

Abstract
Protein production in yeasts is related to the specific growth rate μ. To elucidate on this correlation, we studied the transcriptome of Pichia pastoris at different specific growth rates by cultivating a strain secreting human serum albumin at μ = 0.015 to 0.15 h–1 in glucose-limited chemostats. Genome-wide regulation revealed that translation-related as well as mitochondrial genes were upregulated with increasing μ, while autophagy and other proteolytic processes, carbon source-responsive genes and other targets of the TOR pathway as well as many transcriptional regulators were downregulated at higher μ. Mating and sporulation genes were most active at intermediate μ of 0.05 and 0.075 h–1. At very slow growth (μ = 0.015 h–1) gene regulation differs significantly, affecting many transporters and glucose sensing. Analysis of a subset of genes related to protein folding and secretion reveals that unfolded protein response targets such as translocation, endoplasmic reticulum genes, and cytosolic chaperones are upregulated with increasing growth rate while proteolytic degradation of secretory proteins is downregulated. We conclude that a high μ positively affects specific protein secretion rates by acting on multiple cellular processes.
Keywords: Filamentous growth, Recombinant protein secretion, Specific growth rate, Unfolded protein response, Yeast
1 Introduction
The demand for recombinant industrial enzymes and pharmaceutical proteins is rapidly increasing. To further improve process efficiency it is of key importance to understand the relation between cellular growth and protein productivity.
Yeasts are a well-established platform for the production of recombinant proteins. It has been demonstrated that protein production is positively correlated with specific growth rate (μ) in Saccharomyces cerevisiae [1] and in Pichia pastoris [2]. Growth coupling of protein synthesis may be based on growth dependent regulation of the promoter used to express the recombinant gene. It can be anticipated that the transcriptional strength of glycolytic promoters (which are commonly used for protein production in yeasts [3]) is directly correlated with glycolytic flux and thus with specific growth rate. Indeed, transcript levels of the human serum albumin (HSA) gene in P. pastoris under control of the glyceraldehyde 3-phosphate dehydrogenase (GAP) promoter increased steadily with the same rate as actin transcript levels at increasing μ [4]. In contrast, HSA transcript levels under control of the translation elongation factor EF-1 (TEF1) promoter showed increased abundance relative to actin with increasing μ, indicating a strong positive correlation.
The majority of pharmaceutical proteins and many technical enzymes are produced by secretion into the culture supernatant. Thus, besides transcription and translation, the secretory pathway plays a major role in controlling and potentially limiting productivity. The biological control of the secretory pathway in relation to growth is of great interest to increase our understanding of what triggers protein synthesis and secretion.
Therefore we have studied transcriptome regulation of a P. pastoris strain secreting HSA over a wide range of μ in chemostat cultures. Similar transcriptome studies were done with S. cerevisiae with different aims. Brauer et al. [5] compared cultures grown under limitation of different nutrients, investigating cell cycle control, stress response and metabolic activity. Castrillo et al. [6] compared cultures at four different nutrient limitations and three different growth rates at the levels of transcriptome, proteome, and metabolome, mainly to understand growth regulation at the different levels of gene expression and metabolic flux control. Induction of ribosomal and metabolic genes with increasing growth rate was related to control by the repressor/activator Rap1 and a positional effect of genes on the chromosomes near replication origins [7]. A factorial design of experiments was used to identify “general” growth regulated genes comparing carbon, nitrogen and oxygen limitation in chemostat [8].
While these studies provide ample information on cell growth control and stress regulation, there is only limited information concerning protein synthesis and secretion. The main general result is that S. cerevisiae upregulates translation-related genes with increasing growth rate which is not really surprising as the cells need a higher concentration of metabolic enzymes and have to reproduce their total cellular protein content at higher rate. More recently, correlation of recombinant protein synthesis with growth was investigated at the transcriptome level in S. cerevisiae [1] and in Trichoderma reesei [9]. Productivity of insulin precursor and α-amylase in S. cerevisiae increased with μ, accompanied by upregulation of unfolded protein response (UPR) and other stress related genes. In T. reesei, protein productivity correlated more with cell density than with specific growth rate.
In the present study we cover a very broad range of specific growth rates, ranging from 0.015 h–1 (the lowest setpoint we could maintain in chemostat) to 0.15 h–1 (nearly μmax). As standard fed-batches reach very low μ at extended process times with high biomass concentrations [3] we were especially interested to gain understanding of the reaction of P. pastoris to such very low growth rates. Gene regulation at near-zero growth is not well understood. Boender et al. [10] studied transcriptome regulation of anaerobic S. cerevisiae cultures at μ below 0.01 h–1, comparing them to higher growth rates. Main results were an upregulation of many mitochondrial genes at very slow growth, although cultures were grown anaerobically, and a decoupling of ribosomal genes (remaining active) from those involved in the translation process which are downregulated near zero growth.
By analyzing transcriptome regulation of P. pastoris cultures grown at a broad range of μ we aimed to understand growth regulated cellular processes in this yeast with a special emphasis on the protein folding and secretory pathway, including its quality control, and on peculiarities of very low specific growth rate.
2 Materials and methods
2.1 Pichia pastoris strain
The P. pastoris SMD1168H (Δpep4) strain secreting HSA under control of the strong glycolytic GAP promoter employing the native HSA leader sequence for secretion was generated as described in [4]. The selected production clone was determined to have three copies of the expression cassette integrated in the genome.
2.2 Cultivations
Chemostat cultivations were carried out at a working volume of 1 L in a 3.5 L bench-top bioreactor (Minifors, Infors, CH). Cells were grown at dilution rates of 0.015, 0.025, 0.05, 0.075, 0.100, 0.125, and 0.150 h–1 (in triplicates). As preculture, a one liter shake flask containing 100 mL of YPG-Zeo medium (per liter: 10 g yeast extract, 10 g peptone, 10 g glycerol, 25 mg Zeocin) was inoculated with 1.2 mL cryostock of the P. pastoris strain and incubated for approximately 24 h at 25°C and 180 rpm. This culture was used to inoculate the bioreactor to an optical density (OD600) of 1.0. Cultivation temperature was kept constant at 25°C, pH was controlled at 5.85 with 25% ammonia and the dissolved oxygen concentration was kept above 20% by controlling the stirrer speed between 600 and 1200 rpm at a constant airflow of 252 L h–1. After batch end was reached (indicated by a sharp peak in dissolved oxygen concentration), continuous cultivation was initiated at a dilution rate of 0.1 h–1 for three resident times, followed by three different dilution rate setpoints. Samples were taken after at least five resident times when steady state conditions were attained. Viability was measured as described previously [11] on the Gallios™ flow cytometer (Beckman Coulter) using the Cell Viability Kit from BD Biosciences. Batch and chemostat media composition was described in ref. [2].
2.3 RNA extraction, microarray hybridization, and data analysis
RNA isolation, cRNA synthesis, hybridization to the P. pastoris DNA microarrays (Agilent platform) as well as scanning was done according to the Agilent protocol for 2-color expression arrays. The design and general processing of P. pastoris microarrays used in this study was described in ref. [12]. Samples were labeled in 2-color technical duplicates and hybridized against a reference pool generated of cells grown at various culture conditions. For each dilution rate setpoint, log2 fold change (FC) values were calculated against the fastest specific growth rate of 0.15 h–1, using the limma package of the R-project [13]. p-Value correction for multiple testing was done using the false discovery rate controlling method of Benjamini and Yekutieli [14]. To analyze the specific regulation at the lowest setpoint, comparisons of the FCs between 0.015 and 0.025 h–1 to those of 0.025 to 0.05 h–1 were performed.
2.4 Reporter metabolites
Reporter metabolites were calculated by the method published by Patil and Nielsen [15] using the BioMet toolbox [16]. For each condition, adjusted p-values out of the transcriptional comparison (each dilution rate setpoint against the highest specific growth rate of 0.15 h–1) were used for the calculation of Z scores for each reporter metabolite. The published genome-scale metabolic model for P. pastoris iLC915 [17] was applied as underlying metabolic model.
2.5 Cluster analysis and Gene Ontology (GO) term enrichment
Grouping of genes with similar expression patterns across different growth rates was performed with log2 FC values using k-means clustering analysis with Euclidian distance carried out with the Genesis software tool [18]. Criteria for the selection of regulated genes are described below in the Section 3. Gene Ontology (GO) term enrichment analysis for the obtained clusters was done using the GO term finder and Saccharomyces Genome Database (SGD) annotations [19]. The corrected p-value cut-off was set to 0.05 and a background list composed of annotated genes and genes with unknown function of P. pastoris was provided.
2.6 Analytical methods
HSA concentration was determined using the Human Albumin ELISA Quantitation Set (Cat. No. E80-129, Bethyl Laboratories, TX) as described in ref. [20].
For protein gel analysis of supernatants the NuPAGE® Novex® Bis–Tris system was used and carried out according to the manufacturer instructions. Proteins were either visualized by silver staining or transferred to a nitrocellulose membrane for western blot analysis. HSA was probed by using a 1:30 000 dilution in PBS with 0.1% Tween and 2% bovine serum albumin (BSA) of the HRP conjugated Human Albumin detection Antibody (A80-129P, Bethyl) and detection was performed using the SuperSignal™ West Pico Chemiluminescent Substrate (34079, Thermo Scientific).
3 Results and discussion
3.1 Product formation at different specific growth rates
To study the impact of growth rate on cell physiology, gene expression, and recombinant protein secretion, the HSA producing P. pastoris strain was grown in aerobic glucose limited chemostat cultures over a broad range of different dilution rates (D = μ = 0.015, 0.025, 0.05, 0.075, 0.1, 0.125, and 0.15 h–1). Initially, cultivations were also attempted at a dilution rate of 0.175 h–1, but cells were washed out before steady state was reached. Samples were analyzed for biomass concentration, cell viability, product concentration and quality as well as yield coefficients (Table 1). In accordance with a higher proportion of maintenance energy requirements on the overall energy budget, biomass yields decreased with slower growth [21]. The specific product formation rate (qP), SDS–PAGE, and western blot analysis of supernatants are displayed in Fig. 1. A positive correlation between HSA productivity and μ could be observed over the full range of analyzed setpoints. We did not see any degradation of the secreted product at μ below 0.02 h–1, different to a similar setup for production of an antibody Fab fragment [2].
Table 1.
Overview of growth and product formation parameters at the different dilution rates studied
| D (h–1) | YDM (g L–1) | Product (mg L–1) | YXS (g g–1) | YPS (mg g–1) | YPX (mg g–1) | Viability (%) |
|---|---|---|---|---|---|---|
| 0.015 | 18.25 ± 0.35 | 9.69 ± 1.05 | 0.37 ± 0.007 | 0.19 ± 0.021 | 0.53 ± 0.048 | 94.1 |
| 0.025 | 20.43 ± 0.27 | 8.44 ± 1.39 | 0.41 ± 0.005 | 0.57 ± 0.028 | 1.39 ± 0.074 | 95.9 |
| 0.05 | 24.27 ± 0.55 | 26.70 ± 2.07 | 0.49 ± 0.011 | 0.53 ± 0.041 | 1.10 ± 0.076 | 99.5 |
| 0.075 | 25.74 ± 0.47 | 21.57 ± 2.44 | 0.51 ± 0.009 | 0.43 ± 0.049 | 0.84 ± 0.081 | n.a |
| 0.1 | 26.56 ± 0.98 | 20.65 ± 1.64 | 0.53 ± 0.020 | 0.41 ± 0.033 | 0.78 ± 0.091 | 99.0 |
| 0.125 | 26.29 ± 0.27 | 23.25 ± 1.97 | 0.53 ± 0.005 | 0.46 ± 0.039 | 0.88 ± 0.065 | 98.8 |
| 0.15 | 25.95 ± 0.52 | 30.04 ± 1.77 | 0.52 ± 0.010 | 0.60 ± 0.035 | 1.16 ± 0.075 | 98.7 |
Values represent the means of three cultivations ± standard error of the mean. YDM, yeast dry mass; YXS, biomass per substrate yield; YPS, product per substrate yield; YPX, product per biomass yield.
Figure 1.
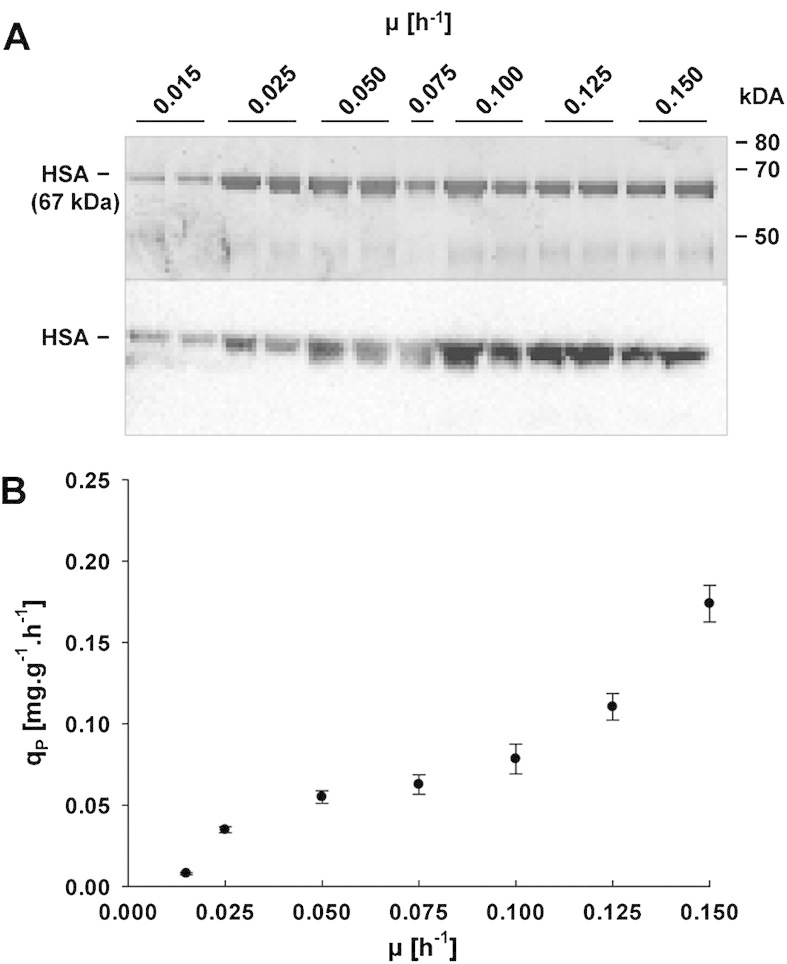
Recombinant HSA production by P. pastoris grown at different specific growth rates in glucose-limited chemostat cultures. (A) Representative SDS–PAGE (top) and corresponding anti-HSA western blot (bottom) under reducing conditions. Equal volumes of undiluted culture supernatants were loaded and visualized by silver staining or transferred to a nitrocellulose membrane for western blot analysis developed with HRP conjugated Human Albumin detection Antibody (A80-129P, Bethyl). For each growth rate setpoint (except for μ = 0.075 h–1) two samples from individual cultivations were analyzed. (B) Specific HSA secretion rate qP plotted against specific growth rate μ. The specific HSA secretion rate was calculated using mean product concentrations and yeast dry mass from three independent chemostat cultivations. All samples were analyzed in technical duplicates. Error bars represent standard error of the mean.
3.2 Genome-wide transcriptional analysis at different specific growth rates
Changes in genome-wide gene expression were analyzed using in-house designed P. pastoris specific microarrays. Analysis of transcriptome data derived from different specific growth rates may be biased due to the fact that the majority of the so-called house-keeping genes have linearly increasing specific transcription rates with increasing μ (own unpublished data). To ensure that the expression values of these house-keeping genes do not change with μ [10], the same amounts of RNA were applied, leading to constant expression values of genes involved in glycolysis and other house-keeping functions, as illustrated in Fig. 2. Of 5354 ORFs represented on the microarrays, 2886 genes were differentially regulated at least at one growth rate when compared to the highest μ of 0.15 h–1 (adjusted p-value < 0.02). No minimal log2 FC was applied since also small changes in gene expression were considered important in order to obtain a global view on transcriptional regulation. Growth rate responsive genes were grouped into 12 different clusters according to their expression profile by k-means cluster analysis (see Fig. 3 and Supporting information, Data S1). Of the 2886 differentially regulated genes, expression of 1226 genes was positively correlated to growth rate (clusters 6, 11, and 12) and 1080 genes showed a negative correlation (clusters 3, 5, 7, and 9). Transcription of the remaining 579 genes was regulated in a more complex way, showing up- or downregulation only at very low growth rates (clusters 1, 4, and 10) or peaking in expression between μ of 0.05 and 0.075 h–1 (clusters 2 and 8).
Figure 2.
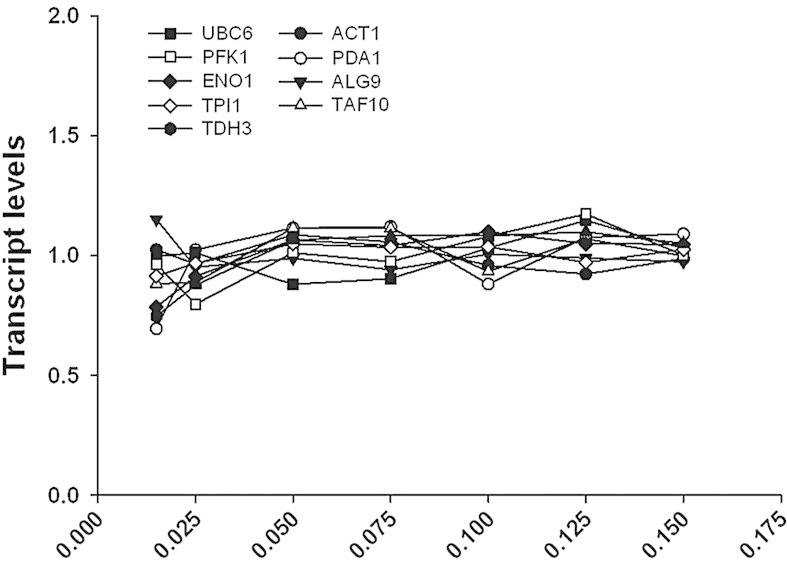
Expression levels of house-keeping genes in P. pastoris grown at different specific growth rates in glucose-limited chemostat cultures. House-keeping genes were selected according to ref. [10] and supplemented with genes encoding glycolytic enzymes. Gene expression values were normalized by dividing by the respective average expression values of a given gene. For every growth rate setpoint samples were collected from three individual cultivations and microarray experiments were performed in 2-color technical duplicates for every sample. UBC6, ubiquitin conjugating enzyme; PFK1, subunit of phosphofrucokinase; ENO1, enolase I; TPI1, triose phosphate isomerase; TDH3, glyceraldehyde-3-phosphate dehydrogenase; ACT1, actin; PDA1, subunit of pyruvate dehydrogenase; ALG9, mannosyltransferase; TAF10, subunit of TFIID and SAGA complexes.
Figure 3.

Global gene expression profiles in P. pastoris grown at different specific growth rates in glucose-limited chemostat cultures. Genes that were differentially expressed when compared to the highest μ of 0.15 h–1 at least at one growth rate setpoint (adjusted p-value < 0.02, see Section 2) were grouped into 12 clusters using the k-means clustering algorithm. Relative expression levels (log2 scale, y-axis) are displayed for each gene at the different growth rate setpoints (x-axis) as well as the mean FC (black line). For every growth rate setpoint samples were collected from three individual cultivations and microarray experiments were performed in 2-color technical duplicates for every sample.
Significantly enriched GO terms for the obtained clusters were determined using the GO Term Finder web tool and SGD annotations [19]. A comprehensive list of all determined GO terms for the categories biological process (BP), molecular function (MF), and cellular component (CC) can be found in Supporting information, Data S1. Enriched GO terms for the category BP are listed in Table 2.
Table 2.
Enriched GO terms for the category “biological process” of clusters including all regulated genes
| Cluster number | Number of genes without a homolog in S. cerevisiae among total number of genes within cluster | GO term | Corrected p-value |
|---|---|---|---|
| 1 | 69 of 170 | Carbohydrate metabolic process | 2.67E-02 |
| 2 | 19 of 34 | Cellular process involved in reproduction | 2.35E-08 |
| Response to pheromone | 1.67E-07 | ||
| Reproduction | 2.99E-07 | ||
| Multi-organism cellular process | 4.68E-07 | ||
| Multi-organism process | 1.02E-06 | ||
| Cytogamy | 1.42E-06 | ||
| Cell surface receptor signaling pathway | 3.86E-03 | ||
| Cellular developmental process | 2.38E-02 | ||
| 3 | 22 of 29 | Unknown | 6.75E-04 |
| 4 | 93 of 242 | – | – |
| 5 | 59 of 141 | – | – |
| 6 | 35 of 162 | rRNA metabolic process | 9.51E-19 |
| Ribonucleoprotein complex biogenesis | 1.18E-17 | ||
| ncRNA metabolic process | 1.61E-13 | ||
| Gene expression | 4.10E-11 | ||
| Cytoplasmic translation | 8.08E-10 | ||
| Translation | 3.92E-08 | ||
| RNA phosphodiester bond hydrolysis | 6.36E-06 | ||
| Cellular biosynthetic process | 1.65E-05 | ||
| Oganic substance metabolic process | 2.74E-05 | ||
| Cellular metabolic process | 5.22E-05 | ||
| Primary metabolic process | 5.41E-05 | ||
| Metabolic process | 2.39E-04 | ||
| Macromolecule metabolic process | 1.56E-03 | ||
| Nitrogen compound metabolic process | 5.23E-03 | ||
| RNA 5'-end processing | 1.00E-02 | ||
| Cellular amino acid biosynthetic process | 2.31E-02 | ||
| Biosynthetic process | 2.63E-02 | ||
| Methylation | 2.65E-02 | ||
| Cellular process | 3.27E-02 | ||
| 7 | 149 of 411 | Cell communication | 1.96E-07 |
| Response to stimulus | 4.85E-05 | ||
| Signaling | 6.25E-04 | ||
| Peroxisome organization | 4.84E-03 | ||
| Response to chemical stimulus | 5.05E-03 | ||
| Mitochondrion degradation | 1.28E-02 | ||
| Catabolic process | 1.33E-02 | ||
| 8 | 39 of 66 | Unknown | 2.41E-02 |
| 9 | 159 of 499 | Response to stimulus | 4.69E-02 |
| 10 | 42 of 67 | Unknown | 1.73E-03 |
| Amino acid transmembrane transport | 9.52E-03 | ||
| Glutamine family amino acid catabolic process | 3.93E-02 | ||
| 11 | 153 of 582 | Gene expression | 5.85E-06 |
| Mitochondrion organization | 1.30E-05 | ||
| Biosynthetic process | 1.84E-05 | ||
| Cellular metabolic process | 1.99E-05 | ||
| Organic substance biosynthetic process | 2.42E-05 | ||
| Cellular process | 2.54E-05 | ||
| tRNA metabolic process | 5.38E-05 | ||
| Metabolic process | 2.27E-04 | ||
| Translation | 2.36E-04 | ||
| 11 | 153 of 582 | Organic substance metabolic process | 6.44E-04 |
| Amino acid activation | 5.65E-03 | ||
| Macromolecule metabolic process | 6.45E-03 | ||
| Primary metabolic process | 3.56E-02 | ||
| 12 | 83 of 483 | Ribosome biogenesis | 3.01E-40 |
| ncRNA metabolic process | 2.59E-36 | ||
| Gene expression | 2.89E-35 | ||
| Cellular metabolic process | 8.87E-23 | ||
| Primary metabolic process | 2.96E-22 | ||
| Organic substance metabolic process | 1.61E-21 | ||
| Metabolic process | 4.99E-20 | ||
| Macromolecule metabolic process | 5.89E-18 | ||
| Cytoplasmic translation | 5.63E-16 | ||
| Cellular process | 9.97E-16 | ||
| Cellular biosynthetic process | 1.30E-14 | ||
| Biosynthetic process | 2.24E-13 | ||
| Nitrogen compound metabolic process | 3.04E-13 | ||
| RNA phosphodiester bond hydrolysis | 1.84E-10 | ||
| Alpha-amino acid biosynthetic process | 4.40E-10 | ||
| Cellular aromatic compound metabolic process | 6.14E-10 | ||
| Cellular component organization or biogenesis | 2.12E-09 | ||
| Cellular nitrogen compound metabolic process | 2.68E-09 | ||
| Heterocycle metabolic process | 3.35E-09 | ||
| Mitochondrial translation | 3.55E-09 | ||
| RNA 5'-end processing | 5.88E-09 | ||
| Organic cyclic compound metabolic process | 8.86E-09 | ||
| rRNA 5'-end processing | 3.96E-08 | ||
| Protein metabolic process | 9.16E-06 | ||
| Nucleic acid phosphodiester bond hydrolysis | 2.84E-04 | ||
| Regulation of translation | 8.81E-04 | ||
| Single-organism biosynthetic process | 9.94E-04 | ||
| Nuclear export | 2.18E-02 | ||
| Methylation | 3.12E-02 |
Redundant GO terms were excluded using the web-based tool REVIGO [51].
For all clusters displaying an expression pattern positively correlated to growth rate (clusters 6, 11, and 12), genes involved in the BPs of “gene expression, ” “translation, ” “biosynthetic processes, ” or related terms were significantly enriched. For example, out of 280 genes associated with the term “ribosome biogenesis, ” 196 genes could be found in one of the respective clusters, showing that faster growing cells cover their increased demand in novel cell material by boosting their translational capacity. Similar findings have been described for S. cerevisiae [5–7] and other organisms like Escherichia coli [22], so that upregulation of translation can be regarded as a conserved process in response to growth. Using the common TF algorithm of Genomatix MatInspector software [23], Rap1 binding sites were identified in the promoters of 68 out of 77 annotated ribosomal protein (RP) genes, additionally all RP genes contain binding sites for the ribosomal RNA processing element (RRPE)-binding protein Stb3 in their promoter regions.
For clusters 11 and 12 a large fraction of genes was related to mitochondrial functions, including not only “mitochondrial translation” but also several members of the TOM and TIM complexes as well as genes important for cytochrome c oxidase activity. Upregulation of mitochondrial genes with increasing carbon-limited growth rate has been described for S. cerevisiae as well [5, 24]. These results likely reflect increased energy requirements and therefore elevated respiratory activity at faster growth.
In cluster 6, the genes with the highest downregulation at low growth rate can be found: Flo5-2, a lectin-like cell wall protein involved in flocculation and the hypothetical gene PAS_chr2-1_0002.
Genes that show lower expression levels at higher growth rates can be found in clusters 3, 5, 7 and 9, albeit with different degrees of regulation (Fig. 3), and will thus be discussed together. In particular a large number of transcription factors (TFs) and transcriptional regulators is present in cluster 9 (42 TFs; corresponding to more than 8% of total genes), and to a lesser extend also in clusters 5 and 7 (9 and 27 TFs, respectively, corresponding to approximately 6.5% of total genes present in the clusters). Most of these transcriptional regulators are involved in stress response, response to nutrient levels and cell organization based on their function in S. cerevisiae. Out of 22 TFs without homologs in S. cerevisiae, only one TF has been assigned a function (Mpp1 is an activator of peroxisome biogenesis and function in Hansenula polmorpha [25]). The regulatory targets of the other TFs are unidentified yet.
Important enriched GO terms for all genes negatively correlated to increasing growth rate (cluster 3, 5, 7, and 9 together) are “cellular catabolic processes, ” “response to external stimulus, ” “response to nutrient levels, ” “intracellular signal transduction” and “biological regulation.” Many genes with a role in cellular catabolic processes are involved in autophagy as well as transport to the peroxisome and mitochondrial degradation, which have been reported to be under control of target of rapamycin (TOR) signaling [26] and are similarly regulated in response to growth rate in S. cerevisiae [6, 7]. Additionally, proteolysis related processes such as transport to the vacuole, ER-associated degradation (ERAD), proteasomal degradation, and a high number of genes involved in ubiquitination are among the downregulated genes. However, the regulated gene set is distinct from the one observed in S. cerevisiae by Fazio et al. [8]. Some of the genes involved in ubiquitination are also responsible for cell cycle regulation, in particular G1/S transition of the mitotic cell cycle and the anaphase promoting complex (APC). No clear correlation of subunits of the proteasome could be determined, although the TF that stimulates expression of proteasome genes, Rpn4, is part of cluster 7 (see also discussion below). Many of the transcriptional regulators are involved in transcriptional control of carbon-source responsive genes. For example Aca1, Azf1, Cat8-2, Hap5, Mig1-1, and associated regulatory proteins such as Grr1 and Reg1 play a role in glucose repression and induction. A homolog of Adr1 clusters in this group which activates transcription of several glucose-repressed genes including peroxisomal proteins and genes involved in methanol utilization [27]. Furthermore Gal4/Lac9 is found here which probably regulates a different set of target genes, as none of the other GAL genes (GAL1-3, GAL7, and GAL80) is present in P. pastoris.
Interestingly, although we monitored gene expression changes in relation to growth rate in carbon-limited conditions, also nitrogen-responsive regulators and their corresponding transcription targets are downregulated with increasing growth rate (see Supporting information, Data S1). As this behavior was not observed in the studies employing S. cerevisiae upon carbon limitation [6, 24], differences in the regulation of nutrient responsive regulons between the two yeasts can be concluded.
In this respect, TOR complex subunits are among the growth rate regulated genes in our study, contrary to the data presented for S. cerevisiae by Castrillo et al. [6]. It should be noted that P. pastoris (similar to most other fungi and higher eukaryotes) has just one TOR gene named TOR2 (compared to the two highly similar TOR genes TOR1 and TOR2 in S. cerevisiae) [26], which probably acts in both TOR complexes. While TORC1 which consists of Tor1/2, Kog1, Lst8, and Tco89 is a key regulator of cell growth in response to nutrient availabilty, TORC2 is built of Tor2, Avo1, Avo2, Tsc11, Lst8, Bit61, and Slm1/2 and responsible for regulation of cell wall integrity, actin cytoskeleton polarization and receptor endocytosis [28]. Along with TOR2, KOG1, TSC11, and AVO2 are downregulated with increasing growth rate. No homologs of Bit61 and Slm2 can be found in the P. pastoris genome.
While TOR signaling usually exhibits its regulatory function by affecting the cellular localization of transcription regulatory proteins, we also see significant transcriptional control of TOR responsive TFs in P. pastoris. A high degree of transcriptional regulation in P. pastoris as compared to S. cerevisiae has been described previously for regulation of glycolytic genes and seems to be related to the high affinity glucose uptake system of the Crabtree negative yeast [29].
We see transcriptional induction of glyoxlate shunt enzymes at low growth rate (corresponding to lower glucose concentration), similar to the observations by Regenberg et al. [7] in S. cerevisiae, although the growth rates employed in our study are significantly lower and respiratory growth is maintained throughout all growth rates in P. pastoris. Although it might be assumed that this is due to the lower glucose concentrations at lower growth rate, we do not observe glucose repression of ICL1 and MLS1 when analyzing promoter activity (our unpublished data). As also described for S. cerevisiae in Gutteridge et al. [24], genes responsible for the degradation of the storage carbohydrates glycogen and trehalose have lower expression levels at increasing growth rate, while glycogen and trehalose biosynthetic genes do not show a growth dependent regulation pattern (see Supporting information, Data S1). Regarding sugar transporters [30], the gene encoding the single plasma membrane glucose sensor SNF2 is downregulated with increasing growth rate, along with transporters of alternative carbon sources such as glycerol and maltose. Only one of the two P. pastoris high affinity glucose transporters (PAS_chr3_0023) is upregulated at low growth rates, while expression of the second one (GHT1) seems not to be affected by growth rate. P. pastoris low affinity glucose transporters and hexokinase HXK1 are downregulated at low specific growth rate, which leads to the induction of the alcohol oxidase genes AOX1 and AOX2 as reported by Zhang et al. [31]. None of the other genes involved in the methanol utilization pathway are found among the growth regulated genes in our study.
TOR signaling also influences the expression of nitrogen responsive genes through the GATA transcription activators Gat1 and Gln3 in reponse to different nitrogen sources [32], however, the correlation to growth rate was not reported previously. Although upstream activators of TORC signaling are largely unknown, glutamate and glutamine have been established to be important indicators of nutrient status [33]. In this respect, glutamate is one of the highest scoring reporter metabolites (see Supporting information, Table S3) at low growth rates in our study which indicates high transcriptional changes of metabolic genes around this metabolite.
TORC1 signaling also couples the highly energy consuming step of ribosome biogenesis with nutrient availability by controlling the transcription of RP genes and other components of the translation machinery through the localization of RP specific TFs [34]. Under good nutrient conditions, TOR inhibits catabolic processes such as autophagy [26].
Other TFs with expression negatively correlated to growth rate are involved in stress response (e.g. general stress response regulator Msn4; Aft2, and Cth1 involved in iron homeostasis, oxidative stress response TF Yap1, zinc-responsive TF Zap1, stress regulatory proteins Sko1 and Skn7; Rfx1 regulating genes in reponse to DNA damage) or regulation of cell wall organization in response to various stimuli (e.g. regulators of cell wall integrity pathway Rlm1 and Ssd1; Rim101 and Nrg1 involved in pH response and cell wall reorganization; Mit1, Hms1, and Flo8 conferring the distinction between pseudohyphal and vegetative growth and the expression of cell surface flocculins), some of which have been shown to physically interact with the TOR complexes. It should be highlighted that many of the other genes present in clusters 3, 5, 7, and 9 are indeed under transcriptional control of the above-mentioned transcriptional regulators (see Supporting information, Data S1).
Additionally, the MCM2-7 complex that binds chromosomal replication origins and assembles as part of the prereplicative complex during the G1 phase of the cell cycle is enriched in the clusters 7 and 9.
Genes with a bell shaped expression pattern peaking at an intermediate growth rate of 0.05 h–1 were grouped into the clusters 2 and 8. GO analysis for the category BP showed that a high proportion of genes from cluster 2 corresponds to mating related terms like “cellular process involved in reproduction” and “response to pheromone” (among them genes encoding the pheromone receptors Ste2 and Ste3, the G-protein α subunit Gpa1 and the scaffold protein Far1). In case of cluster 8 only genes with unknown function were significantly enriched. However, several further key elements of the mating and fusion pathway (STE4, STE12, and FUS3-1) as well as other mating related genes were located in this cluster.
A common strategy of yeasts to survive unfavorable growth conditions is the formation of spores. While natural isolates of S. cerevisiae usually occur in a diploid or polyploid state [35], P. pastoris is most stable in its vegetative haploid form and needs to mate in order to allow sporulation [36]. Contrary to S. cerevisiae, which mates spontaneously in rich media, P. pastoris (like Schizosaccharomyces pombe and Kluyveromyces lactis) enters its sexual life cycle only under certain conditions such as nitrogen starvation [37]. It is therefore not surprising that the mating pathway is induced in this yeast when nutrients become more limiting. The drop in expression levels below specific growth rates of 0.05 h–1 may reflect a change in the survival strategy of P. pastoris, as the risk of being unable to complete the sexual life cycle may become too high at scarce nutrient supply.
For K. lactis it has been shown that the TF RME1 plays a important role in the regulation of mating genes in response to nutrional signals [38] and for fission yeast the TF Ste11 (not to be confused with its S. cerevisae homonym) has been described as a key element in the activation of mating in response to starvation (reviewed in [37]). Expression of RME1 in P. pastoris shows a comparable pattern to the genes found in clusters 2 and 8 but is less strongly regulated (cluster 1). However, no putative RME1 binding motif was identified when the promotor regions of 22 genes connected to mating were analyzed (data not shown) and there seems to be no homolog to S. pombe Ste11 in the P. pastoris genome, leaving the question unanswered how P. pastoris concerts nutritional and mating signaling.
Cluster 1 is comprised of genes which are upregulated at intermediate growth rates between 0.025 and 0.075 h–1 (but to a lesser extent than clusters 2 and 8) and partially downregulated at the lowest μ of 0.015 h–1. Genes significantly enriched in this cluster are related to “carbohydrate metabolic processes, ” playing a role in storage carbohydrate synthesis, cell wall remodeling, lipid metabolism, and central carbon metabolism, among them TDH3, the native gene controlled by the GAP promoter. For cluster 4 no significantly enriched GO terms were identified, however the HSA gene fell into this almost unregulated cluster. Both TDH3 and HSA were unregulated except for decreased transcription at the lowest growth rate.
Cluster 10 consists of genes which are upregulated at very slow growth and are discussed below in Section 3.5.
During our study, we could also observe that at growth rates below 0.1 h–1, a considerable fraction of cells changed their morphological appearance. At a growth rate of 0.015 h–1 most of the cells had an elongated shape and formed ocassionally branched pseudohyphae (Supporting information, Fig. S1). Interestingly, if the growth rate was increased again above 0.075 h–1, some cells remained in the elongated state. In S. cerevisiae, expression of Flo11, a GPI-anchored cell surface glycoprotein (flocculin) required for filamentous growth, is subjected to epigenetic regulation and the epigenetic state of FLO11 is stable over several generations [39, 40]. It is therefore possible, that also in P. pastoris the transition between the yeast and the filamentous form is under epigenetic control.
Filamentous growth in yeast is triggered by different extracellular stimuli, nutrient limitation being the most common. In S. cerevisae at least four signaling cascades have been decribed which regulate filamentous growth: the mitogen activated protein kinase (MAPK) pathway, the rat sarcoma/protein kinase A (RAS/PKA) pathway, the TOR pathway, and the sucrose nonfermentable (SNF) pathway (extensively reviewed by [41]). However, how these different pathways exactly work together in order to adminstrate filamentous growth is a subject of current research. Homologs of most genes taking part in regulation of filamentous growth were identified in P. pastoris. Although their mode of action is mainly based on kinase activity, we observed growth rate dependent transcriptional regulation of many of the respective genes (Supporting information, Data S1), showing mostly stronger expression at more limiting conditions.
3.3 Core set of growth regulated genes
By comparing their data with previous studies Fazio et al. [8] have identified a core set of growth regulated genes of S. cerevisiae. The 21 common upregulated genes include 11 involved in translation (mainly ribosomal) and 3 related to sphingolipid synthesis. Among the 10 common downregulated genes 4 are involved in regulation of fructose 1, 6-bisphosphatase, the key regulatory enzyme of gluconeogenesis. Of these 31 core growth regulated genes 26 have homologs in P. pastoris, and of these 21 (81%) were regulated in the same manner (Supporting information, Tables S1 and S2). It is not surprising that ribosomal genes are a majority among the common upregulated genes. Another common regulatory pattern seems to be directed towards upregulation of gluconeogenesis at faster growth. In S. cerevisiae, Pfk26 (6-phosphofructo-2-kinase) leads to inhibition of gluconeogenesis, so that its downregulation with increasing growth will enable an upregulation of gluconeogenesis. Interestingly P. pastoris has two homologs of PFK26 which are reversely regulated. The major form, PFK26-1 is downregulated with increasing growth rate as in S. cerevisae. The core growth regulated genes include a second regulatory pathway of gluconeogenesis, involving the glucose induced degradation (GID) complex which is responsible for 1, 6-fructose bisphosphatase (Fbp1) degradation. Thus downregulation of VID genes as parts of the GID complex will result in higher Fbp1 activity which will thus trigger more gluconeogenesis at faster growth. We observe the same pattern of gluconeogenesis upregulation in both yeasts, with less representation of VID gene regulation in P. pastoris, but a more complex PFK26 regulation.
This large overlap of conserved growth regulated genes across the species border is certainly surprising, considering the evolutionary distance between these two yeasts and their different physiology [42]. We can thus postulate that these genes represent something like an evolutionary conserved core set of genes directly regulated together with growth rate.
3.4 Protein folding and secretion
Apart from the observed upregulation of genes associated to biosynthesis, translation, and ribosomes in particular, it is of major importance to understand how processes related to protein folding and secretion are regulated with growth. We have therefore analyzed a subset of genes related to these processes in more detail. Recently, we have investigated the genomic setup of eight yeast species (including P. pastoris) concerning the secretory pathway [43]. Genes were identified by reciprocal BLAST search of S. cerevisiae genes annotated to ER, protein folding, glycosylation, ERAD, Golgi, SNAREs, and vesicle-mediated transport, backed up by manual curation. This list was further supplemented with genes for vacuolar proteins, yielding a final list of 542 P. pastoris genes, about 10% of the genome. All of these genes excluding the unregulated ones (adjusted p-value < 0.02) were grouped according to their log2 FC transcription values comparing each μ setpoint with the highest setpoint by k-means clustering into six clusters (Supporting information, Data S2). The genes fall essentially into four categories (Fig. 4A): steadily upregulated (cluster A and C), steadily downregulated (cluster B and E), upregulated only at the lowest μ setpoint (cluster D), and genes with a more complex regulation pattern with a maximum at intermediate μ around 0.05 h–1 (cluster F). These clusters were then mapped on the folding and secretion pathway to visualize gene regulation (Fig. 4B). Genes related to translocation, glycosylation, and protein folding and the proteasome are mainly upregulated at higher μ, while about 20% of regulated genes related to ERAD and the vacuole are downregulated with increasing μ. Among the downregulated vacuolar genes 10 are related to autophagy. Genes related to vesicular transport show a more complex regulation pattern. Figure 4C indicates tabularly which BPs of folding and secretion are highly regulated.
Figure 4.

Transcriptional regulation of genes encoding proteins with a role in protein folding and secretion in P. pastoris grown at different specific growth rates in glucose-limited chemostat cultures. (A) Genes related to the secretory pathway that were at least at one growth rate setpoint differentially expressed (adjusted p-value < 0.02, see Section 2) when compared to the highest μ of 0.15 h–1 were grouped into six clusters using the k-means clustering algorithm. Relative expression levels (log2 scale, y-axis) are displayed for each gene at the different growth rate setpoints (x-axis) as well as the mean FC (magenta line). For every growth rate setpoint samples were collected from three individual cultivations and microarray experiments were performed in 2-color technical duplicates for every sample. (B) Genes of the clusters in (A) were mapped on the secretory pathway, using the color code of the clusters. For genes without a homolog in S. cerevisiae or a different yeast, the P. pastoris GS115 ORF number was assigned. If P. pastoris contained more than one ortholog, a number was added, e.g. MNN4-1, MNN4-2. (C) Overview of total numbers of genes allocated to different biological processes related to the secretory pathway, and relative numbers of regulated genes. Color intensities reflect the degree of regulation of the respective groups.
Among the genes showing upregulation with increasing μ we found many UPR regulated genes of translocation, protein folding, and glycosylation as well as cytosolic chaperones, which have been shown to be induced upon overexpression of the UPR regulator HAC1 [12]. In P. pastoris UPR is triggered by induction of HAC1 transcription rather than by posttranscriptional splicing of the HAC1 mRNA [44]. As HAC1 expression is upregulated with μ in our experiment (cluster 6 in Fig. 2) as are its primary targets, we can conclude that UPR induction with increasing specific growth rate is one major regulatory reaction to increasing cellular proliferation.
Positive correlation of recombinant protein secretion rate may thus be inferred to increasing efficiency of protein translocation to the ER, and to enhanced folding assistance in the ER. The observed upregulation of glycosylation per se is not connected to secretion. At closer sight this relates mainly to early N-glycosylation steps in the ER, which have important functions in protein quality control via the calnexin cycle [45]. It should be noted, however, that HSA is not glycosylated so that this folding assistance is not effective for this product.
Intracellular proteolytic degradation has been described to be a considerable sink for recombinant protein in P. pastoris [46, 47]. While we observe mainly upregulation of proteasomal genes, more ERAD genes are downregulated than upregulated with increasing μ. Faster growth causes a need for proteasomal activity to turn over cell cycle regulators and other regulatory proteins [48], while ERAD – although connected to proteasomal degradation – is devoted to degrade misfolded secretory proteins, so that these two functions diverge at fast growth. The yeast vacuole is the second main intracellular container for protein disposal. Twenty percent of all genes related to the vacuole were downregulated with increasing growth, indicating that P. pastoris reduces the need for disposal (and turnover) of cellular proteins at increasing μ. The downregulation of 9 out of 11 ATG (autophagy related) genes with increasing μ shows clearly a decrease of protein turnover at faster growth, which may come to the benefit of recombinant protein production as well.
Vesicular transport genes are less regulated, which may reflect the fact that only part of the proteins entering the secretory pathway are dedicated to leave the cell. In more detail, intracellular vesicle transport (COPI and COPII vesicles) are rather upregulated than downregulated, while genes related to exocytosis are mainly downregulated. This may constitute a bottleneck for (recombinant) protein secretion at least at higher growth rates. Overexpression of KIN2 (which is downregulated at faster growth) increased the secretion of a recombinant protein in P. pastoris, while overexpression of SSO2 (upregulated at high growth rate) did not [49], supporting the hypothesis of an exocytosis bottleneck.
3.5 Gene regulation at very slow growth
To get a better understanding of transcriptional regulation in response to very slow growth, we compared pairwise the growth rate setpoints 0.015 and 0.025 h–1 (comparison A) as well as 0.025 and 0.05 h–1 (comparison B) and identified 732 genes which were at least in one comparison differentially expressed (adjusted p-value < 0.05). The quotient between FC values of regulated genes was calculated in order to discriminate between genes that showed a similar trend in expression between comparison A and B and those with a divergent expression profile. Genes with a quotient between 1.5 and 0.67 were excluded and the remaining 494 genes were ranked in four different groups (Supporting information, Data S3): Group 1 contained 226 genes which were downregulated in comparison A and less downregulated, not regulated or upregulated in comparison B. Group 2 was composed of 238 genes which showed the opposite expressional trend. Group 3 (11 genes) was comprised of genes which were stronger downregulated in comparison B than in comparison A and group 4 of 19 genes with stronger upregulation in comparison B than in comparison A. The gene sets of the groups were analyzed for enriched GO terms (Table 3).
Table 3.
Enriched GO terms for the categories biological process (BP), molecular function (MF), and cellular component (CC) of genes regulated at very low μ
| Group | Number of genes without a homolog in S. cerevisiae among total number of genes within cluster | GO term | Corrected p-value |
|---|---|---|---|
| 1 | 79 of 224 | BP sexual reproduction | 3.14E-04 |
| BP multi-organism cellular process | 1.75E-02 | ||
| BP cellular process involved in reproduction | 1.85E-02 | ||
| BP filamentous growth of a population of unicellular organisms | 1.86E-02 | ||
| BP filamentous growth | 2.82E-02 | ||
| BP multi-organism process | 4.41E-02 | ||
| BP reproductive process | 4.57E-02 | ||
| MF nucleic acid binding transcription factor activity | 4.67E-03 | ||
| MF sequence-specific DNA binding transcription factor activity | 4.67E-03 | ||
| MF amino acid transmembrane transporter activity | 1.78E-02 | ||
| MF cation transmembrane transporter activity | 1.82E-02 | ||
| MF ion transmembrane transporter activity | 2.65E-02 | ||
| MF substrate-specific transmembrane transporter activity | 3.95E-02 | ||
| CC plasma membrane | 1.03E-05 | ||
| CC cell periphery | 2.33E-04 | ||
| CC plasma membrane part | 2.32E-02 | ||
| 2 | 92 of 239 | BP arginine metabolic process | 2.18E-02 |
| BP arginine biosynthetic process | 3.43E-02 | ||
| BP single-organism metabolic process | 4.92E-02 | ||
| CC plasma membrane | 3.56E-04 | ||
| CC cell periphery | 5.23E-03 | ||
| 3 | 6 of 11 | MF hydrolase activity, hydrolyzing O-glycosyl compounds | 9.07E-03 |
| MF hydrolase activity, acting on glycosyl bonds | 1.32E-02 | ||
| CC intrinsic to plasma membrane | 1.58E-02 |
In case of group 1, genes enriched for the category BP were either related to “filamentous growth” or to “sexual reproduction” and neighboring terms emphasizing again that cells refrain from mating and sporulation as a survival strategy when conditions become very scarce. The most significantly enriched term in the category MF was “nucleic acid binding TF activity.” While some genes related to this term (HMLALPHA, KAR4, and RME1) are involved in the sexual life cycle, others play a role in the regulation of filamentous growth (HMS1, MGA1, NRG1, and PDH1). Group 1 was also enriched for genes with a transmembrane transport activity being around half of the genes involved in amino acid and polyamine transport.
For group 2, three terms for the category BP were enriched – “arginine metabolic process, ” “arginine biosynthetic process, ” and “single-organism metabolic process.” In S. cerevisiae transcription of genes related to arginine biosynthesis is regulated by the repressor Arg81 [50], and its P. pastoris homolog is downregulated with increasing growth rate. At μ = 0.015 h–1, however, a subset of arginine synthesis genes is upregulated compared to the next higher setpoints, and this does not coincide with ARG81 regulation. A closer look at the pathway revealed that specifically all genes involved in the urea cycle (CPA1, ARG3, ARG1, ARG4, and CAR1) are upregulated at the lowest growth rate, contrary to the majority of the upstream synthesis process from glutamate to ornithine (ARG2, ARG7, and ARG8). This regulation pattern is also reflected in the reporter metabolites (Supporting information, Table S3). For the comparison 0.015–0.025 h–1 metabolic genes which convert all the intermediates of the urea cycle (ornithine, citrulline, argininosuccinate, fumarate, and arginine) are subjected to regulation, contrary to the comparison of the next two setpoints, 0.025–0.05 h–1. We conclude that arginine synthesis follows an Arg81 driven upregulation with increasing growth rate, while at very slow growth another regulatory pathway induces the urea cycle. In mammalian cells the urea cycle serves the disposal of surplus ammonium into urea, but its activity has not been discussed in yeasts. Under normal growth conditions it appears not reasonable for a single cellular organism to spend energy on the removal of a nitrogen source. It will be interesting in future to investigate whether very slow growth leads to a surplus of ammonium which may be caused, e.g. by amino acid degradation.
4 Concluding remarks
We have studied the genome-wide transcriptional regulation of P. pastoris from very slow growth to almost μmax with special attention to protein folding and secretion. Upregulation of ribosomal genes and other genes involved in translation appears as a general pattern as observed also in other organisms. Also mitochondrial genes are generally upregulated in P. pastoris – even stronger than in S. cerevisiae, which is probably due to the fact that P. pastoris as a Crabtree negative yeast maintains a fully respiratory metabolism under all studied conditions. Transcriptional upregulation at lower μ pertains a large number of transcriptional regulators, indicating that P. pastoris reacts to different growth rates by tuning the expression levels of many TFs, like those regulating stress response as well as carbon source and nitrogen responsive genes. Apparently P. pastoris represses mating and sporulation both at very slow and fast growth for different reasons. At fast growth, equivalent to rich nutrient supply, there is no need for sexual reproduction which increases genetic variability and is thus advantageous in less favorable conditions. At very low growth rates, indicative of scarce nutrient supply, the cells seem to avoid the risk not to be able to finish the sexual cycle.
A closer look on the regulation of the secretory pathway reveals that genes related to protein translocation into the ER and to folding in the cytosol and ER are mostly upregulated at higher growth rates, implying that these processes are necessary for higher protein turnover rates at fast growth. Downregulation of vacuolar genes indicates that proteolytic degradation is less prevalent at higher growth rates. Most of the proteins entering the secretory pathway in yeasts fulfill a function inside the cell rather than being secreted outside. This may explain why genes related to vesicular transport and exocytosis are not clearly upregulated at fast growth. We conclude that higher growth rates bring benefits for the production of secreted recombinant proteins, which must be balanced however with process parameters like oxygen and heat transfer, total process time and productivity.
Acknowledgments
This work was funded by the Austrian Science Fund (FWF): Doctoral Program BioToP-Biomolecular Technology of Proteins (FWF W1224). Further support by the Federal Ministry of Economy, Family and Youth (BMWFJ), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol and ZIT – Technology Agency of the City of Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG is acknowledged. The authors thank Frederik Hoppe for his excellent technical support during bioreactor cultivations.
The authors declare no financial or commercial conflict of interest.
Glossary
- BP
biological process
- ERAD
ER associated degradation
- FC
fold change
- GO
gene ontology
- HSA
human serum albumin
- RP
ribosomal proteins
- TF
transcription factor
- TOR
target of rapamycin
- UPR
unfolded protein response
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
suppinfo_data_S1
suppinfo_data_S2
suppinfo_data_S3
suppinfo
References
- 1.Liu Z, Hou J, Martinez JL, Petranovic D, Nielsen J. Correlation of cell growth and heterologous protein production by Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2013;97:8955–8962. doi: 10.1007/s00253-013-4715-2. [DOI] [PubMed] [Google Scholar]
- 2.Maurer M, Kuhleitner M, Gasser B, Mattanovich D. Versatile modeling and optimization of fed batch processes for the production of secreted heterologous proteins with Pichia pastoris. Microb. Cell Fact. 2006;5:37. doi: 10.1186/1475-2859-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mattanovich D, Branduardi P, Dato L, Gasser B, et al. Recombinant protein production in yeasts. Methods Mol. Biol. 2012;824:329–358. doi: 10.1007/978-1-61779-433-9_17. [DOI] [PubMed] [Google Scholar]
- 4.Stadlmayr G, Mecklenbräuker A, Rothmüller M, Maurer M, et al. Identification and characterisation of novel Pichia pastoris promoters for heterologous protein production. J. Biotechnol. 2010;150:519–529. doi: 10.1016/j.jbiotec.2010.09.957. [DOI] [PubMed] [Google Scholar]
- 5.Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, et al. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castrillo JI, Zeef LA, Hoyle DC, Zhang N, et al. Growth control of the eukaryote cell: A systems biology study in yeast. J. Biol. 2007;6:4. doi: 10.1186/jbiol54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regenberg B, Grotkjaer T, Winther O, Fausboll A, et al. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006;7:R107. doi: 10.1186/gb-2006-7-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fazio A, Jewett MC, Daran-Lapujade P, Mustacchi R, et al. Transcription factor control of growth rate dependent genes in Saccharomyces cerevisiae: A three factor design. BMC Genomics. 2008;9:341. doi: 10.1186/1471-2164-9-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arvas M, Pakula T, Smit B, Rautio J, et al. Correlation of gene expression and protein production rate – a system wide study. BMC Genomics. 2011;12:616. doi: 10.1186/1471-2164-12-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boender LG, van Maris AJ, de Hulster EA, Almering MJ, et al. Cellular responses of Saccharomyces cerevisiae at near-zero growth rates: Transcriptome analysis of anaerobic retentostat cultures. FEMS Yeast Res. 2011;11:603–620. doi: 10.1111/j.1567-1364.2011.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hohenblum H, Borth N, Mattanovich D. Assessing viability and cell-associated product of recombinant protein producing Pichia pastoris with flow cytometry. J. Biotechnol. 2003;102:281–290. doi: 10.1016/s0168-1656(03)00049-x. [DOI] [PubMed] [Google Scholar]
- 12.Graf A, Gasser B, Dragosits M, Sauer M, et al. Novel insights into the unfolded protein response using Pichia pastoris specific DNA microarrays. BMC Genomics. 2008;9:390. doi: 10.1186/1471-2164-9-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smyth GK, Limma . In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Heidelberg: Springer; 2005. pp. 397–420. [Google Scholar]
- 14.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 15.Patil KR, Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. USA. 2005;102:2685–2689. doi: 10.1073/pnas.0406811102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cvijovic M, Olivares-Hernandez R, Agren R, Dahr N, et al. BioMet Toolbox: Genome-wide analysis of metabolism. Nucleic Acids Res. 2010;38:W144–W149. doi: 10.1093/nar/gkq404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caspeta L, Shoaie S, Agren R, Nookaew I, Nielsen J. Genome-scale metabolic reconstructions of Pichia stipitis and Pichia pastoris and in silico evaluation of their potentials. BMC Syst. Biol. 2012;6:24. doi: 10.1186/1752-0509-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sturn A, Quackenbush J, Trajanoski Z. Genesis: Cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 19.Boyle EI, Weng S, Gollub J, Jin H, et al. GO::TermFinder – open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prielhofer R, Maurer M, Klein J, Wenger J, et al. Induction without methanol: Novel regulated promoters enable high-level expression in Pichia pastoris. Microb. Cell Fact. 2013;12:5. doi: 10.1186/1475-2859-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pirt SJ. The maintenance energy of bacteria in growing cultures. Proc. R. Soc. Lond. B Biol. Sci. 1965;163:224–231. doi: 10.1098/rspb.1965.0069. [DOI] [PubMed] [Google Scholar]
- 22.Kaczanowska M, Ryden-Aulin M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol Mol Biol Rev. 2007;71:477–494. doi: 10.1128/MMBR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cartharius K, Frech K, Grote K, Klocke B, et al. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 24.Gutteridge A, Pir P, Castrillo JI, Charles PD, et al. Nutrient control of eukaryote cell growth: A systems biology study in yeast. BMC Biol. 2010;8:68. doi: 10.1186/1741-7007-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leao-Helder AN, Krikken AM, van der Klei IJ, Kiel JA, Veenhuis M. Transcriptional down-regulation of peroxisome numbers affects selective peroxisome degradation in Hansenula polymorpha. J. Biol. Chem. 2003;278:40749–40756. doi: 10.1074/jbc.M304029200. [DOI] [PubMed] [Google Scholar]
- 26.Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy. 2007;3:106–116. doi: 10.4161/auto.3595. [DOI] [PubMed] [Google Scholar]
- 27.Lin-Cereghino GP, Godfrey L, de la Cruz BJ, Johnson S, et al. Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris. Mol. Cell Biol. 2006;26:883–897. doi: 10.1128/MCB.26.3.883-897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loewith R, Jacinto E, Wullschleger S, Lorberg A, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 29.Baumann K, Dato L, Graf AB, Frascotti G, et al. The impact of oxygen on the transcriptome of recombinant S. cerevisiae and P. pastoris – a comparative analysis. BMC Genomics. 2011;12:218. doi: 10.1186/1471-2164-12-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattanovich D, Graf A, Stadlmann J, Dragosits M, et al. Genome, secretome and glucose transport highlight unique features of the protein production host Pichia pastoris. Microb. Cell Fact. 2009;8:29. doi: 10.1186/1475-2859-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang P, Zhang W, Zhou X, Bai P, et al. Catabolite repression of Aox in Pichia pastoris is dependent on hexose transporter PpHxt1 and pexophagy. Appl. Environ. Microbiol. 2010;76:6108–6118. doi: 10.1128/AEM.00607-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper TG. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: Connecting the dots. FEMS Microbiol. Rev. 2002;26:223–238. doi: 10.1111/j.1574-6976.2002.tb00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crespo JL, Powers T, Fowler B, Hall MN. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc. Natl. Acad. Sci. USA. 2002;99:6784–6789. doi: 10.1073/pnas.102687599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powers T, Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol. Biol. Cell. 1999;10:987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ezov TK, Boger-Nadjar E, Frenkel Z, Katsperovski I, et al. Molecular-genetic biodiversity in a natural population of the yeast Saccharomyces cerevisiae from “evolution canyon:” Microsatellite polymorphism, ploidy and controversial sexual status. Genetics. 2006;174:1455–1468. doi: 10.1534/genetics.106.062745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cregg J, Sheny S, Johnson M, Waterham H. Classical genetic manipulation. In: Higgins DR, Cregg JM, editors. Pichia Protocols. New York: Humana Press; 1998. pp. 17–26. [DOI] [PubMed] [Google Scholar]
- 37.Merlini L, Dudin O, Martin SG. Mate and fuse: How yeast cells do it. Open Biol. 2013;3:130008. doi: 10.1098/rsob.130008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Booth LN, Tuch BB, Johnson AD. Intercalation of a new tier of transcription regulation into an ancient circuit. Nature. 2010;468:959–963. doi: 10.1038/nature09560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halme A, Bumgarner S, Styles C, Fink GR. Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell. 2004;116:405–415. doi: 10.1016/s0092-8674(04)00118-7. [DOI] [PubMed] [Google Scholar]
- 40.Octavio LM, Gedeon K, Maheshri N. Epigenetic and conventional regulation is distributed among activators of FLO11 allowing tuning of population-level heterogeneity in its expression. PLoS Genet. 2009;5:e1000673. doi: 10.1371/journal.pgen.1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cullen PJ, Sprague GF., Jr The regulation of filamentous growth in yeast. Genetics. 2012;190:23–49. doi: 10.1534/genetics.111.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dujon B. Yeast evolutionary genomics. Nat. Rev. Genet. 2010;11:512–524. doi: 10.1038/nrg2811. [DOI] [PubMed] [Google Scholar]
- 43.Delic M, Valli M, Graf AB, Pfeffer M, et al. The secretory pathway: Exploring yeast diversity. FEMS Microbiol. Rev. 2013;37:872–914. doi: 10.1111/1574-6976.12020. [DOI] [PubMed] [Google Scholar]
- 44.Guerfal M, Ryckaert S, Jacobs PP, Ameloot P, et al. The HAC1 gene from Pichia pastoris: Characterization and effect of its overexpression on the production of secreted, surface displayed and membrane proteins. Microb. Cell Fact. 2010;9:49. doi: 10.1186/1475-2859-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parlati F, Dominguez M, Bergeron JJ, Thomas DY. Saccharomyces cerevisiae CNE1 encodes an endoplasmic reticulum (ER) membrane protein with sequence similarity to calnexin and calreticulin and functions as a constituent of the ER quality control apparatus. J. Biol. Chem. 1995;270:244–253. doi: 10.1074/jbc.270.1.244. [DOI] [PubMed] [Google Scholar]
- 46.Pfeffer M, Maurer M, Kollensperger G, Hann S, et al. Modeling and measuring intracellular fluxes of secreted recombinant protein in Pichia pastoris with a novel 34S labeling procedure. Microb. Cell Fact. 2011;10:47. doi: 10.1186/1475-2859-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfeffer M, Maurer M, Stadlmann J, Grass J, et al. Intracellular interactome of secreted antibody Fab fragment in Pichia pastoris reveals its routes of secretion and degradation. Appl. Microbiol. Biotechnol. 2012;93:2503–2512. doi: 10.1007/s00253-012-3933-3. [DOI] [PubMed] [Google Scholar]
- 48.Humphrey T, Pearce A. Cell cycle molecules and mechanisms of the budding and fission yeasts. Methods Mol. Biol. 2005;296:3–29. doi: 10.1385/1-59259-857-9:003. [DOI] [PubMed] [Google Scholar]
- 49.Gasser B, Sauer M, Maurer M, Stadlmayr G, Mattanovich D. Transcriptomics-based identification of novel factors enhancing heterologous protein secretion in yeasts. Appl. Environ. Microbiol. 2007;73:6499–6507. doi: 10.1128/AEM.01196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubois E, Messenguy F. Isolation and characterization of the yeast ARGRII gene involved in regulating both anabolism and catabolism of arginine. Mol. Gen. Genet. 1985;198:283–289. doi: 10.1007/BF00383008. [DOI] [PubMed] [Google Scholar]
- 51.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
suppinfo_data_S1
suppinfo_data_S2
suppinfo_data_S3
suppinfo