., Webb R. C. (2009) Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J. Am. Soc. Hypertens. 3, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Touyz R. M., Schiffrin E. L. (1997) Angiotensin II regulates vascular smooth muscle cell pH, contraction, and growth via tyrosine kinase-dependent signaling pathways. Hypertension 30, 222–229 [DOI] [PubMed] [Google Scholar]
- 11. Zhang F., Hu Y., Xu Q., Ye S. (2010) Different effects of angiotensin II and angiotensin-(1–7) on vascular smooth muscle cell proliferation and migration. PLoS One 5, e12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun J. J., Kim H. J., Seo H. G., Lee J. H., Yun-Choi H. S., Chang K. C. (2008) YS 49,1-(α-naphtylmethyl)-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, regulates angiotensin II-stimulated ROS production, JNK phosphorylation and vascular smooth muscle cell proliferation via the induction of heme oxygenase-1. Life Sci. 82, 600–607 [DOI] [PubMed] [Google Scholar]
- 13. Bascands J. L., Girolami J. P., Troly M., Escargueil-Blanc I., Nazzal D., Salvayre R., Blaes N. (2001) Angiotensin II induces phenotype-dependent apoptosis in vascular smooth muscle cells. Hypertension 38, 1294–1299 [DOI] [PubMed] [Google Scholar]
- 14. Ji Y., Liu J., Wang Z., Liu N. (2009) Angiotensin II induces inflammatory response partly via Toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell Physiol. Biochem. 23, 265–276 [DOI] [PubMed] [Google Scholar]
- 15. Min L. J., Mogi M., Iwai M., Horiuchi M. (2009) Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing Res. Rev. 8, 113–121 [DOI] [PubMed] [Google Scholar]
- 16. Toro L., Amador M., Stefani E. (1990) ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. Am. J. Physiol. 258, H912–H915 [DOI] [PubMed] [Google Scholar]
- 17. Lu T., Zhang D. M., Wang X. L., He T., Wang R. X., Chai Q., Katusic Z. S., Lee H. C. (2010) Regulation of coronary arterial BK channels by caveolae-mediated angiotensin II signaling in diabetes mellitus. Circ. Res. 106, 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sorensen C. M., Giese I., Braunstein T. H., Holstein-Rathlou N. H., Salomonsson M. (2011) Closure of multiple types of K+ channels is necessary to induce changes in renal vascular resistance in vivo in rats. Pflugers Arch. 462, 655–667 [DOI] [PubMed] [Google Scholar]
- 19. Guyton A. C. (1991) Blood pressure control: special role of the kidneys and body fluids. Science 252, 1813–1816 [DOI] [PubMed] [Google Scholar]
- 20. Ruilope L. M., Lahera V., Rodicio J. L., Carlos Romero J. (1994) Are renal hemodynamics a key factor in the development and maintenance of arterial hypertension in humans? Hypertension 23, 3–9 [DOI] [PubMed] [Google Scholar]
- 21. Meera P., Wallner M., Song M., Toro L. (1997) Large conductance voltage- and calcium-dependent K+ channel, a distinct member of voltage-dependent ion channels with seven N-terminal transmembrane segments (S0–S6), an extracellular N terminus, and an intracellular (S9–S10) C terminus. Proc. Natl. Acad. Sci. U.S.A. 94, 14066–14071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li M., Tanaka Y., Alioua A., Wu Y., Lu R., Kundu P., Sanchez-Pastor E., Marijic J., Stefani E., Toro L. (2010) Thromboxane A2 receptor and MaxiK-channel intimate interaction supports channel trans-inhibition independent of G-protein activation. Proc. Natl. Acad. Sci. U.S.A. 107, 19096–19101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hachet-Haas M., Converset N., Marchal O., Matthes H., Gioria S., Galzi J. L., Lecat S. (2006) FRET and colocalization analyzer: a method to validate measurements of sensitized emission FRET acquired by confocal microscopy and available as an ImageJ Plug-in. Microsc. Res. Tech. 69, 941–956 [DOI] [PubMed] [Google Scholar]
- 24. Pérez G. J., Bonev A. D., Nelson M. T. (2001) Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am. J. Physiol. Cell Physiol. 281, C1769–C1775 [DOI] [PubMed] [Google Scholar]
- 25. Wu Y., Eghbali M., Ou J., Lu R., Toro L., Stefani E. (2010) Quantitative determination of spatial protein-protein correlations in fluorescence confocal microscopy. Biophys. J. 98, 493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li M., Zhang Z., Koh H., Lu R., Jiang Z., Alioua A., Garcia-Valdes J., Stefani E., Toro L. (2013) The β1-subunit of the MaxiK channel associates with the thromboxane A2 receptor and reduces thromboxane A2 functional effects. J. Biol. Chem. 288, 3668–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park W. S., Ko E. A., Jung I. D., Son Y. K., Kim H. K., Kim N., Park S. Y., Hong K. W., Park Y. M., Choi T. H., Han J. (2008) APE1/Ref-1 promotes the effect of angiotensin II on Ca2+-activated K+ channel in human endothelial cells via suppression of NADPH oxidase. Arch. Pharm. Res. 31, 1291–1301 [DOI] [PubMed] [Google Scholar]
- 28. Toro L., Ramos-Franco J., Stefani E. (1990) GTP-dependent regulation of myometrial KCa channels incorporated into lipid bilayers. J. Gen. Physiol. 96, 373–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scornik F. S., Codina J., Birnbaumer L., Toro L. (1993) Modulation of coronary smooth muscle KCa channels by Gsα independent of phosphorylation by protein kinase A. Am. J. Physiol. 265, H1460–H1465 [DOI] [PubMed] [Google Scholar]
- 30. Kume H., Graziano M. P., Kotlikoff M. I. (1992) Stimulatory and inhibitory regulation of calcium-activated potassium channels by guanine nucleotide-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 89, 11051–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou X. B., Wulfsen I., Lutz S., Utku E., Sausbier U., Ruth P., Wieland T., Korth M. (2008) M2 muscarinic receptors induce airway smooth muscle activation via a dual, Gβγ-mediated inhibition of large conductance Ca2+-activated K+ channel activity. J. Biol. Chem. 283, 21036–21044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Twitchell W. A., Rane S. G. (1994) Nucleotide-independent modulation of Ca2+-dependent K+ channel current by a mu-type opioid receptor. Mol. Pharmacol. 46, 793–798 [PubMed] [Google Scholar]
- 33. Matsumoto T., Ishida K., Taguchi K., Kobayashi T., Kamata K. (2010) Losartan normalizes endothelium-derived hyperpolarizing factor-mediated relaxation by activating Ca2+-activated K+ channels in mesenteric artery from type 2 diabetic GK rat. J. Pharmacol. Sci. 112, 299–309 [DOI] [PubMed] [Google Scholar]
- 34. Bollag W. B., Kent P., White S., Wilson M. V., Isales C. M., Calle R. A. (2008) Phorbol ester increases mitochondrial cholesterol content in NCI H295R cells. Mol. Cell Endocrinol. 296, 53–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sausbier M., Arntz C., Bucurenciu I., Zhao H., Zhou X. B., Sausbier U., Feil S., Kamm S., Essin K., Sailer C. A., Abdullah U., Krippeit-Drews P., Feil R., Hofmann F., Knaus H. G., Kenyon C., Shipston M. J., Storm J. F., Neuhuber W., Korth M., Schubert R., Gollasch M., Ruth P. (2005) Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation 112, 60–68 [DOI] [PubMed] [Google Scholar]
- 36. Sumners C., Fleegal M. A., Zhu M. (2002) Angiotensin AT1 receptor signalling pathways in neurons. Clin. Exp. Pharmacol. Physiol. 29, 483–490 [DOI] [PubMed] [Google Scholar]
- 37. Sailer C. A., Kaufmann W. A., Kogler M., Chen L., Sausbier U., Ottersen O. P., Ruth P., Shipston M. J., Knaus H. G. (2006) Immunolocalization of BK channels in hippocampal pyramidal neurons. Eur. J. Neurosci. 24, 442–454 [DOI] [PubMed] [Google Scholar]
- 38. Marrion N. V., Tavalin S. J. (1998) Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905 [DOI] [PubMed] [Google Scholar]