

Abstract
The Ebola virus (EBOV) genome only encodes a single viral polypeptide with enzymatic activity, the viral Large (L) RNA-dependent RNA polymerase protein. However, currently there is limited information about L protein, which has hampered development of antivirals. Therefore, antifiloviral therapeutic efforts must include additional targets such as protein-protein interfaces (PPIs). Viral protein 35 (VP35) is multifunctional and plays important roles in viral pathogenesis, including viral mRNA synthesis and replication of the negative-sense RNA viral genome. Previous studies revealed that mutation of key basic residues within the VP35 interferon inhibitory domain (IID) results in significant EBOV attenuation, both in vitro and in vivo. In the current study, we use an experimental pipeline that includes structure-based in silico screening, biochemical and structural characterization, along with medicinal chemistry to identify and characterize small molecules that target a binding pocket within VP35. NMR mapping experiments and high resolution x-ray crystal structures show that select small molecules bind to a region of VP35 IID that is important for replication complex formation through interactions with the viral nucleoprotein (NP). We also tested select compounds for their ability to inhibit VP35 IID-NP interactions in vitro as well as VP35 function in a minigenome assay and EBOV replication. These results confirm the ability of compounds identified in this study to inhibit VP35-NP interactions in vitro and to impair viral replication in cell-based assays. These studies provide an initial framework to guide development of antifiloviral compounds against filoviral VP35 proteins.
Keywords: in silico drug discovery, filoviral inhibitors, antivirals, VP35
Introduction
Ebolavirus (EBOV) and Marburgvirus (MARV) are members of the Filoviridae family of single stranded, non-segmented negative sense RNA viruses. EBOV and MARV infections are characterized by severe hemorrhagic fever, that have led to fatality rates nearing 90% in some outbreaks1. The virulence of filoviruses makes anti-filoviral therapeutics a priority. However, these viruses encode only one protein, large protein (L), which exhibits enzymatic activity. Therefore, efforts to target non-enzymatic viral proteins with small molecules are an important antiviral development strategy. However, targeting non-enzymatic proteins at protein-protein interfaces (PPIs) is challenging and often requires detailed structural and functional information about protein-protein complexes and interfaces. Unlike enzymatic active sites, PPIs are often shallow with limited molecular features that can facilitate ligand binding.
High fatality rates have been attributed, in part, to the ability of EBOV and MARV to efficiently subvert host innate immune responses as well as subsequent adaptive immune responses1; 2; 3. Among the strategies employed are suppression of interferon (IFN)-α/β production and inhibition of IFN-α/β induced antiviral signaling4; 5; 6; 7; 8; 9. Of particular note, the suppression of IFN-α/β responses is critical for EBOV virulence10; 11. Use of multiple strategies for immune suppression by filoviruses underscores the importance of targeting conserved viral factors.
The filoviral VP35 is an attractive potential therapeutic target because it carries out multiple functions critical for viral replication and its structure is known to high resolution. It is an important multifunctional protein that can antagonize host immune responses, including IFN production initiated by retinoic-acid inducible gene-I (RIG-I) like receptors (RLRs), and it functions as a co-factor in the viral polymerase complex (Fig 1a). VP35 consists of an N-terminal coiled-coil domain12 and a C-terminal domain termed the IFN inhibitory domain (IID)13; 14; 15; 16; 17. In the IID, there are two basic patches: the first basic patch (FBP) is important for interactions with EBOV nucleoprotein (NP) and VP35 polymerase co-factor function and the central basic patch (CBP) is important for VP35 dsRNA binding and IFN inhibition16; 17 (Supplementary Fig. 1). Recombinant viruses with VP35 mutations are greatly attenuated in guinea pigs and protect against subsequent EBOV infection 11. In addition, RNAi against VP35 also attenuated viral growth 18. Together, these observations support the therapeutic potential of VP3511. The availability of high resolution structures of the VP35 IID domains of EBOV Zaire (eIID)16, Reston (rIID)17; 19, and MARV (mIID)20 provides a new opportunity for structure based antiviral development 21.
Fig. 1. Filoviral VP35 proteins are multifunctional targets for therapeutic development.
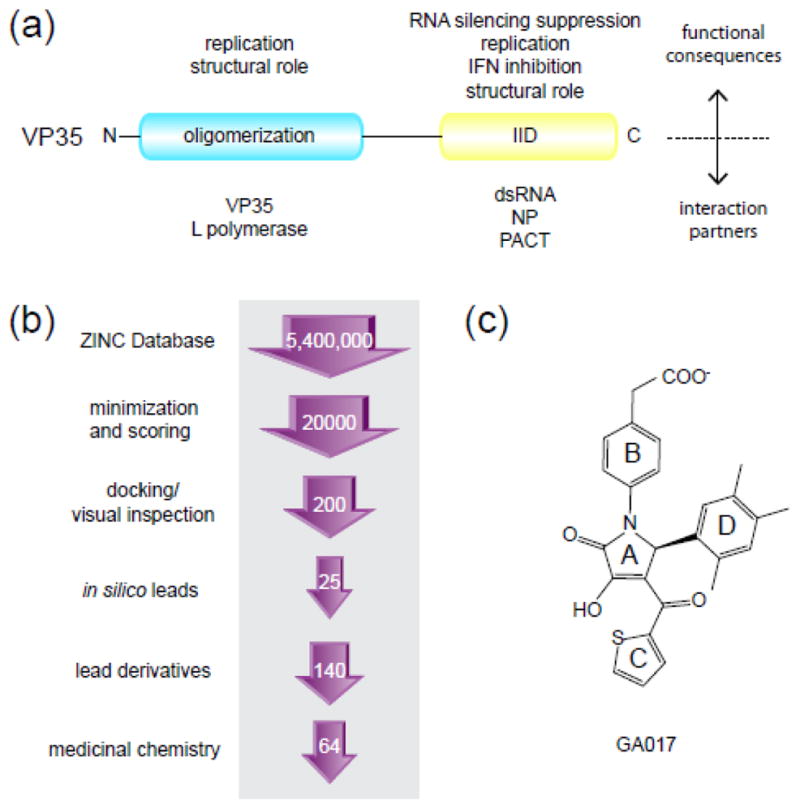
(a) Domain organization of filoviral VP35 based on previous biochemical and structural studies 42. Approximately 140 amino acids at the N-terminus form the oligomerization domain and the C-terminal 125 amino acids form the IID. The different interaction partners for both domains and the corresponding functional consequences are listed. (b) Outline of the structure-based in silico screening approach used in this study. The number of compounds screened at each stage is indicated within the arrows. Experimental screening included 25 initial compounds, followed by 140 GA-series analogs, and 64 optimized compounds (VPL) synthesized using binding and structural data obtained from the GA-series molecules. (c) The chemical structure of GA017 containing the pyrrolidinone scaffold.
Here we describe studies that identify, validate, and functionally characterize small molecule binders that target a key PPI between VP35 and NP. To this end, we employed an in silico screen using the structure of the Zaire EBOV VP35 IID (eIID) as a target for efficient and cost effective lead identification. In the current study, we use an experimental pipeline that includes structure-based in silico screening, biochemical and structural characterization along with medicinal chemistry to identify and characterize small molecules that target a key binding pocket within VP35. High resolution structural studies and NMR mapping experiments show that select small molecules bind within the region of VP35 that is important for interaction with the viral nucleoprotein (NP) and replication complex formation. We also tested select compounds for their ability to inhibit VP35-NP interactions in vitro, to inhibit VP35 function in a cell-based minigenome assay and to inhibit EBOV replication. Altogether, these studies show that small molecules identified through an in silico screen can bind filoviral VP35 proteins and inhibit its replication function. Our studies also provide a framework to guide development of antifiloviral compounds against VP35 proteins.
Results
In silico screening
A total of 5.4 million drug-like small molecules from the ZINC library 22 were docked to the eIID (PDB ID: 3FKE) and the top in silico hits were validated by an NMR-based assay (Fig. 1). Two hundred compounds were selected after the final screening and docked models were subjected to visual inspection and selection. Of these, 25 compounds were subsequently selected for NMR-based binding studies. Compounds used for in vitro validation were selected based on LigScore of 5.5–6.5 and LogP values <5. Of the 25 compounds tested, we identified 5 compounds with dissociation constant (KD) values of 550–1278 μM (Fig. 2a; Supplementary Table 1). All 5 compounds contained common structural elements and are represented by GA017 with a pyrrolidinone scaffold (Fig. 1c). In order to optimize binding, we identified additional compounds that contained the pyrrolidinone scaffold of GA017 through structural interaction fingerprint (SiFT) analysis23, and these were either purchased or synthesized (complete results of the chemical synthesis process, including NMR validation, are described in Supplementary Materials and Methods). In silico findings were validated by a series of biochemical, structural, and functional characterizations of VP35-compound interactions (Fig. 1b). Overall, these efforts resulted in an 18-fold enhancement in binding affinity.
Fig. 2. Chemical structures of representative compounds that bind VP35 IID.
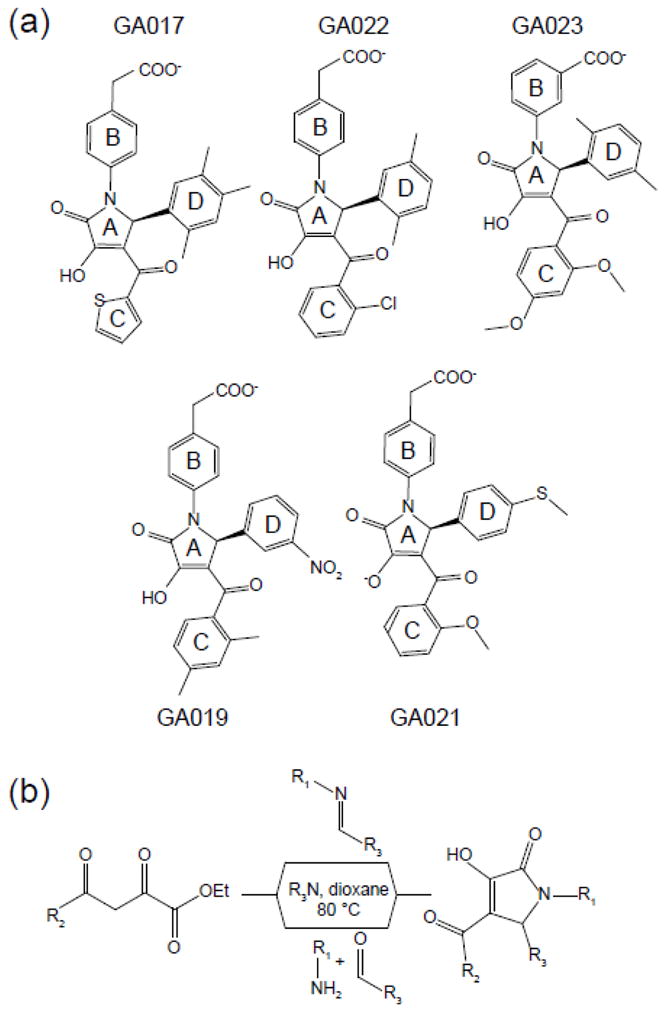
(a) Compounds with methyl substitutions in the phenyl D ring bind with higher affinity compared to nucleophilic substitutions. (b) Outline of the chemical synthesis scheme to generate pyrrole-based compounds.
Compounds containing pyrrolidinone scaffold bind eIID
NMR-based binding studies revealed that eIID-compound interactions occur in the fast exchange regime relative to the chemical shift time scale as judged by the full titration data for GA017 (Fig. 3, Supplementary Fig. 2). Of note, while most compound binding resulted in linear chemical shift changes upon titration, indicative of a single free and bound conformation, there were several peaks that deviated. While the exact cause of this deviation is unknown, we suspect that a combination of the influence of the small molecule chemistry and exchange regime of binding are likely contributors to this phenomenon. Therefore, 1H-15N HSQC titration data from only residues that displayed linear peak migration patterns between a single free and bound state were used to determine the binding affinity. We were able to extract dissociation constants using two methods by plotting the normalized change in chemical shift versus concentration. We carried out titrations between 0–1000 μM for select compounds and KD values were calculated using the chemical shifts of 2–3 residues (Fig. 3). Among these, Ile303 on VP35 IID was the best reporter of small molecule binding, due to the large chemical shift difference between free and bound form as well as proximity to the binding site. Therefore, it was selected to estimate binding affinities based on the magnitude of the chemical shift, proximity to the binding site. Therefore, the estimate of dissociation constants for each small molecule was determined using the chemical shift differences derived from the averaged titration curves using Ile303 (Supplementary Fig. 3). Following this method, we tested 146 compounds, termed the GA-series, obtained through a commercial vendor (Enamine Inc., Kiev, Ukraine), and an additional 64 compounds, termed the VPL-series, which were synthesized at UT Southwestern Medical Center at Dallas (Fig. 2b and Supplementary Materials and Methods). Structures and the estimated binding affinities defined through our NMR-based binding assay are reported in Supplementary Table 1 (arranged by potency) and in Supplementary Table 2 (arranged by functional groups). These results indicate that we have compounds with an estimated KD range from ~30 μM to ~10 mM and have at least one compound containing the pyrrolidinone scaffold with no observable binding (VPL-64). A summary of our structure activity relationships, shown in Fig. 4, reveal a number of important trends that provide insight into eIID-compound interactions. For example, binding requires a meta-CH2CO2H or a para-CH2CO2H in the B-ring (Supplementary Table 2) and a thiophene or a substituted phenyl group at the C-ring. In contrast, a number of different aromatic groups can be accommodated at the D-ring. Interestingly, the largest variations in binding affinities are due to changes in the D-ring (Supplementary Table 2). For example, replacing a phenyl D-ring with a thiophenyl ring containing a halogen atom, such as Cl or Br as a substituent, results in over 10-fold KD enhancement. Substituting this thiophenyl ring for other substituents, or even removing the halogen atom alone results in a 10-fold higher KD (see GA229 vs. VPL-36), which further highlights the importance of this interaction. Chemical groups that enhance binding at the D-ring are interchangeable with the C-ring. A meta-trifluoromethyl group present in several of the tightest binding compounds at the D-ring shows high affinity binding when present at both the C- and D-ring positions (e.g. VPL-58). Finally, compounds lacking an aromatic group at the Cring or containing large chemical groups at the B-, C-, or D-rings displayed near loss of binding (e.g. VPL-56, VPL-62, GA017-F2, GA219).
Fig. 3. NMR-based studies reveal SAR for pyrrolidinone scaffold containing ligands that bind with high affinity to the first basic patch of VP35 IID.

Titration of VPL-36 (green) or GA017 (purple) into 15N-labeled eIID resulted in a large number of chemical shift perturbations. (a) VPL-36 titration monitored at residues Ile295 and Ile303. (b) Plot of normalized chemical shift change versus compound concentration for two residues, Ile295 and I303, when VPL36 is titrated into 15N-labelled eIID. (c) GA017 titration as monitored at residues Ile295 and Ile303. (d) Plot of normalized chemical shift change versus concentration for two residues, Ile295 and I303, when GA017 is titrated into 15N-labelled eIID. Dissociation constants were measured by fitting average chemical shift deviation (see Methods) at each concentration of titrant.
Fig. 4. Summary of key functional groups that impact ligand binding to eIID.
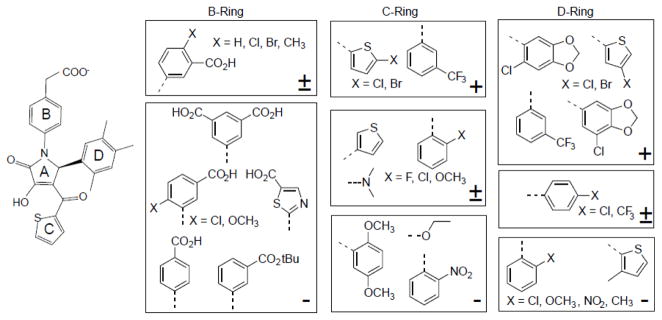
The relative impact of functional groups are represented with respect to either GA017 or a closely related compound and the ranking is indicated by +, +/−, − for enhanced binding, no impact, or diminished binding, respectively.
Compounds bind near the eIID first basic patch
In order to structurally characterize eIID-compound interactions, we generated co-crystal structures of select compounds by soaking native eIID crystals (Table 1). Consistent with our docking predictions (data not shown) and NMR chemical shift mapping (discussed above), all pyrrolidinone compounds bind near a pocket formed by residues from the alpha-helical and beta-sheet subdomains, including Ala221, Arg225, Gln241, Leu242, Lys248, Lys251, Pro293, Ile295, Ile297, Asp302, and Phe328 (Fig. 5a–b). In all, about 20 residues from the eIID protein make up the pocket. Correspondingly, more than 50% of accessible surface area of all compounds is buried at the VP35-NP interface in our structures. These 20 residues are distributed evenly between the alpha helical and beta sheet subdomains. LigPlot24 analysis of the eIID bound structures of GA017, VPL-42, and VPL-60 (Fig. 5c) show that highly conserved hydrophobic residues, including Val245, Ile295, and Phe328, form critical interactions with the compounds. The most expansive hydrophobic interactions are with the D-ring (Fig. 5c). In addition to these common interactions, all compounds show at least two hydrogen bonds between the compound and eIID, including H-bonds with Lys251 and Gln241, which contribute to the binding energy. Removal of either the carboxylic acid or ketone moieties of the ligands completely eliminates binding based on our chemical shift based binding assay. Interestingly, Lys251 is important for the VP35 polymerase co-factor function, as mutations at Lys251 lead to loss of function25. Although most compound atoms make contact with eIID, notable absences include the hydroxyl and ketone groups from the A-ring of the pyrrolidinone scaffold, which were solvent exposed (Fig. 5). Comparison of individual compound bound structures with their respective in silico predictions displayed remarkable similarity in the location and local contacts by the compounds (Supplementary Fig. 4). Further confirmation of compound binding was carried out with the use of two IID mutants, Lys248A and Ile295A. Mutation of these two critical residues individually resulted in near complete loss of compound binding (Fig. 6), while residues outside the binding pocket had no impact on ligand binding. Next, we tested the ability of select compounds to bind rIID and mIID because of the high structural similarity among these IID domains and eIID. Results of these titrations reveal that select compounds, such as GA017, bind in a pocket in mIID and rIID similar to eIID (Fig. 7). Although complete chemical shift assignments are currently not available for rIID and mIID and therefore, we are unable to confirm the exact residues involved in the binding pocket.
Table 1.
Data collection, structure solution, and refinement statistics.
Crystallographic data collection, refinement, and validation statistics
| VP35 IID– GA017 4IBB |
VP35 IID– GA246 4IBC |
VP35 IID– VPL27 4IBD |
VP35 IID– VPL29 4IBE |
VP34 IID– VPL42 4IBF |
VP35 IID– VPL48 4IBG |
VP35 IID– VPL51 4IBH |
VP35 IID– VPL57 4IBI |
VP35 IID– VPL58 4IBJ |
VP35 IID– VPL60 4IBK |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Data collection | ||||||||||
| Space Group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Unit cell parameters | ||||||||||
| a, b, c (Å) | 51.47, 65.78, 72.22 | 51.95, 65.90, 72.04 | 51.56, 65.49, 72.60 | 51.57, 65.51, 72.16 | 51.47, 65.95, 72.00 | 51.47, 65.94, 72.48 | 51.38, 65.80, 72.79 | 51.30, 65.45, 72.65 | 51.26, 66.35, 72.66 | 51.22, 65.69, 72.83 |
| Resolution range (Å)* | 40.00 – 1.75 (1.78 – 1.75) | 50.00 – 1.74 (1.77 – 1.74) | 50.00 – 1.84 (1.87 – 1.84) | 50.00 – 1.95 (1.98 – 1.95) | 50.00 – 2.29 (2.33 – 2.29) | 50.00 – 1.41 (1.43 – 1.41) | 50.00 – 1.88 (1.91 – 1.88) | 50.00 – 1.46 (1.49 – 1.46) | 50.00 – 1.54 (1.57 – 1.54) | 50.00 – 1.85 (1.88 – 1.85) |
| Unique reflections | 24,103 | 25,426 | 21,924 | 17,475 | 11,548 | 47,377 | 20,598 | 40,626 | 36,721 | 21,610 |
| Redundancy | 5.2 (4.1) | 6.4 (3.9) | 8.4 (7.3) | 4.4 (3.3) | 6.9 (7.2) | 7.6 (2.1) | 7.3 (5.8) | 7.9 (4.7) | 6.7 (5.1) | 6.9 (5.6) |
| Completeness (%) | 95.3 (83.3) | 98.7 (90.3) | 99.8 (96.8) | 93.6(94.5) | 99.9 (100.0) | 98.8 (87.3) | 99.9 (100.0) | 93.9 (33.3) | 98.1 (83.1) | 99.9 (99.5) |
| Rmerge (%) | 11.2 (46.1) | 12.5 (69.6) | 11.7 (92.7) | 11.5 (86.2) | 12.1 (50.3) | 10.0 (74.0) | 14.8 (95.9) | 8.0 (54.1) | 8.2 (65.2) | 12.5 (48.1) |
| <I>/<σ> | 15.4 (1.7) | 23.7 (2.1) | 23.9 (2.0) | 12.7 (2.1) | 15.6 (4.3) | 30.8 (2.1) | 20.4 (2.3) | 32.8 (1.9) | 22.6 (2.3) | 31.2 (6.3) |
| Structure solution and refinement | ||||||||||
| Resolution (Å) | 35.11 – 1.75 (1.80 – 1.75) | 32.95 – 1.74 (1.81 – 1.74) | 35.38 – 1.84 (1.92 – 1.84) | 48.50 – 1.94 (2.00 – 1.94) | 40.58 – 2.29 (2.52 – 2.29) | 31.76 – 1.41 (1.44 – 1.41) | 29.98 – 1.88 (1.98 – 1.88) | 41.91 – 1.47 (1.51 – 1.47) | 35.42 – 1.54 (1.56 – 1.54) | 41.90 – 1.85 (1.93 – 1.85) |
| No. of reflections | 21,174 | 25,243 | 21,830 | 15,087 | 10,367 | 47,217 | 20,156 | 40,482 | 31,662 | 21,347 |
| Completeness (%) | 83.7(55.0) | 97.8 (88.0) | 99.3 (95.0) | 80.2(40.0) | 89.9(67.0) | 98.5 (85.0) | 97.6 (95.0) | 96.3 (76.0) | 84.7 (41.0) | 98.7 (97.0) |
| non-hydrogen atoms | 2,141 | 2,163 | 2,206 | 2,104 | 2,068 | 2,205 | 2,154 | 2,201 | 2,259 | 2,234 |
| Rwork/Rfree (%) | 18.1/22.7 | 19.0/25.0 | 17.7/23.3 | 18.3/23.3 | 18.0/26.0 | 17.8/20.0 | 18.1/22.7 | 19.0/23.7 | 18.3/22.9 | 19.3/24.1 |
| R.m.s. deviations | ||||||||||
| Bond lengths (Å) | 0.006 | 0.006 | 0.007 | 0.006 | 0.008 | 0.007 | 0.007 | 0.007 | 0.006 | 0.007 |
| Bond angles (°) | 1.05 | 1.025 | 1.239 | 1.200 | 1.145 | 1.162 | 1.218 | 1.346 | 1.092 | 1183 |
| B-factors (Å2) | ||||||||||
| Protein | ||||||||||
| (chain A) | 23.8 | 23.8 | 20.3 | 30.0 | 35.8 | 15.9 | 18.0 | 21.5 | 21.2 | 17.5 |
| (chain B) | 23.8 | 22.7 | 18.8 | 27.5 | 38.8 | 15.5 | 17.7 | 22.0 | 21.2 | 17.2 |
| Small molecule | ||||||||||
| (chain C) | 38.6 | 40.2 | 23.4 | 32.4 | 46.3 | 17.2 | 22.7 | 20.1 | 25.7 | 21.0 |
| (chain D) | 36.4 | 40.1 | 23.4 | 33.2 | 48.4 | 16.3 | 26.0 | 24.4 | 28.0 | 23.0 |
| Water | 29.7 | 31.4 | 42.3 | 34.1 | 41.3 | 29.5 | 25.4 | 31.6 | 33.3 | 28.0 |
| Ramachandran plot outliers (%) | 0 | 0 | 0.4 | 0 | 0 | 0.8 | 0.4 | 0.8 | 0 | 0 |
| Molprobity score | 1.60 | 1.30 | 1.36 | 1.37 | 1.30 | 1.24 | 1.27 | 1.31 | 1.56 | 1.20 |
| Molprobity clashscore | 4.43 | 5.48 | 6.53 | 6.67 | 5.59 | 4.63 | 5.16 | 5.73 | 11.15 | 4.22 |
Values in parentheses are for the highest resolution shell.
Fig. 5. N-aryl pyrrole containing compounds make substantial contacts with VP35 IID near the first basic patch.

Structure of eIID bound to VPL compound is shown in (a) surface representation (cyan) with a 1σ electron density (sigmaA weighted 2Fo-Fc) mesh for small molecule (VPL-42, PDB 4IBG). (b) Residue-specific contacts between eIID and VPL-42 are highlighted by the amino acid sidechains (orange) within 5 Å of the small molecule. (c) LigPlots showing key hydrophobic and hydrogen bonding contacts for GA017 (PDB 4IBB), VPL-42 (PDB 4IBG), and VPL-60 (PDB 4IBK).
Fig. 6. Mutating residues in the binding pocket abrogates small molecule binding.
1H-15N HSQC spectra are shown for: (a) 15N-enriched zVP35 IID WT in the absence (black) or presence of GA017 (green) and (b) 15N-enriched eVP35 IID I295A in the absence (black) or presence of GA017 (red).
Fig. 7. N-aryl pyrrole containing small molecules are pan-filoviral VP35 IID binders.
1H/15N HSQC spectra of (a) eIID in the absence (dark green) or presence of GA017 (light green), (b) rIID in the absence (dark blue) or presence of GA017 (light blue), and (c) mIID in the absence (dark red) or presence of GA017 (light red).
Pyrrolidinone compounds disrupt PPIs in vitro
Given the overlap between residues important for VP35-NP binding from our previous study 25 and those observed in small molecule-VP35 IID structures (Fig. 5a), we tested the ability of select compounds to disrupt VP35-NP interaction by in vitro pull down assay. As shown in Fig. 8a, select compounds that bind eVP35 IID with affinities (KD) <100 μM can inhibit VP35-NP interaction. While it is difficult to ascertain how different compounds impact this PPI in a quantitative manner, our data suggests that affinity alone may not be the determinant driving binding as demonstrated with GA017, is a relatively high affinity binder, showing less inhibition than VPL42 and VPL60 (compare lanes in Fig. 8a). These observations confirm the ability of the pyrrolidinone compounds to antagonize PPIs. However the lack of correlation between affinity and inhibitory activity of the VP35-NP interaction suggests that, as expected, interface interaction is likely a complex function of binding KD and physico-chemical properties of the ligand.
Fig. 8. Select compounds inhibit VP35-NP interaction in vitro and minigenome activity in cells.

(a) Select compounds can inhibit VP35-NP interaction in vitro as measured by a pull down assay. Differential levels of VP35 noted on the GA017 relative to VPL42 and VPL60 may reflect relative inhibition levels. (b) Select compounds were tested for their ability to inhibit a minigenome assay, and the resulting data show that some compounds do and others do not inhibit minigenome activity in a dose-dependent manner. Solid triangle represents (25, 50 and 100 mM concentrations of compounds as indicated in the figure. Positive controls are replication alone, +/− DMSO as well as +/− VP35 in the absence of DMSO. Maximum DMSO co-solvent concentration was 0.05%.
Pyrrolidinone compounds inhibit viral minigenome activity
Select compounds were tested for their ability to inhibit the function of the viral polymerase complex by using a previously described minigenome assay25. In this system, the viral polymerase complex is reconstituted by transfecting cells with expression plasmids for the EBOV NP, VP35, VP30 and L proteins and for a model viral genome encoding only a GFP-chloramphenicol acetyltransferase (CAT) reporter gene. These results, shown in Fig. 8b for several compounds, including that for GA017, show that our pyrrolidinone compounds can antagonize viral minigenome function in a dose-dependent manner. In addition to the minigenome activity, we also measured the levels of firefly luciferase produced from co-transfected constitutively-expressed mammalian expression plasmids. At 0, 25, 50 and 100 μM compound, no statistical difference between relative levels of firefly luciferase activity were detected (Supplementary Fig. 5), suggesting that these compounds do not specifically influence the luciferase activity nor do they cause significant cell toxicity.
Select compounds inhibit EBOV
Compounds were tested for their ability to inhibit a replication competent EBOV in a cell-based assay. This is a recombinant virus that encodes a GFP reporter but is otherwise similar to wild type virus for infection and pathogenicity26. Assays were performed at BSL4 containment. Both initial infection efficiency as well as virus produced from treated cells was tested (Figure 9). As measured by GFP reporter expression, GA248, VPL-42 and VPL-57 each inhibited virus infection relative to the untreated (DMSO only) control. In contrast, VP-15, which has a lower binding affinity showed weak inhibition (Fig. 9a). VP35 is involved in virus assembly and RNA replication complex activity and so its inhibition should have a greater impact on late stages of the replication cycle. To measure this effect, supernatants from BSRT7 cells infected at 0.05 MOI for 48 h were collected and used to infect a new monolayer of BSRT7 cells. For this assay, VPL-42 and VPL-57 were still active but now GA17, GA240, VPL-60, VPL-51, and VPL-58 also showed strong inhibition of virus production. Again, each was more active than VPL-15, but even VPL-15 showed some activity (50% inhibition compared to control). This inhibition suggests that despite lack of biochemical evidence for a strong interaction with VP35, in cells the compound can still interfere with virus replication. However, it is unclear if this is due to a direct effect on viral VP35 protein. Interestingly, GA248, which inhibited virus infection in the GFP-reporter assay, did not inhibit infectious virus release. This suggests that it may preferentially affect VP35 during an early phase of replication.
Figure 9. Select compounds inhibit EBOV replication and release.
(a) Inhibition of viral replication was monitored by the infection efficiency, which was calculated by dividing the number of infected, GFP expressing cells, by the total number of cells at 48 hours post challenge with EBOV-GFP. BRST7 cells were treated with indicated compounds at 125 μM in triplicate. (b) Inhibition of virus release was measured by the ability of supernatant to infect fresh cells. Cells treated with the indicated compounds and challenged with virus and supernatants collected after 48 hours. The supernatants from these viral infected cells (with or without compound treatment) were diluted and a portion transferred to fresh cells. After an additional 24 h the fresh cells were fixed and the infection efficiency calculated as for (A). Only those compounds showing a significant (P<0.05) decrease from DMSO treated control are shown.
Discussion
Using a computational approach, we screened over 5.4 million potential compounds and, through further computational analysis and NMR-based screening methods, we identified several compounds capable of binding VP35 IID with high affinity and specificity. Structures of several compounds in complex with VP35 IID highlight several hydrophobic interactions between compound functional groups and side chains from key residues important for VP35 function in vivo. Therefore these inhibitors can potentially function as leads for development of panfiloviral therapeutics.
Targeting PPIs is a challenge for therapeutic development for several reasons, including the high energy barrier to identify lead candidates, availability for binding interactions as well as a general lack of curvature limiting the number of available binding pockets with sufficient surface area for ligand binding. In contrast, active site analogs often serve as good starting points to inhibitor optimization for enzymatic targets. Using an in silico screening approach allows for rapid, high efficient and low cost methods for computational determination of potential binders and, when paired with a method for structural determination, allows for the optimization of binding interactions to maximize affinity and specificity. When structural knowledge regarding PPIs is available, this can be applied as an additional filter to identify in silico hits likely to disrupt function.
One aspect of drug design that was not addressed in our in silico modeling is drug access to sites where virus replication takes place. We believe this accounts for many of the differences seen in terms of biochemical measurement of compound affinity to VP35 and activity in the minigenome assay or with replicating virus. The ability of a compound to partition into water versus the membranes of the cell dictates ability to access these sites and reach concentrations where it can be active. For many virus types, different stages of replication occur in membranous structures or in the aqueous phase of the cytoplasm. This means that chemical partitioning is predicted to have a large impact on anti-viral activity. The assembling virus also adds an additional layer of complexity, requiring the cooperative interaction of many structural components, including NP, VP40 and VP35. While not studied here, our observation that compounds are better able to inhibit virus production than initial infection likely reflects disruption of such assembly and will be investigated in future work.
While the functional importance of filoviral VP35 is well appreciated, there is a clear gap in utilizing that knowledge to target VP35 for antiviral inhibitor development. Using a combination of in silico docking, biochemical and structural characterization and functional validation, we now provide important information required for developing therapeutics against filoviral VP35s. In particular, the use of available structural and functional data to define the small molecule binding site near the NP binding region and the ability of select small molecules to inhibit VP35-NP interaction at a PPI that is critical for replication function. Our results using dose dependent minigenome analysis further supports targeting of select inhibitors to the VP35-NP PPI. Finally, the ability of select compounds to inhibit GFP-expressing EBOVs further highlights the potential utility of the compounds we have identified and characterized. However, the affinities of the compounds identified are less than optimal for lead compounds. Efforts to optimize the chemical scaffold identified here and to identify more potent inhibitors of filoviruses is underway. In addition to providing a class of small molecules that target a PPI, this study also validates the use of in silico docking, where functional data can guide the selection of target sites in proteins by computational methods to provide chemical leads against PPIs of high priority pathogens that encode few enzymatic antiviral targets.
Materials and Methods
In silico screening
Autodock 4.027 automated by the DOVIS pipeline28; 29 was used to screen 5.4 million compounds from the ZINC22 against the FBP groove in the crystal structure of eVP35 IID (PDB ID code 3FKE; Supplementary Fig. 1a). Energy minimization was performed on protein-compound complexes of the 20,000 top-ranked molecules using CHARMM (v. 35b2)30 with the MMFF force field module31. Minimized complexes were re-ranked by LigScore232. Visual inspection of the top 100 complexes led to the purchase of 25 compounds (Enamine Inc., Kiev, Ukraine). 130 more compounds that had the most similar structural interacting fingerprints (SIFt)23 to those hits were also selected and purchased.
Protein expression, purification, and crystallization
eVP35 IID proteins were purified as described previously16; 17; 33. Crystallization screens (Hampton Research) were used to identify initial conditions using purified VP35 IID protein and further optimized using in-house reagents with 10% DMSO. After 24–48 hours, 0.4 μL of 20–50 mM compound in 100% DMSO was added to crystals and incubated at 25°C for 1–24 hours. Diffraction data were collected at the Advanced Photon Source Beamline 19ID (Argonne National Laboratory) at 100 K. Ligand bound structures were solved by isomorphous replacement, using the structure of ligand free eVP35 IID (PDB ID code 3FKE) as the search model, and refined using REFMAC534 or Phenix35.
Optimization of initial hits and synthesis
In order to optimize key functional groups, compounds were synthesized based on initial SAR data obtained by NMR chemical shift mapping and structural data, including charge/surface complementation (Detailed methods are described in the Supplementary Methods).
NMR spectroscopy
50 mM stock solutions of small molecules were prepared by dissolving compounds in 100% d6-DMSO. Samples containing 4–10 compounds were screened by NMR and 1H-15N HSQC spectra were analyzed for changes in chemical shift. Up to 80 μM of each compound was added to 80 μM 15N-labeled eVP35 IID samples in 10 mM HEPES (pH 7.0), 150 mM NaCl, 2 mM TCEP, 5% D2O, and 5% d6-dimethylsulfoxide (DMSO). All experiments were acquired at 25°C on a Bruker Avance II spectrometer operating at 700.13 MHz with 5 mm z axis field gradient and inverse detection TXI (1H/13C/15N) cryogenic probe, using the following parameters: 16 scans, 128 complex points (t1), and 2048 total points (t2). NMR data were processed using NMR Pipe and NMRDraw36. NMR spectra were analyzed with NMRView37.
Initial hits were identified by comparing 1H-15N eVP35 IID chemical shifts in the absence and presence of compound. Chemical shifts >5x digital resolution were considered significant. Initial hits were validated by titration studies as described below. Chemical shifts were obtained from 1H-15N HSQC experiments using the following equation:
Estimated binding constants (KD, estimate, Supplementary Table 1–2) were obtained by the following method. Twenty different compounds with varying affinities and the same pyrollidinone scaffold structure were titrated in a concentration range of 0–1000 μM (5–8 data points). Using non-linear least square fits, maximum chemical shift difference were obtained. For this analysis, only peaks that display maximum shifts > 3 x digital resolution were selected. For single point estimations, residue I303 was selected based on several parameters, including proximity and response to binding as well as correlation between KD values obtained using single point data and titrations (Supplementary Fig. 3). Moreover, I303 displayed good resolution with no chemical shift overlap. Maximum chemical shift differences, Δδ, and KD values were obtained by fitting the following equation to the data:
Bmax values obtained by fitting were used to normalize Δ δ for comparison of different residues within the same small molecule data set. All single point titrations were carried out with 80 μM small molecule ligand and the KD values were estimated by the following equation,
where [Ligand] was 80 μM and Bmax was the average value of the fit parameter from the 20 titrations for I303. The use of I303 as an indicator of binding was validated through the correlation coefficient between the measured KD obtained by I303 titrations.
Optimization of initial hits
Initial compounds containing the pyrrolidinone scaffold were purchased from Enamine, Inc (See compounds with GA prefixes, Supplementary Table 1 and 2). In order to optimize key functional groups, compounds were synthesized based on initial SAR data obtained by NMR chemical shift mapping and structural data, including charge/surface complementation (See Supplementary Methods for detailed synthesis of VPL compounds).
Structural studies
Commercial crystallization screens (Hampton Research) were used to identify initial conditions using purified eVP35 IID protein and further optimized using in-house reagents with 10% DMSO. These conditions defined as condition 1 (7 mg/mL protein solutions diluted 1:1 with 100 mM magnesium acetate (pH 7.5), 15% (wt/vol) PEG 3350 and 10% (v/v) DMSO) or condition 2 (7 mg/ml protein solutions diluted 1:1 with 100 mM sodium citrate (pH 5.5), 15% (wt/vol) PEG 3350 and 10% (v/v) DMSO) were used to obtain crystals grown at 25°C using the hanging-drop vapor-diffusion method. After 24–48 hours, 0.4 μL of 20–50 mM compound in 100% DMSO was added to drops containing single crystals and incubated at 25°C for 1–24 hours. Condition 1 was used to soak GA-series compounds and condition 2 was used to obtain complexes of VPL compounds. All crystals were back-soaked in reservoir solution containing 25% glycerol (wt/vol) and plunge frozen in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source Beamline 19ID (Argonne National Laboratory) at 100 K. X-ray data was processed using the HKL300038. Ligand bound structures were solved by molecular replacement, using the structure of ligand free eVP35 IID (PDB ID: 3FKE) as the search model, and refined using REFMAC534 or Phenix35. Small molecule structures were generated by the PRODRG2 server and manually built into the complex structures using Coot39. Water molecules were added using Coot and the model was further refined with REFMAC5 or Phenix. Structures were validated using MolProbity40. Data collection, refinement and validation statistics for 10 structures corresponding to molecules GA017, GA246, VPL48, VPL51, VPL57, VPL60, VPL58, VPL27, VPL29, and VPL42 are shown in Table 1.
NP pulldown assay
Pulldown assays were performed in buffer containing 10 mM HEPES (pH 7.0), 150 mM NaCl, 5% Dimethyl sulfoxide (DMSO), and 5 mM 2-mercaptoethanol at 25 °C. MBP-His tagged eVP35 IID WT protein was immobilized on amylose resin, incubated with purified His-tagged NP protein, and subsequently washed. For small molecule competition assays, 500 μM small molecules was incubated with MBP-His tagged eVP35 IID prior to incubation with His-tagged NP. Following washout of unbound material, beads containing MBP-His tagged eVP35 IID WT protein (or His tagged eVP35 IID/His-tagged NP complex) was resolved on SDS-PAGE and Western blotted with mouse anti-His antibody (Santa Cruz biotechnology), followed by horseradish peroxidase (HRP) conjugated goat anti-mouse antibody (Bio-Rad). Membranes were developed using Millipore Immobilon Western Chemiluminescence HRP substrate and recorded on a ChemiDoc (Bio-Rad).
Ebola minigenome studies
Minigenome (MG) assays were performed as previously described25; 41 with the following modifications. For the MG assay, the BSRT7 cells were transfected with expression plasmids for EBOV L, VP30, NP and VP35, which reconstitute the EBOV RNA polymerase complex. Co-transfected was the MG plasmid that produces the MG RNA, which contains cis-acting sequences allowing its recognition by the reconstituted EBOV polymerase complex. The MG RNA encodes only a renilla luciferase. A co-transfected RNA polymerase II-driven firefly luciferase expression plasmid served as a control for transfection efficiency and for cytotoxicity. 4 hrs after transfection, compounds were added diluted in 250ul of the media (dilutions were made for respective concentrations). 18h later a Dual luciferase assay (Promega) was performed following manufacturer’s protocols and read by Promega plate reader. Luciferase activity was normalized to firefly luciferase for each group, with the negative control (without VP35) set to 1.
EBOV infections, replication and release assays
For all EBOV infection assays 3000 BSRT7 cells per well of a 384 well plate (Greiner bio-one) were grown overnight in DMEM (HyClone) supplemented with 10% FBS. The 80% confluent monolayer was treated with the indicated concentration of each compound for 1 hour in triplicate. Cells were then transferred to the BSL-4 laboratory and infected with EBOV-GFP at an MOI of 0.05. Infected cells were kept in a humidified CO2 incubator for 48 h. At this time, 10 μL supernatant from each well was used to infect a fresh monolayer of BSRT7 cells seeded in 96 well plates. This gave a 10-fold dilution of drug containing medium, which meant that all drugs would be inactive. Cells in the 384 well plate were fixed by immersing in neutral buffered formalin overnight. 96 well plates were fixed 24 hours post infection by immersing in neutral buffered formalin for 24 h. Fixed cells were washed three times with PBS and nuclei stained with Hoecst-33342 (Life-technologies- 1:50,000 dilution). Cells were imaged on a Nikon Ti Eclipse microscope. Infected cells (green) and total cells (blue) were counted using CellProfiler software (Broad Institute) using customized pipelines which are available upon request to R. Davey. Data was analyzed and plotted using GraphPad Prism software.
Supplementary Material
Highlights.
Ebola VP35 is a multifunctional viral protein critical for viral replication.
VP35 IFN inhibitory domain (IID) structure revealed several drugable pockets.
An in silico screen identified potential binders near functionally important patches.
Binding site, near a region important for viral replication, was validated by NMR chemical shift mapping.
High resolution complexes of small molecule/VP35 IID were obtained.
Select small molecules inhibit VP35 functions in vitro and in cell-based studies, including pathogenic Zaire Ebola virus infection of tissue culture cells.
Acknowledgments
We thank Drs. S. Ginell, N. Duke, F. Rotella, K. Lazarski, and J. Lazarz at Argonne National Laboratory Structural Biology Center Beamlines (SBC). SBC is supported by the U.S. D.O.E. under contract DE-AC02-06CH11357. DMB and ZO are supported in part by the Center for Structural Genomics of Infectious Diseases (CSGID) under the NIAID/NIH/DHS Contracts HHSN272200700058C and HHSN272201200026C Anderson (PI). This work is supported in part by NIH grants (R01AI081914 to G.K.A. and R01AI059536, R56AI089547, R56AI093786 and R21AI097568 to C.F.B.), MRCE Developmental Grant (U54AI057160-Virgin (PI) to G.K.A.), and the Welch Foundation (I-1612 to J.M.R.). This study made use of the National Magnetic Resonance Facility at Madison, which is supported by National Institutes of Health grants P41RR02301 (Biomedical Research Technology Program, National Center for Research Resources) and P41GM66326 (National Institute of General Medical Sciences). Equipment in the facility was purchased with funds from the University of Wisconsin, the National Institutes of Health (P41GM66326, P41RR02301, RR02781, RR08438), the National Science Foundation (DMB-8415048, OIA-9977486, BIR-9214394), and the U.S. Department of Agriculture.
Footnotes
Accession numbers:
Coordinates and structure factors for small molecule-VP35 IID complexes have been deposited in the Protein Data Bank under the PDB codes: 4IBB (GA017), 4IBC (GA246), 4IBD (VPL27), 4IBE (VPL29), 4IBF (VPL42), 4IBG (VPL48), 4IBH (VPL51), 4IBI (VPL57), 4IBJ (VPL58), and 4IBK (VPL60).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2011;377:849–62. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bosio CM, Aman MJ, Grogan C, Hogan R, Ruthel G, Negley D, Mohamadzadeh M, Bavari S, Schmaljohn A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J Infect Dis. 2003;188:1630–8. doi: 10.1086/379199. [DOI] [PubMed] [Google Scholar]
- 3.Bray M, Geisbert TW. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol. 2005;37:1560–6. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. 2006;80:5156–67. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol. 2007;81:13469–77. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valmas C, Grosch MN, Schumann M, Olejnik J, Martinez O, Best SM, Krahling V, Basler CF, Muhlberger E. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010;6:e1000721. doi: 10.1371/journal.ppat.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:182–92. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schumann M, Gantke T, Muhlberger E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol. 2009;83:8993–7. doi: 10.1128/JVI.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106:2886–91. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartman AL, Dover JE, Towner JS, Nichol ST. Reverse genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J Virol. 2006;80:6430–40. doi: 10.1128/JVI.00044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prins KC, Delpeut S, Leung DW, Reynard O, Volchkova VA, Reid SP, Ramanan P, Cardenas WB, Amarasinghe GK, Volchkov VE, Basler CF. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J Virol. 2010;84:3004–15. doi: 10.1128/JVI.02459-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reid SP, Cardenas WB, Basler CF. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology. 2005;341:179–89. doi: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–56. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardenas WB, Loo YM, Gale M, Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80:5168–78. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartman AL, Towner JS, Nichol ST. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology. 2004;328:177–84. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Leung DW, Ginder ND, Fulton DB, Nix J, Basler CF, Honzatko RB, Amarasinghe GK. Structure of the Ebola VP35 interferon inhibitory domain. Proc Natl Acad Sci U S A. 2009;106:411–6. doi: 10.1073/pnas.0807854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung DW, Shabman RS, Farahbakhsh M, Prins KC, Borek DM, Wang T, Muhlberger E, Basler CF, Amarasinghe GK. Structural and functional characterization of Reston Ebola VP35 Interferon Inhibitory Domain. J Mol Biol. 2010 doi: 10.1016/j.jmb.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haasnoot J, de Vries W, Geutjes EJ, Prins M, de Haan P, Berkhout B. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007;3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimberlin CR, Bornholdt ZA, Li S, Woods VL, Jr, MacRae IJ, Saphire EO. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc Natl Acad Sci U S A. 2010;107:314–9. doi: 10.1073/pnas.0910547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bale S, Julien JP, Bornholdt ZA, Kimberlin CR, Halfmann P, Zandonatti MA, Kunert J, Kroon GJ, Kawaoka Y, Macrae IJ, Wilson IA, Saphire EO. Marburg Virus VP35 Can Both Fully Coat the Backbone and Cap the Ends of dsRNA for Interferon Antagonism. PLoS Pathog. 2012;8:e1002916. doi: 10.1371/journal.ppat.1002916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basler CF, Amarasinghe GK. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res. 2009;29:511–20. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng Z, Chuaqui C, Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem. 2004;47:337–44. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–86. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 25.Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol. 2010;84:10581–91. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Towner JS, Paragas J, Dover JE, Gupta M, Goldsmith CS, Huggins JW, Nichol ST. Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology. 2005;332:20–7. doi: 10.1016/j.virol.2004.10.048. [DOI] [PubMed] [Google Scholar]
- 27.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang X, Kumar K, Hu X, Wallqvist A, Reifman J. DOVIS 2.0: an efficient and easy to use parallel virtual screening tool based on AutoDock 4.0. Chem Cent J. 2008;2:18. doi: 10.1186/1752-153X-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Kumar K, Jiang X, Wallqvist A, Reifman J. DOVIS: an implementation for high-throughput virtual screening using AutoDock. BMC Bioinformatics. 2008;9:126. doi: 10.1186/1471-2105-9-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comp Chem. 1996;17:490–519. [Google Scholar]
- 32.Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. LigScore: a novel scoring function for predicting binding affinities. J Mol Graph Model. 2005;23:395–407. doi: 10.1016/j.jmgm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, Otwinowski Z, Liu G, Huh J, Basler CF, Amarasinghe GK. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–52. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscilation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J Virol. 1999;73:2333–42. doi: 10.1128/jvi.73.3.2333-2342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leung DW, Prins KC, Basler CF, Amarasinghe GK. Ebolavirus VP35 is a multifunctional virulence factor. Virulence. 2010;1:526–31. doi: 10.4161/viru.1.6.12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.