

SUMMARY
The P7C3 class of aminopropyl carbazole chemicals fosters the survival of neurons in a variety of rodent models of neurodegeneration or nerve cell injury. To uncover its mechanism of action, an active derivative of P7C3 was modified to contain both a benzophenone for photo-crosslinking and an alkyne for CLICK chemistry. This derivative was found to bind nicotinamide phosphoribosyltransferase (NAMPT), the rate limiting enzyme involved in the conversion of nicotinamide into nicotinamide adenine dinucleotide (NAD). Administration of active P7C3 chemicals to cells treated with doxorubicin, which induces NAD depletion, led to a rebound in intracellular levels of NAD and concomitant protection from doxorubicin-mediated toxicity. Active P7C3 variants likewise enhanced the activity of the purified NAMPT enzyme, providing further evidence that they act by increasing NAD levels through its NAMPT-mediated salvage.
INTRODUCTION
No substantive therapeutics are available for the treatment of almost any form of disease entailing nerve cell death. Patients suffering from any of a wide spectrum of neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, fronto-temporal dementia and Huntington’s disease are condemned to progressive demise of the CNS by virtue of nerve cell death. It is likewise the case that no effective treatments exist for injuries to the brain or peripheral nervous system, including traumatic or concussive brain injury, spinal cord injury, or peripheral nerve injury. Any chemical having the capacity to safely impede nerve cell death in the context of these varied diseases or injuries would offer the opportunity for transformative impact in modern medicine.
Previously we performed a target-agnostic, in vivo screen in search of chemicals that might enhance hippocampal neurogenesis in adult mice (Pieper et al., 2010). The screen was simple in concept. We selected 1,000 drug-like chemicals from the 250,000 compounds in the UTSWMC high throughput screening center. The compounds were selected to preserve chemical diversity, enhance the representation of chiral molecules and avoid untoward chemical properties such as reactive moieties. The 1,000 molecules were randomly pooled into groups of ten, and each pool was administered directly into the left ventricle of two adult mice. Intracranial delivery was facilitated by stereotactic positioning of a canula fed directly by an Alzet mini-pump containing the mixture of ten chemicals. The drug mixture was administered over a week-long period at concentrations anticipated to deliver mid-nanomolar levels of the ten test compounds. Daily injections of the thymidine analog, bromodeoxyuridine (BrdU), were co-administered in order to monitor the formation of new hippocampal nerve cells. Following compound administration, animals were sacrificed such that brain tissue could be recovered, sectioned and stained with antibodies to BrdU. This two-year screen led to the discovery of a handful of pools that enhanced neurogenesis in both test mice that had been exposed to the pool. Breakdown of the active pools allowed the individual testing of each of the ten chemicals in the pool, leading to the discovery of eight pro-neurogenic compounds (Pieper et al., 2010).
Among the eight, pro-neurogenic chemicals, pharmacological testing gave evidence that only one of the compounds had favorable pharmacological properties. Pool seven (P7) contained an aminopropyl carbazole as its active, third compound (C3). When administered to mice via intraperitoneal (IP), intravenous (IV) or oral routes, the P7C3 compound revealed favorable half-life, volume of distribution and brain penetration. It was also found that P7C3 could be safely administered to mice and rats for prolonged periods at concentrations well above those required to stimulate hippocampal neurogenesis, giving evidence that the molecule was not overtly toxic to rodents.
Although it was initially anticipated that pro-neurogenic compounds would act by stimulating the mitotic birth of newborn nerve cells in the subgranular layer of the dentate gyrus, P7C3 revealed no such activity. The two-fold enhanced level of BrdU-positive neurons observed over a week-long dose of P7C3 was absent when animals were pulsed with BrdU for only 24 hours. More strikingly, when BrdU was pulsed for only one day, and animals were subsequently administered P7C3 for a month, we observed a far larger enhancement in BrdU-positive hippocampal neurons (500%). Instead of stimulating the mitotic division of neuronal stem cells, these observations gave evidence that P7C3 mitigated the death of newborn neurons. Under the conditions of our study only 10–20% of newborn neurons survive the month-long differentiation process to become properly wired hippocampal neurons. Prolonged administration of P7C3 significantly mitigated the death of newborn neurons, such that upwards of half of the cells survive the month-long “differentiation gauntlet” taking place between stem cell mitosis and terminal nerve cell differentiation. Indeed, localized expression of pro-apoptotic proteins in newborn neurons has been found to limit neurogenesis, whereas expression of anti-apoptotic proteins enhances neurogenesis in adult mice (Kim et al., 2009) (Kuhn et al., 2005) (Sun et al., 2004). It has likewise been found that the pro-neurogenic benefits of wheel running and/or enriched environment for caged rodents is primarily attributable to neuroprotective effects on newborn hippocampal neurons (Kempermann et al., 1997; Kim et al., 2010) (van Praag et al., 1999).
Having discovered the aminopropyl carbazole chemical in an unbiased, in vivo screen, and having found that it protects newborn neurons from death, we use methods of medicinal chemistry to improve the potency and pharmacological properties of P7C3 (MacMillan et al., 2011; Naidoo et al., 2014; Pieper et al., 2014). Using these improved, active derivatives of P7C3 we have observed neuroprotective activity in animal models of Parkinson’s disease (De Jesus-Cortes et al., 2012), amyotrophic lateral sclerosis (Tesla et al., 2012), as well as concussive injury to the rodent brain (Yin et al., Cell Reports, in press). Of significant concern, however, was the fact that we have been ignorant of the mechanism of action of the P7C3 class of neuroprotective chemicals. Here we provide evidence that these chemicals function by enhancing the activity of the rate-limiting enzyme in the salvage of nicotinamide adenine dinucleotide (NAD) from nicotinamide.
RESULTS
Active variants of P7C3 protect cultured cells from doxorubicin-mediated toxicity
Progress on studies aimed towards resolving the mode of action of P7C3 has been hampered by the fact that we have been limited to studies of drug action in living animals. Our standard assay of neurogenesis requires, under optimal conditions, a one month turn-around from the synthesis of a new derivative to a score for its neuroprotective activity. In efforts to break this logjam, we tested whether P7C3 might have cell-protective activity to any of eight generic toxins. The active A20 variant of P7C3 was found to protect cultured U2OS cells from doxorubicin-mediated toxicity (Figure 1A). By contrast, no evidence of drug-mediated protection was observed for any of seven other toxins (Figure S1). Furthermore, as judged by the DNA damage marker – phosphorylation of H2AX, and the hallmark of apoptosis – caspase-3 activation, co-administration of A20 blocked doxorubicin-induced apoptosis, but not DNA damage response (Figure S2).
Figure 1. Protection of cultured U2OS cells from doxorubicin-mediated toxicity by active derivatives of P7C3.
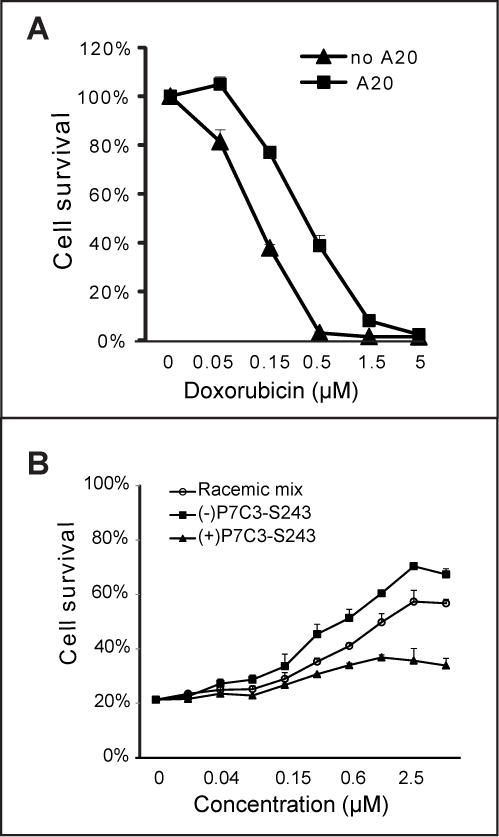
(A) U2OS cells were treated with 5μM P7C3-A20 for 2 h prior to incubation with the indicated concentrations of doxorubicin for 72h. (B) Comparison of the P7C3-S243 enantiomers in protection of cells from doxorubicin revealed that (−)-P7C3-S243 was more active than (+)-P7C3-S243, consistent with their respective neuroprotective activities in vivo (see text). In all cell survival graphs, data are expressed as mean ± standard deviation (SD) of experimental duplicates. See also Figure S1, S2 and S3, and Table S1.
In order to assess whether a correlation might exist between pro-neurogenic activity in living mice and doxorubicin toxicity (dox:tox) protection, we assayed 224 compounds that have been synthesized and assayed for pro-neurogenic activity over the past 4–5 years (Table S1). These include hundreds of structure:activity relationship (SAR) derivatives of P7C3, as well as a number of tricyclic antidepressants reported in the literature to have pro-neurogenic activity (Malberg et al., 2000). P7C3 derivatives were the only molecules having dox:tox protective activity (Figure S3 and Table S2). Further inspection of the data revealed that the chemical features of the carbazole ring, length of the aliphatic linker, and nature of the aromatic ring were all important determinants for both pro-neurogenic activity and dox:tox protection. Perhaps most compellingly, it was observed that the (−)-P7C3-S243 enantiomer that was active in the in vivo neurogenesis assay (Naidoo et al., 2014), the MPTP model of Parkinson’s disease (Naidoo et al., 2014), and the concussive blast injury model (co-submitted manuscrip) was also protective in the dox:tox assay (Figure 1B). By contrast, the (+)-P7C3-S243 enantiomer that was far less active in the three in vivo assays of neuroprotection was also considerably less active in the dox:tox protection assay. From these observations we conclude that active derivatives of P7C3 function by a related mechanism to protect neurons in living animals as well as cultured U2OS cells exposed to doxorubicin.
Identification of p70 and p55 as intracellular targets of P7C3
Various methods have been employed in efforts to identify the intracellular target of P7C3, including biotinylation followed by affinity purification, radiolabeling followed by filter binding assays, and photo-crosslinking. Among these strategies, the photo-crosslinking approach has been uniquely successful. P7C3-S326 was prepared to contain both a benzophenone for photo-crosslinking and an alkyne moiety for use in click chemistry (Figure 2A). This derivative was found to be active in both the in vivo neurogenesis assay and the dox:tox assay. When incubated with cultured cells, the P7C3-S326 derivative became covalently attached to roughly a dozen proteins. Among the covalently modified proteins, only one could be competed when the cells were co-incubated with a 30X excess of the active A20 variant of P7C3. This polypeptide exhibited an apparent molecular mass of 70kD (Figure 2B).
Figure 2. Identification of the P7C3 binding protein p70 using the P7C3-S326 photo-crosslinking probe.
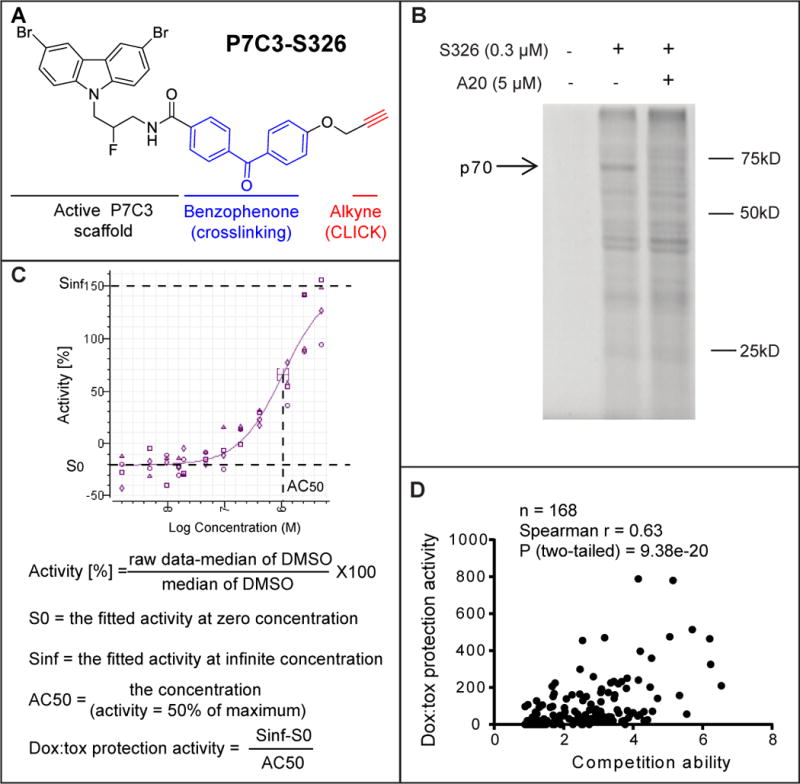
(A) Chemical structure of P7C3-S326. (B) Full gel image of photo-crosslinking in H2122 cells with P7C3-S326 followed by click chemistry with Alexa 532 dye. The active analog P7C3-A20 competed away the UV-dependent binding of p70 to P7C3-S326. See also Figure S4. (C) Formulas used for calculating doxorubicin toxicity (dox:tox) protection activity. Protection activity is expressed by (Sinf-S0)/AC50 in the twelve-point dose response curve (DRC) corresponding to each compound, wherein both efficacy and potency of compounds were considered. Sinf represents the expected maximal protection. S0 is the baseline. AC50 is the concentration of a compound where 50% of its maximal protection effect was observed. See also Figure S3 and Table S2. (D) Scatter plot of 168 derivatives of P7C3 revealed a significant correlation between protective activity of P7C3 analogs from doxorubicin-mediated toxicity and their ability to compete for photo-crosslinking of P7C3-S326 to p70. In the photo-crosslinking assays, the p70 band intensity was quantified by Image J, and compared the sample of P7C3-S326 alone with that of P7C3-S326 plus competitor. Pearson correlation coefficient (r) and two tailed P value were determined by GraphPad Prism 6 and Spearman Rank Correlation (v1.0.1) software. See also Table S1.
Binding competition assays were conducted with 168 chemical derivatives of P7C3 as a means of testing whether there might be a correlation between dox:tox protection and binding competition. The scatter plot shown in Figure 2D revealed a statistically significant correlation between the two activities, yielding a P value of 9.4 × 10−20. It can thus be predicted with a high level of confidence that active variants of P7C3 are able to compete for P7C3-S326 crosslinking, but that inactive variants cannot. Armed with this knowledge, cell fractionation experiments were performed in order to enrich for the p70 band. During the course of these studies it was periodically observed that P7C3-S326 could also be crosslinked to a second band running with an apparent molecular weight of 55kD. Just as was the case for p70, active variants of P7C3 competed for p55 binding, whereas inactive variants did not. Moreover, both proteins co-purified with a cellular fraction following centrifugation at 12,000G.
The p70 and p55 targets of P7C3 correspond to NAMPT
Partially purified protein preparations were resolved by 2D gel electrophoresis. In order to discriminate between protein spots competed by an excess of the active, A20 variant of P7C3, and those that were not, separate lysates were prepared from cells that had been co-exposed to the P7C3-A20 competitor or not. Click chemistry was used to append a red fluorescent dye onto P7C3-S326-modified proteins of the former lysates (Kolb et al., 2001). The latter lysates were modified by a green dye, such that in imaging 2D gels, we could focus on green protein spots and avoid spots labeled by both green and red dyes. Such efforts led to the identification of green-only spots migrating at either 70kD or 55kD (Figure 3A–C). Proteins of both molecular weights migrated in the isoelectric focusing dimension at four closely spaced pH positions. This electrophoretic behavior might be consistent with post-translational modifications of the same protein leading to different isoforms bearing slight differences in ionic charge. Perplexingly, the pattern of charge distribution was identical for the 70kD and 55kD polypeptides.
Figure 3. Identification of p70 and p55 targets of P7C3 by two-dimensional gel electrophoresis and mass spectrometry.
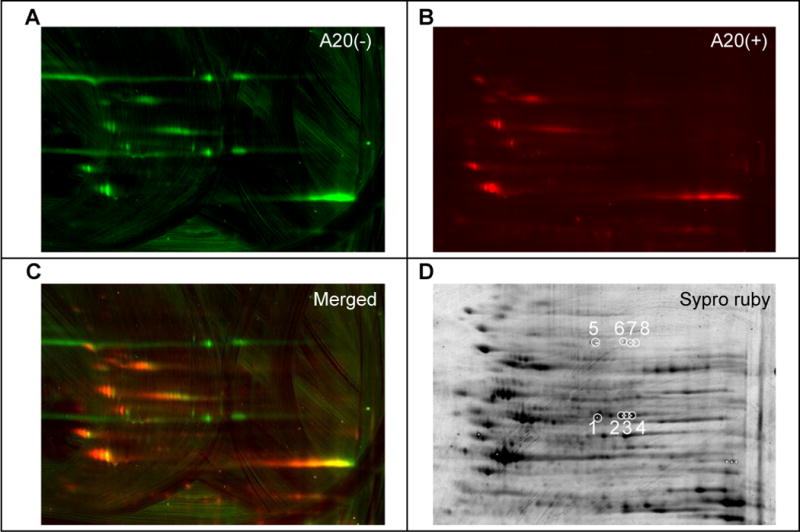
Lysates from cells exposed to 0.3 μM P7C3-S326 were CLICKed with a green dye, Alexa 532, and those from cells co-exposed to 0.3 μM P7C3-S326 and 5 μM of the active competitor P7C3-A20 were CLICKed with a red dye Cy5. CLICK reacted lysates were combined, subjected to two-dimensional gel electrophoresis, and scanned in the green channel (A) and the red channel (B). Images from both channels were merged to reveal green-only spots (C). After spot picking, total proteins were visualized by Sypro Ruby staining (D). White circles in (D) indicate spots excised for shotgun mass spectrometry analysis. See also Table S3.
The eight green-only spots, four from p70 and four from p55, were then picked and subjected to shotgun mass spectrometry for protein identification. All four of the p55 spots yielded by far the highest number of spectral counts for nicotinamide phosphoribosyltransferase (NAMPT) (Table S3). Both the predicted size (55kD) and isoelectric point (pH 6.7) of NAMPT match spots 1–4 picked from the 2D gel. Likewise, mass spectrometry based analysis gave substantive percentage coverage levels of 12%, 82%, 38% and 62% for the NAMPT enzyme in spots 1–4 (Table S3). By contrast, no proteins could be identified above the false discovery rate for the four p70 spots. Post-staining of the 2D gel with sypro ruby dye showed prominent protein staining for all four of the p55 spots, and no staining for any of the four p70 spots (Figure 3D). Apparently, despite equivalent photo-crosslinking by P7C3-S326, the molar ratio of drug modification of p70 must be at least three orders of magnitude greater than p55.
The perplexing relationship between p70 and p55 was clarified upon adding 7M urea to the SDS PAGE gels used to resolve the two bands. In the absence of urea, almost all of the S326-modified material migrated as the p70 band. Addition of urea to the gel converted all of the labeled material to the p55 band (Figure 4A). By running a gel crafted to contain a horizontal gradient of urea, 7M on the right and no urea on the left, it was possible to observe graded conversion of p70 into p55 (Figure 4B). These observations give evidence that when NAMPT is covalently bound by P7C3-S326, it can adopt an anomalous pattern of electrophoretic migration under gel conditions that are not fully denaturing.
Figure 4. Conversion of p70 into p55 in the presence of urea.
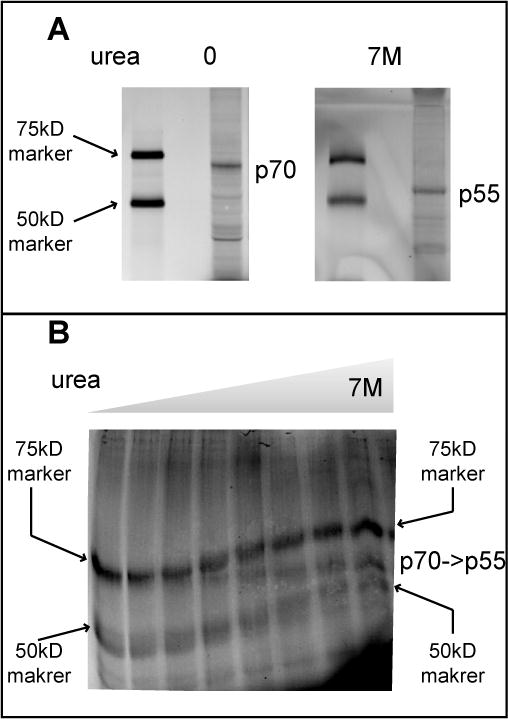
(A) CLICK reacted lysates from H2122 cells treated with P7C3-S326 were separated on a standard 8% SDS-PAGE gel (left panel) or one supplemented with 7M urea (right panel), and visualized on a Typhoon scanner. Asterisk indicates the crosslinked protein that migrates as 70KDa in a standard SDS-PAGE gel and 55KDa in the SDS-PAGE gel supplemented with 7M urea. (B) The same lysates were resolved on a horizontal urea gradient gel co-loaded with crosslinked p70 and the same 75kD and 50kD size standard proteins displayed in panel A. The horizontal urea gradient proceeds from zero denaturant on the left to 7M denaturant on the right.
Active derivatives of P7C3 restore NAD levels in doxorubicin-treated cells
Having observed P7C3-S326 crosslinking to both the p70 and p55 forms of NAMPT, we wondered whether active variants of P7C3 might modify the activity of the enzyme. The NAMPT enzyme controls the rate limiting step in the NAD salvage pathway (Preiss and Handler, 1958). NAMPT is an obligate dimer whose molecular structure has been resolved by X-ray crystallography (Khan et al., 2006) (Wang et al., 2006). On the basis of molar ratio of crosslinking, P7C3-S326 strongly prefers the p70 isoform of NAMPT relative to the p55 isoform. Hypothesizing that p70 represents a drug-altered form of NAMPT, we tested whether the highly active P7C3-A20 variant might either enhance or reduce NAD levels in living cells.
Doxorubicin is a DNA-damaging toxin known to activate the poly ADP-ribose polymerase (PARP) enzyme as a central feature of DNA repair (Munoz-Gamez et al., 2009; Pacher et al., 2002). PARP uses NAD as the donor for poly ADP-ribose synthesis and hence lowers cellular levels of NAD. U2OS cells exposed to sub-lethal levels of doxorubicin were subjected to metabolite analysis by mass spectrometry, allowing simultaneous, quantitative analysis of hundreds of intracellular metabolites (Tu et al., 2007) (Wang et al., 2009). As expected, doxorubicin treatment led to a significant decline in NAD levels relative to all other metabolites assayed. Co-administration of the A20 variant of P7C3 led to a dose-dependent replenishment of NAD (Fig 5A). The mass spectrometry methods used to quantitate metabolite levels measured NAD relative to hundreds of other intracellular metabolites. As such, the ability of the A20 variant of P7C3 to restore intracellular levels of NAD does not simply reflect the generalized health of cells, but instead reflects the relative increase in NAD levels compared with hundreds of other metabolites.
Figure 5. Active variants of P7C3 compensate NAD exhaustion induced by doxorubicin.
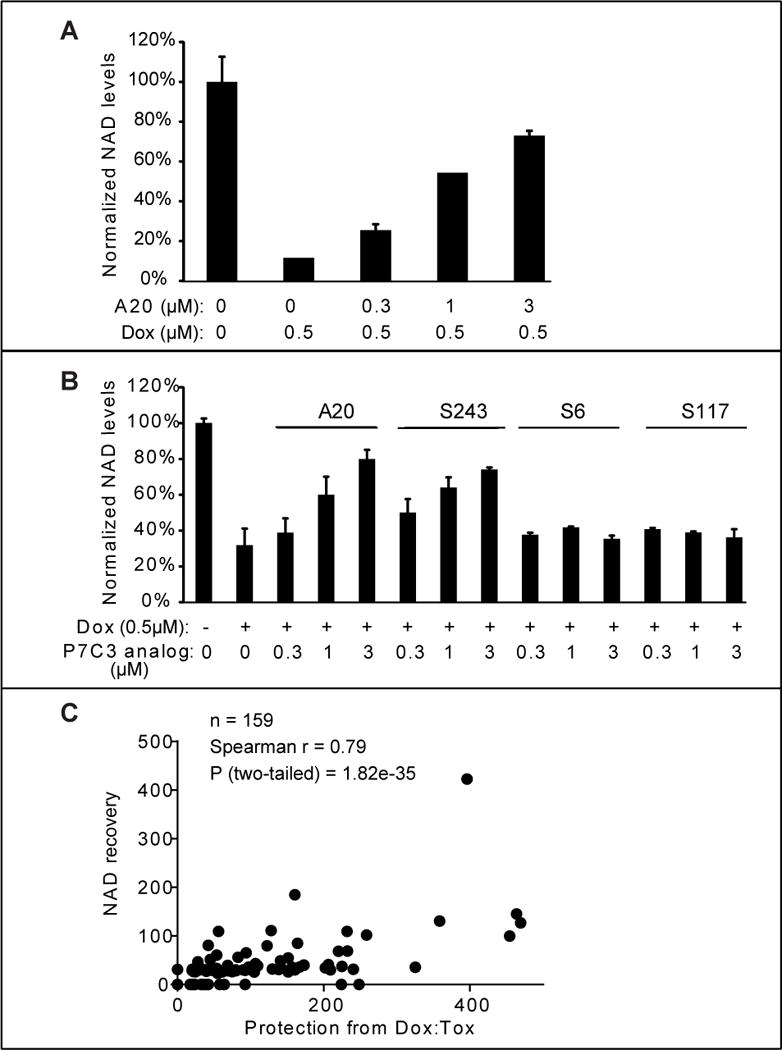
(A) U2OS cells were treated with the indicated concentration of P7C3-A20. Cells were harvested and NAD metabolites were measured by LC-MS/MS. Abundance of NAD was normalized relative to total metabolites (Experimental Procedures). The data are represented as the mean ± SD of experimental duplicates. (B) Active derivatives P7C3-A20 and P7C3-S243, but not inactive derivatives P7C3-S6, or P7C3-S117, facilitate replenishment of NAD levels in doxorubicin-treated cells. Cells were grown in 96-well plates and treated with the indicated concentrations of P7C3-A20, P7C3-S243, P7C3-S6 or P7C3-S117 together with 0.5μM doxorubicin for 45h. Cellular NAD abundance was determined by NAD/NADH Glo assay kit. See also Figure S5 and Table S2. (C) Scatter plots revealed a strong correlation between dox:tox protective activities of 159 compounds and their relative abilities to replenish NAD levels. The activities in both assays are represented by (Sinf-S0)/AC50 in dose response curves of test compounds. See also Table S1.
A more facile method of quantifying the NAD analyte, employing the commercially available “NAD/NADH-Glo” kit, confirmed P7C3-A20-mediated rebound of NAD levels in doxorubicin-treated U2OS cells. We also observed NAD rebound for the racemic mix of the active P7C3-S243 compound, but no rebound at all for the inactive P7C3-S6 and P7C3-S117 compound variants (Fig 5B). This method of assay was used to test 159 variants of P7C3 that had been used to query the correlative relationship between dox:tox protection and competition in the P7C3-S326 crosslinking assay (Table S1 and Figure 2D). Each chemical variant of P7C3 was co-incubated with doxorubicin on cultured U2OS cells at five doses, and cells were assayed for intracellular levels of NAD (Figure S5 and Table S2). The scatter plot shown in Figure 5C reveals exceptional concordance between the dox:tox protective activities of the 159 compounds and their relative abilities to enhance NAD levels (correlative P value of 1.8 × 10−35).
NAMPT is the rate limiting enzyme in the salvage pathway whereby cells sequentially convert nicotinamide into nicotinamide adenine mononucleotide (NMN) and NAD (Preiss and Handler, 1958). If active variants of P7C3 indeed bind to the NAMPT dimer in a manner fostering its activity, then these compounds should enhance the flux of nicotinamide through the salvage pathway. To test this hypothesis, 14C-labeled nicotinamide was administered to cultured U2OS cells that were pre-treated with doxorubicin, and assayed by thin layer chromatography (TLC) followed by autoradiography. As shown in Figure 6, enhanced conversion of radiolabeled nicotinamide into NMN and NAD was observed in a dose-responsive manner upon exposure of cultured U2OS cells to the active, A20 variant of P7C3. Upon examining if P7C3 derivatives might elevate NAD levels in naïve cells (without doxorubicin pretreatment), we failed to observe any compound-mediated effect on NAD levels.
Figure 6. P7C3-A20 enhances the flux of nicotinamide through the salvage pathway.
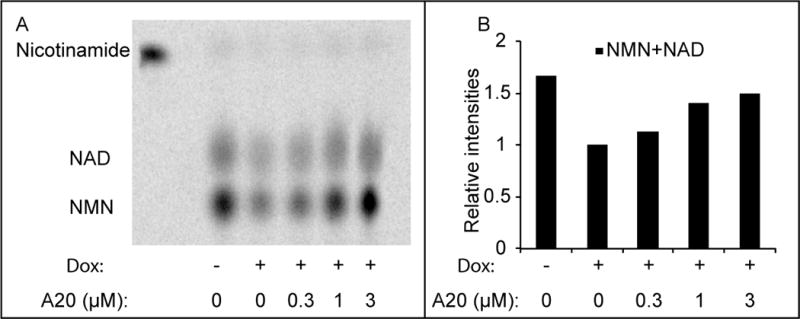
(A) Cells were pre-treated with 0.5μM doxorubicin for 48 hours, followed by 6 hours treatment with 14C-nicotinamide in the presence of the indicated amount of P7C3-A20. Metabolites were extracted and analyzed by thin layer chromatography (a representative result from three experiments is shown here). Free 14C-nicotinamide was loaded (the first lane) as a standard. (B) Quantification of the relative intensities of NAD and NMN from the thin layer chromatogram.
Active variants of P7C3 enhance the activity of the purified NAMPT enzyme
A recombinant form of the human NAMPT enzyme was cloned, expressed and purified according to published conditions (Khan et al., 2006) (Wang et al., 2006). The enzymatic activity of recombinant NAMPT was monitored in a coupled assay wherein nicotinamide conversion to NMN was followed by sequential production of NAD (via the NMNAT enzyme) and NADH (via alcohol dehydrogenase). As shown in Figure 7A, the A20 variant of P7C3 activated the enzyme reaction in a dose dependent manner. No activation of NADH production was observed when NAMPT was eliminated from the reaction, giving evidence that enhancement did not result from compound effects on either NMNAT or alcohol dehydrogenase (Figure S6).
Figure 7. Active variants of P7C3 enhance the activity of purified NAMPT enzyme.
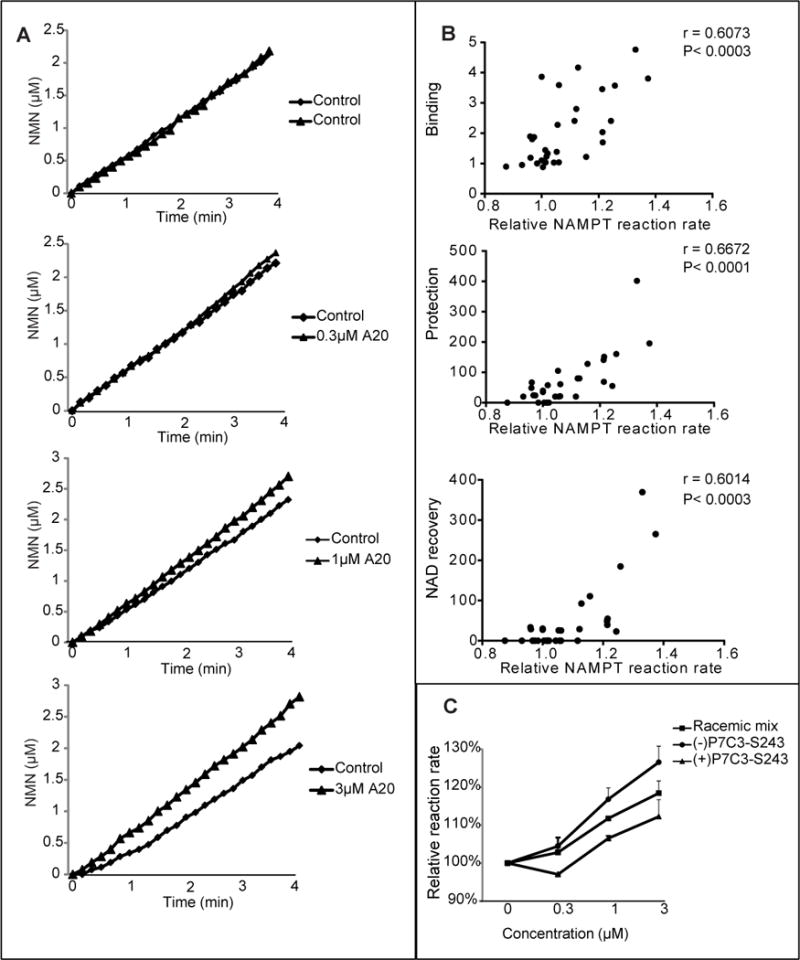
(A) P7C3-A20 was incubated at indicated concentrations in a reaction coupled with three enzymes; NAMPT, NMNAT, and ADH (Experimental Procedures). NAMPT activity was recorded for the indicated period of time at OD340nm by measuring NADH appearance, leading to the indicated concentration-time plot. Each assay was repeated three independent times with similar results. See also Figure S6. (B) Analysis of the activities of thirty P7C3 analogs was performed to assess their effects on NAMPT enzymatic activity. The reaction rate was calculated as the slope of the concentration-time curve. Relative reaction rate was normalized by the control reaction run prior to compound addition. Data represents the mean of experimental duplicates. Scatter graphs were plotted to compare the ability of compounds to activate NAMPT relative to their ability to compete away P7C3-S326 crosslinking (top scatter plot), ability to protect cells from doxorubicin-mediated toxicity (middle scatter plot), or ability to facilitate NAD restoration in doxorubicin-treated cells (bottom scatter plot). Significant correlations were observed from all three sets of data. See also Table S1. (C) Comparison of P7C3-S243 enantiomers showed that (−)-P7C3-S243 was superior to (+)-P7C3-S243 in activating the purified NAMPT enzyme. Data are expressed as mean ± SD of duplicate independent assays. See also Figure S7.
Two additional experiments were conducted to investigate activation of the recombinant NAMPT enzyme. First, thirty derivatives of P7C3 having varied activity were tested for enzyme activation. The ability of the thirty variants to activate NAMPT in the coupled enzyme assay was then correlated on scatter plots to three other measures of compound activity: (i) ability to protect cultured cells from doxorubicin toxicity; (ii) ability to compete with photo cross-linking by P7C3-S326 in living cells; and (iii) ability to restore NAD levels in doxorubicin treated cells (Figure 7B). In all cases statistically significant evidence of correlation was observed. Second, we compared the activities of the racemic mix of P7C3-S243, the (−)P7C3-S243 enantiomer and the (+)P7C3-S243 enantiomer in NAMPT enzyme assay. As shown in Figure 7C, the (−)P7C3-S243 enantiomer was more active in stimulating NAMPT than the racemix mix, which – in turn – was more active than the (+)P7C3-S243 enantiomer. This same hierarchy of activity has been observed in all in vivo assays tested to date, including adult neurogenesis, and the MPTP model of Parkinson’s disease (Naidoo et al., 2014), concussive blast injury (Yin et al., Cell Reports, in press), protection of cultured cells from doxorubicin-mediated activity (Figure 1) and rebound of NAD levels in doxorubicin-treated cells (Table S1).
Finally, we tested whether the recombinant NAMPT enzyme could be photo-cross linked to P7C3-S326. As shown in Figure S7, P7C3-S326 binds directly to NAMPT in a manner competed by active variants of P7C3 (P7C3-A20 and P7C3-S243), but not by the inactive P7C3-S87 variant.
DISCUSSION
Why is it that active variants of P7C3 are able to protect cultured cells from doxorubicin-mediated toxicity, in the face of no protective activity against seven other toxins? Three observations now help understand this selectivity. First, doxorubicin reduces intracellular levels of NAD, likely via activation of poly-ADP ribose polymerase (PARP). Second, active variants of P7C3 protect cultured cells from dox:tox, likely by stimulating a rebound in the intracellular levels of NAD. Third, active variants of P7C3 enhance the activity of the purified, recombinant NAMPT enzyme. We conclude that the dox:tox protective activity of our aminopropyl carbazole chemicals is directly reflective of their ability to enhance the salvage pathway whereby nicotinamide is converted to NAD. Our findings are consistent with earlier reports that programmed enhancement in the expression of the NAMPT enzyme protects mammalian cells against genotoxic stress (Yang et al., 2007) and extends the life span of budding yeast (Anderson et al., 2002). The target-agnostic in vivo screen performed years ago led, in an unbiased manner, to the P7C3 class of neuroprotective chemicals (Pieper et al., 2010). We now hypothesize that the neuroprotective efficacy of our aminopropyl carbazole chemicals can be attributed to their ability to foster salvage of NAD from nicotinamide. This interpretation is concordant with an equally unbiased series of forward genetic studies performed over the past three decades. In the 1980’s scientists working in Oxford, England described a variant strain of mice that impedes Wallerian degeneration, designated the Wallerian degeneration slow (wlds) strain (Lunn et al., 1989). In simple terms, Wallerian degeneration can be described as the demise of axonal processes distal from the cell body to a site of nerve crush. Axons of wlds mice survive a nerve crush far longer than those of wild type mice. The wlds gene was positionally cloned in the 1990’s and found to exist as a triplicated fusion gene encoding the first 70 amino acids of Ufd2a, a ubiquitin-chain assembly factor, linked directly to the complete coding sequence of nicotinamide mononucleotide adenylyl-transferase 1 (NMNAT1) (Coleman et al., 1998) (Conforti et al., 2000) (Mack et al., 2001). Whereas NAMPT initiates salvage from nicotinamide, NMNAT1 is one of three enzymes that convert the NMN intermediate into NAD.
In a seminal paper published in 2004 in Science, Dr. Jeff Milbrandt and colleagues provided evidence that the wlds phenotype can be attributed primarily to the overexpression of NMNAT1 (Araki et al., 2004). The closing sentence of their manuscript is unusually prescient, reading – “It is possible that the alteration of NAD levels by manipulation of the NAD biosynthetic pathway, Sir2 protein activity, or other downstream effectors will provide new therapeutic opportunities for the treatment of diseases involving axonopathy and neurodegeneration.” We offer the conclusion that this is exactly what active variants of the P7C3 category of neuroprotective drugs do – they enhance the intracellular production of NAD by agonizing the activity of the rate limiting enzyme in the NAD salvage pathway.
It has recently been reported that NAD production attenuates as a function of aging in the hippocampus of rodents (Stein and Imai, 2014). The latter study further reported age-related decline in the expression of NAMPT in the hippocampus, along with evidence that selective elimination of the NAMPT gene in neuronal stem cells significantly impairs hippocampal neurogenesis. Now that we understand that active members of the P7C3 class of compounds function by enhancing the activity of the NAMPT enzyme, the observations of Stein and Imai lend credibility to our original discovery of P7C3 as a chemical stimulant of hippocampal neurogenesis.
In addition to offering progress towards an understanding of the mode of action of the P7C3 class of neuroprotective chemicals, this work also points to a number of important, unanswered questions. First and foremost, where on the NAMPT enzyme do our chemicals bind, and how do they facilitate agonistic activation? Given that there are only about a dozen known examples of allosteric chemical activators of enzymes (Zorn and Wells, 2010), understanding the mechanism of NAMPT activation by P7C3 represents an important challenge for future studies. Second, why is it that covalent binding of P7C3-S326 causes NAMPT to migrate in an anomalous manner in SDS PAGE gels? Third, is it indeed the case that NAD levels may be compromised as a function of nerve cell injury or neuronal dysfunction as a consequence of neurodegenerative disease? Fourth, might NAMPT be regulated in vivo by an endogenous metabolite that is either mimicked or counteracted by active variants of the P7C3 class of neuroprotective chemicals? Fifth, is it possible that the modest levels of compound-elevated activation of the purified, recombinant NAMPT enzyme can account for the full in vivo efficacy of active variant of the P7C3 class of neuroprotective chemicals? Several arguments may be invoked to speculate on this question. For example, it is possible that P7C3 displaces the function of an inhibitory metabolite within the cell, or that the native NAMPT enzyme is regulated by post-translational modification, intracellular localization, or any of a wide spectrum of variables not reconstituted in the context of the purified, recombinant enzyme.
We close with two observations that may or may not be related. Studies of aged mice have led to the reported decline in tissue NAD levels, and that certain aspects of age-related decline in overall health can be reversed by nutritional supplementation of NMN (Gomes et al., 2013). In our initial studies of P7C3 we reported compound-mediated abrogation in cognitive decline in aged Fisher rats (Pieper et al., 2010). Included in such studies was the unanticipated observation that compound treated rats maintained body weight relative to the frailty of vehicle-treated controls. These distinct studies offer the possibility of considering the P7C3 class of neuroprotective chemicals as potential treatments for generalized frailty typical of aged animals and humans.
Experimental procedures
Cell culture
Osteosarcoma cell line U2OS cells were grown in DMEM medium (Sigma) supplemented with 10% FBS. Lung cancer cell line H2122 cells were cultured in RPMI-1640 medium (HyClone) supplemented with 5% FBS. These adherent cell lines were cultured employing standard procedures.
Cell survival assay
Cell survival assay was performed in 96-well plates using CellTiter-Glo Luminescent Cell Viability Assay kit (Promega) that measures cellular ATP content. The CellTiter-Glo reagent was diluted by adding 2 volumes of PBS containing 1% Triton X-100. 50 μl of diluted reagent was directly added to 100 μl of cell culture medium per well. The plates were incubated at room temperature for 10 min. Luminescence was recorded by Tecan SPECTRAFluor Plus reader (Tecan). Cell survival was presented as the percentage of viable cells compared with untreated control, and mean ± SD was calculated from duplicates. The dox-tox assay of P7C3 analogs was carried out in the HTS Core (University of Texas Southwestern Medical Center) using EnVison multimode plate reader (Perkin Elmer).
Chemical synthesis of photo-crosslinking probe P7C3-S326
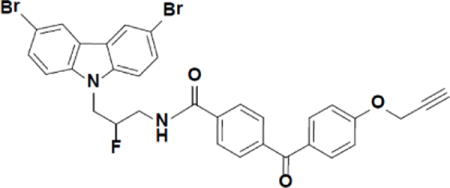
N-(3-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-4-(4-(prop-2-yn-1-yloxy)benzoyl)benzamide (P7C3-S326).
3-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropan-1-amine (Naidoo et al., 2014) (40.2 mg, 0.10 mmol) was added to a solution of 4-(4-(prop-2-yn-1-yloxy)benzoyl)benzoic acid (Bandyopadhyay, 2011) (28.5 mg, 0.10 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (21.2 mg, 0.11 mmol) and 1-hydroxybenzotriazole hydrate (15.0 mg, 0.11 mmol) in dimethylformamide (0.4 ml). The reaction went to completion within an hour. The mixture was diluted with EtOAc and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered and condensed to give a light brown solid in 90% yield. 1H NMR (Acetone-d6, 400 MHz) δ 8.41 (dd, J = 2.0, 0.6 Hz, 2H), 8.07 (d, J = 8.4 Hz, 2H), 7.82 (dd, J = 10.3, 8.6 Hz, 4H), 7.71 – 7.56 (m, 4H), 7.16 (d, J = 8.9 Hz, 2H), 5.39 – 5.10 (dm, 1H), 4.94 (d, J = 2.4 Hz, 2H), 4.91 – 4.85 (m, 1H), 4.82 (d, J = 5.4 Hz, 1H), 4.05 – 3.89 (m, 1H), 3.87 – 3.70 (m, 1H), 3.17 (t, J = 2.4 Hz, 1H). 13C NMR (CD2Cl2, 100 MHz): 194.6, 167.3, 161.7, 141.4, 140.1, 136.9, 132.8, 130.7, 130.2, 129.7, 127.3, 124.1, 123.6, 114.9, 113.0, 111.2 (JC–F = 1.9 Hz), 92.1 (JC–F = 176.2 Hz), 78.2, 76.3, 56.4, 45.8 (JC–F = 22.5 Hz), 42.3 (JC–F = 21.2 Hz). MS (ESI), m/z: calculated 660.01, found 660.7 (M+1).
Live-cell crosslinking with photo-crosslinking derivative S326
S326 were diluted to 0.3 μM in culture medium to treat H2122 cells. After incubation at 37°C for 2 hours, H2122 cells were irradiated for 15 minutes under 306 nm ultraviolet light in a Stratalinker 2400 (Stratagene). Cells were then harvested by dissolving in 1% SDS lysis buffer (50 mM HEPES, pH 8.0, 2 mM MgCl2, 10 mM KCl, 1% SDS, 50 units/ml of Benzonase (Sigma), supplemented with Sigmafast protease inhibitor (Sigma).
Attachment of fluorescent tags via Copper-Catalyzed Azide-Alkyne Cycloaddition (CUAAC) CLICK reaction
For Copper-Catalyzed Azide-Alkyne Cycloaddition CLICK reactions, 43μl of SDS lysate were mixed with 3 μl of 1.7 mM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA), 2 μl of 50 mM CuSO4, 1 μl of 50 mM Tris(2-carboxyethyl)phosphine (TCEP), and 1 μl of 1.25mM Alexa 532-azide. The reaction was incubated at room temperature with constant agitation at 1000rpm on a thermomixer (Eppendorf) for 1 hour. Afterwards, samples were mixed with SDS sample loading buffer and loaded on an 8% SDS-PAGE gel, separated, and imaged using a Typhoon imager (filter settings: excitation 533 nm, emission 555 nm, and high sensitivity).
In vivo binding competition assay
H2122 cells were incubated with the S326 (0.3μM) in the absence or presence of 5μM P7C3 analogs for 2 hours following by UV crosslinking in vivo. Cells were lysed in 1% SDS lysis buffer and lysates were conjugated to the fluorescent Alexa 532 dye using CLICK reaction. The samples were run on SDS-PAGE, and the gel was scanned using a Typhoon Imager. The p70 band intensity was analyzed and quantified by Image J, and the relative intensity in each assay was normalized to that of the DMSO control.
Two-dimensional gel electrophoresis
H2122 cells were incubated with S326 (0.3 μM) in the absence or presence of 5 μM of the active analog A20 for 2 hours followed by UV crosslinking in vivo. Cells were dissolved in 1% SDS lysis buffer and protein concentration was quantified by the BCA assay. For CLICK reactions, 1 mg of lysates prepared from S326 treated cells were reacted with Alexa 532-azide (Red), and 1 mg of lysates from S326 and A20 treated cells were reacted with Cy5-azide (Green). After the CLICK reaction, the two lysates were combined together, mixed with 4 volumes of acetone, incubated at −20°C overnight. Precipitated proteins were collected by centrifugation at 8,000 g, 15 min, 4°C. After two washes with acetone, the pellet was air-dried and resuspended in 500 μl of 2D rehydration buffer (8M urea, 2% CHAPS (wt/vol), 50 mM dithiothreitol (DTT), 2% of IPG buffer). For two-dimensional gel electrophoresis, 500 μg of proteins were loaded onto immobilized pH gradient (IPG) strip of 24 cm pH 3–10 linear (GE Healthcare) by passive rehydration using rehydration chamber for overnight at room temperature. Isoelectric focusing was conducted on an IPG Phor3 manifold (GE healthcare) with the following protocol: 100 V for 3 hours, 300 V for 2 hours, 500 V for 3 hours (Step-and-hold); 1000V for 1 hour (Gradient); 8000V for 3 hours (Gradient); 8000V for 4 hours (Step-and-hold) with total 42,000 V/hour. After isoelectric focusing, the strip was equilibrated for 15 minutes in equilibration buffer I (6 M urea, 2% SDS (wt/vol), 0.375 M Tris; pH 8.8, 20% glycerol (v/v), 100 mM DTT) for 15 min followed by equilibration buffer II (6 M urea, 2% SDS (wt/vol), 0.375 M Tris; pH 8.8, 20% glycerol (v/v), 250 mM iodoacetamide) for 15 minutes. Proteins were separated in the second dimension on 7.5% SDS-PAGE using Ettan Daltsix unit (GE Healthcare) at a constant current of 40 mA/gel for 5–6 hours at 100 W. Gel images were scanned at a resolution of 100 μm with on Typhoon FLA9500 scanner using default settings for CyDyes (GE Healthcare) and preprocessed using Image Quant TL software v8.1 (GE Healthcare). Cropped gel images were analyzed using DeCyder2D Differential Analysis Software v7.1 (GE Healthcare) to detect, normalize and quantify the protein features in the images. The P7C3-A20-competed green spots and several background spots were identified and robotically excised by an Ettan Spot Picker (GE Healthcare).
Mass spectrometry for protein identification
Gel pieces were washed using 50mM TEAB (triethylammonium bicarbonate, pH 8.0) in 50% (v/v) acetonitrile followed by reduction with 20 mM DTT and alkylation with 55 mM iodoacetamide. Gel pieces were dehydrated using 100% (v/v) acetonitrile, air-dried, and then rehydrated in the presence of 250 ng of sequencing grade trypsin (Sigma) in 50mM TEAB. Digests were allowed to proceed overnight at 37°C. Mass spectrometry was performed using Q Exactive mass-spectrometers (Thermo Fischer, Bremen), coupled to identical Ultimate 3000 RSLCnano HPLC systems (Dionex, Sunnyvale CA). Peptides were loaded onto a 75 μm i.d. × 50 cm Thermo Scientific Easy-Spray column packed with 2 μm resin. A 40 min linear gradient of 1%–28% acetonitrile in 0.1% formic acid followed by a 10 min ramp to 98% ACN, using a flow rate of at 350 nl/min, was employed to elute peptides from the column. The Easy-Spray column was heated to 55°C using the integrated heater. HCD analyses were performed on the Q Exactive instrument using a data-dependent top 20 method, with the full-MS scans acquired at 70K resolution (at m/z 200) and MS/MS scans acquired at 17.5K resolution (at m/z 200). The under-fill ratio was set at 0.1%, with a 3 m/z isolation window and fixed first mass of 100 m/z for the MS/MS acquisitions. Charge exclusion was applied to exclude unassigned and charge 1 species, and dynamic exclusion was used with duration of 15 seconds.
Measurement of intracellular NAD abundance
By LC-MS
Cellular metabolites were extracted using 50% methanol by multiple freeze-thaw cycles. The cleared metabolites were dried in a Speed-Vac. The dried materials were dissolved in 0.1% formic acid and subjected to mass spectrometric analysis using a specific method developed for NAD. Relative intracellular NAD levels (NAD ion counts divided by total ion counts) were used for inter-sample comparisons.
By NAD/NADH-Glo assay kit
The assay was performed following the manufacturer’s protocol (Promega). NAD/NADH-Glo reagent contains the NAD Cycling Enzyme that converts NAD+ to NADH. In the presence of NADH, the enzyme Reductase reduces a proluciferin reductase substrate to form luciferin. Cells were grown in 96- or 384-well plates. 50μl or 15 μl of NAD/NADH-Glo™ Detection Reagent were added to each well. The resulting luminescence signals were measured by Tecan plate reader or EnVison multimode plate reader (Perkin Elmer).
Nicotinamide flux analysis with 14C-labeled nicotinamide
Logarithmically growing cells cultured in 6-well plates were treated with doxorubicin (0.5 µM) for 48 hours. Afterwards, 1 µl of 0.5 μCi/ml 14C nicotinamide (specific activity: 50 mCi/mmol; American Radiolabeled Chemicals, Inc.) were added per well. The cells were then washed once with PBS and collected by trypsin digestion. Metabolites were extracted from cells with 50 μl of perchloric acid (0.5 M) by incubating on ice for 20 min. The extracts were then neutralized with 13.75 μl of KCl/KOH (0.5 M/2.0 M) and centrifuged at 2500 × g for 10 min at 4°C. 10 μl of extracted metabolites were separated and identified using silicon thin-layer chromatography plates with 1 M ammonium acetate:ethanol (3:7) as solvent. For autoradiography, the chromatograms were exposed to storage Phospho screen for 3 days. Afterwards, the screen was scanned using the Typhoon scanner, and the 14C-labeled compounds were quantified using Image J.
In vitro NAMPT assay
The NAMPT activity was determined by a coupled-enzyme spectrometric assay as described by Khan et al. (Khan et al., 2006) with minor modifications. Briefly, NAMPT first converted nicotinamide to NMN, which was followed by sequential production of NAD via the nicotinamide mononucleotide adenylyl-transferase (NMNAT). NAD was then reduced to NADH by alcohol dehydrogenase (Sigma) using alcohol as the substrate. The resulting NADH was monitored by OD340 nm. Human NAMPT and NMNAT were overexpressed in E. coli and purified as described by Wang et al. (Wang et al., 2006) and Zhou et al. (Zhou et al., 2002), respectively. The reaction mixture contained 50mM Tris (pH8.0), 0.4 mM phosphoribosylpyrophosphate (PRPP, Sigma) 2.5mM ATP, 12mM MgCl2, 1.5% (v/v) ethanol, 10mM semicarbazide (Sigma), 0.02%(w/v) BSA, 2.4 μg/ml NMNAT, 0.4 unit alcohol dehydrogenase, 1μM NAMPT, and 150 μM nicotinamide. 10μM P7C3 analogs in DMSO were further diluted in ethanol prior to applying to the reaction. To minimize variation, a control reaction was run in each assay prior to compound addition.
Statistics
The correlation data analysis was performed in GraphPad Prism 6. All p values were obtained using Spearman Rank Correlation (v1.0.1) in Free Statistics Software (v1.1.23-r7), Office for Research Development and Education, URL http://www.wessa.net/rwasp_spearman.wasp/
Supplementary Material
Highlights.
The P7C3 class of chemicals protects cells from doxorubicin-mediated toxicity
NAMPT is identified as the intracellular target of P7C3
P7C3 chemicals restore NAD levels in doxorubicin-treated cells
P7C3 chemicals enhance the enzymatic activity of NAMPT in vitro
Acknowledgments
We thank Leeju Wu and Masato Kato for help in the expression and purification of NAMPT; Paula Huntington, Ruth Starwalt, Sandi Terpack, Stephanie Tran and Jeremiah Britt for help with neurogenesis assays on hundreds of chemical derivatives of P7C3; Mike Brown for advice and encouragement; Bruce Posner in the UTSWMC high throughput screening core for assistance in numerous screening assays; and Noelle Williams for help with pharmacological studies. This work was funded by a grant awarded to AP and SLM from the NIMH (5-RO1-MH087986); grants awards from the Welch Foundation (I-1612) and the Edward N. and Della C. Thome Memorial Foundation to JMM; and unrestricted funds provided to SLM by an anonymous donor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.L.M. and J.M.R. designed and supervised the study. G.W. and T.H. designed and performed most of the experiments, interpreted the experimental results, and prepared the figures. D.N., P.T. assisted in the application of photocrosslinking probe for target identification. S.Y., and H.M. performed 2-D electrophoresis and mass spec experiments. J.N. and J.M.R. synthesized all derivatives of P7C3. A.A.P. measured the in vivo neurogenesis activities of P7C3 derivatives. S.L.M. wrote the paper with input from G.W., T.H., and J.M.R.
References
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, Lin SS, Manchester JK, Gordon JI, Sinclair DA. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. The Journal of biological chemistry. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Bong D. Synthesis of trifunctional phosphatidylserine probes for identification of lipid-binding proteins. Eur J Org Chem. 2011;2011:751–758. [Google Scholar]
- Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH. An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9985–9990. doi: 10.1073/pnas.95.17.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesus-Cortes H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17010–17015. doi: 10.1073/pnas.1213956109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386:493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Khan JA, Tao X, Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nature structural & molecular biology. 2006;13:582–588. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- Kim WR, Park OH, Choi S, Choi SY, Park SK, Lee KJ, Rhyu IJ, Kim H, Lee YK, Kim HT, et al. The maintenance of specific aspects of neuronal function and behavior is dependent on programmed cell death of adult-generated neurons in the dentate gyrus. Eur J Neurosci. 2009;29:1408–1421. doi: 10.1111/j.1460-9568.2009.06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SE, Ko IG, Kim BK, Shin MS, Cho S, Kim CJ, Kim SH, Baek SS, Lee EK, Jee YS. Treadmill exercise prevents aging induced failure of memory through an increase in neurogenesis and suppression of apoptosis in rat hippocampus. Exp Gerontol. 2010;45:357–365. doi: 10.1016/j.exger.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angewandte Chemie (International ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Biebl M, Wilhelm D, Li M, Friedlander RM, Winkler J. Increased generation of granule cells in adult Bcl-2-overexpressing mice: a role for cell death during continued hippocampal neurogenesis. Eur J Neurosci. 2005;22:1907–1915. doi: 10.1111/j.1460-9568.2005.04377.x. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. The European journal of neuroscience. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature neuroscience. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, Huntington PJ, Estill SJ, Longgood J, Becker GL, et al. Development of proneurogenic, neuroprotective small molecules. Journal of the American Chemical Society. 2011;133:1428–1437. doi: 10.1021/ja108211m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, Menissier de Murcia J, Almendros A, Ruiz de Almodovar M, Oliver FJ. PARP-1 is involved in autophagy induced by DNA damage. Autophagy. 2009;5:61–74. doi: 10.4161/auto.5.1.7272. [DOI] [PubMed] [Google Scholar]
- Naidoo J, De Jesus-Cortes H, Huntington P, Estill S, Morlock LK, Starwalt R, Mangano TJ, Williams NS, Pieper AA, Ready JM. Discovery of a Neuroprotective Chemical, (S)-N-(3-(3,6-Dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(−)-P7C3-S243], with Improved Druglike Properties. Journal of medicinal chemistry. 2014;57:3746–3754. doi: 10.1021/jm401919s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Bai P, Virag L, Mabley JG, Hasko G, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. The Journal of pharmacology and experimental therapeutics. 2002;300:862–867. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- Pieper AA, McKnight SL, Ready JM. P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chemical Society reviews. 2014 doi: 10.1039/c3cs60448a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen CH, et al. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010;142:39–51. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss J, Handler P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. The Journal of biological chemistry. 1958;233:488–492. [PubMed] [Google Scholar]
- Stein LR, Imai SI. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO journal. 2014;33:937–1085. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Winseck A, Vinsant S, Park OH, Kim H, Oppenheim RW. Programmed cell death of adult-generated hippocampal neurons is mediated by the proapoptotic gene Bax. J Neurosci. 2004;24:11205–11213. doi: 10.1523/JNEUROSCI.1436-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:17016–17021. doi: 10.1073/pnas.1213960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, McKnight SL. Cyclic changes in metabolic state during the life of a yeast cell. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16886–16891. doi: 10.1073/pnas.0708365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2:266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009;325:435–439. doi: 10.1126/science.1173288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zhang X, Bheda P, Revollo JR, Imai S, Wolberger C. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nature structural & molecular biology. 2006;13:661–662. doi: 10.1038/nsmb1114. [DOI] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Kurnasov O, Tomchick DR, Binns DD, Grishin NV, Marquez VE, Osterman AL, Zhang H. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. The Journal of biological chemistry. 2002;277:13148–13154. doi: 10.1074/jbc.M111469200. [DOI] [PubMed] [Google Scholar]
- Zorn JA, Wells JA. Turning enzymes ON with small molecules. Nature chemical biology. 2010;6:179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.