

Abstract
Here it was determined that Chronic Lymphocytic Leukemia (CLL) cells express the α-subunit but not the β-subunit of the granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR/CSF3R). GM-CSFRα was detected on the surface, in the cytosol, and the nucleus of CLL cells via confocal microscopy, cell fractionation, and GM-CSFRα antibody epitope mapping. Because STAT3 is frequently activated in CLL and the GM-CSFRα promoter harbors putative STAT3 consensus binding sites, MM1 cells were transfected with truncated forms of the GM-CSFRα promoter, then stimulated with IL-6 to activate STAT3 to identify STAT3 binding sites. Chromatin immunoprecipitation (ChIP) and an electoromobility shift assay (EMSA) confirmed STAT3 occupancy to those promoter regions in both IL-6 stimulated MM1 and CLL cells. Transfection of MM1 cells with STAT3 siRNA or CLL cells with STAT3 shRNA significantly down-regulated GM-CSFRα mRNA and protein levels. RNA transcripts, involved in regulating cell-survival pathways, and the proteins KAP1 (TRIM28) and ISG15 co-immunoprecipitated with GM-CSFRα. GM-CSFRα-bound KAP1 enhanced the transcriptional activity of STAT3, whereas ISG15 inhibited the NF-κB pathway. Nevertheless, overexpression of GM-CSFRα protected MM1 cells from dexamethasone-induced apoptosis, and GM-CSFRα knockdown induced apoptosis in CLL cells, suggesting that GM-CSFRα provides a ligand-independent survival advantage.
Introduction
B-cell chronic lymphocytic leukemia (CLL), the most common hematologic malignancy in the Western hemisphere, is characterized by a dynamic imbalance between proliferation and apoptosis of neoplastic B-lymphocytes co-expressing CD5 and CD19 antigens (1, 2). Despite recent improvements in managing this disease, CLL remains incurable. Like other lymphoid neoplasms, CLL cells usually express the CD20 antigen. Combining the anti-CD20 antibody rituximab with GM-CSF produced higher response rates than did single-agent rituximab in relapsed follicular B-cell lymphoma (3) and in initial studies in CLL (4).
GM-CSF is produced by a variety of cells, including stromal cells and cells of hematopoietic origin, including B1a cells (5), and regulates the survival, proliferation, differentiation, and activation of hematopoietic cells (6) as well as the function of dendritic cells (7) and T cells (8). GM-CSF regulates by binding to the cell-surface GM-CSF receptor (GM-CSFR). GM-CSFR, first identified on cells of the myelomonocytic lineage by ligand-binding studies (9, 10), belongs to a structurally distinct family of colony-stimulating hematopoietic growth factor receptors that include receptors that bind GM-CSF, M-CSF, or G-CSF (11). The GM-CSFR is a heterodimer comprising GM-CSFRα (12) and GM-CSFRβ (also known as βc) subunits (13). The 80-kDa GM-CSFRα subunit (CD116) is cytokine specific, whereas the 120-kDa CSFRβ subunit (CD131) is nonspecific and is shared with the cytokine-specific β subunits of the IL-3 and IL-5 receptors. GM-CSFR does not have intrinsic tyrosine kinase activity but associates with the tyrosine kinase JAK2, which is required for the initiation of signaling and biological activity. Although the Ig-like domain of GM-CSFRα is a crucial determinant of GM-CSF binding (14), in the absence of GM-CSFRβ, the GM-CSFRα subunit binds GM-CSF with low affinity (11). Both subunits α and β are required for GM-CSF signaling, and the cytoplasmic domains of both GM-CSFRα and β are essential for receptor activation (15, 16); however, only the β domain associates with JAK2 (17).
B-cell CLL cells express CD5, a cell-surface antigen commonly expressed on normal T lymphocytes (2). Although primarily a myeloid growth factor, GM-CSF affects T-cell function (8). Antigen-stimulated CD8+ T cells express GM-CSFRβ (18), and human NK cells, 80% of which express CD8, also express CD160, recently found expressed on CLL cells from 98% of patients (19). Because of the similarities between CLL cells and T lymphocytes, because data suggested that GM-CSF upregulates the expression of CD20 on the surface of CLL cells (20), and because GM-CSF enhanced the effect of anti-CD20 antibodies in follicular lymphoma (3), we sought to explore the effect of GM-CSF on CLL cells.
Consistent with previous reports (21), we found that GM-CSF did not activate GM-CSFR-induced signaling pathways in CLL cells. However, we detected GM-CSFRα, but not GM-CSFRβ, on the cell surface, in the cytoplasm, and in the nucleus of CLL cells. We demonstrated that signal transducer and activator of transcription (STAT)-3, constitutively activated in CLL cells (22), activates the GM-CSFRα promoter and induces GM-CSFRα production, and that GM-CSFRα protects CLL cells from apoptosis.
Materials and Methods
Patients
Peripheral blood (PB) cells were obtained from patients with CLL treated at The University of Texas MD Anderson Cancer Center Leukemia Clinic. Institutional Review Board approval and patients' written informed consent were obtained. PB was obtained from untreated patients. The clinical characteristics of the patients whose PB samples were used in this study are presented in Supplemental Table 1.
B-cell CLL Cell Fractionation
To isolate low-density cells, PB cells were fractionated using Histopaque 1077 (Sigma-Aldrich). More than 95% of the fractionated PB lymphocytes obtained from these patients were CD19+/CD5+, as assessed by flow cytometry.
Cell Culture
Fractionated CLL cells were maintained in DMEM (Sigma-Aldrich) supplemented with 10% FBS (HyClone). The MM1 and the human embryonic kidney (HEK) 293FT cell line were obtained from the American Type Culture Collection. MM1 cells were maintained in RPMI 1640 (Sigma-Aldrich), and 293FT cells were maintained in DMEM supplemented with 10% FBS, both in a humidified atmosphere of 5%carbon dioxide at 37°C. Prior to analysis, MM1 cells were incubated with either recombinant human IL-6 (BioSource International) or tissue culture medium.
Polymerase Chain Reaction (PCR)
Total RNA was extracted using the TRIzol reagent (Life Technologies, Grand Island, NY) and cDNA was synthesized using the SuperScript First-Strand Synthesis System (Life Technologies). Human GM-CSFRα and GM-CSFRβ primers were obtained from Applied Biosystems (Life Technologies). The cell lines OCI/AML3, OCI-2, K562, and Jurkat were used as positive and negative controls. To amplify GM-CSFRα transcripts a regular PCR program was performed with 30 cycles, and to amplify GM-CSFRβ transcripts a 50 cycle touchdown program was executed.
Flow Cytometry Analysis
Cells were suspended in 100 μl of PBS and split into duplicate tubes. Twenty microliters antibody or its isotype were added, and the tubes were incubated in the dark at room temperature for 30 minutes. After incubation, the cells were washed, suspended in 500 μl PBS, and analyzed using the FACSCalibur flow cytometer (Becton Dickinson Immunocytometry Systems). The following antibodies and their corresponding isotype controls were used: mouse anti-human CD116, CD131, CD19, and CD5 (BD Biosciences).
Western Blot Analysis
Cell pellets were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer. The lysed cell pellets were incubated on ice for 5 minutes and the protein concentration was determined using the Micro BCA protein assay reagent kit (Thermo Scientific/Pierce). Supernatant proteins were denatured by boiling for 5 minutes in SDS loading buffer, separated by SDS-PAGE, and transferred to a nitrocellulose membrane.
Following transfer, equal loading was verified by Ponceau S staining. The membranes were blocked with 5% skim milk in Tris-buffered saline and incubated with one of the following antibodies: monoclonal mouse anti-human STAT3 (BD Biosciences and Cell Signaling Technology); monoclonal mouse anti-human phosphotyrosine STAT3, polyclonal rabbit anti-human phosphoserine STAT3, monoclonal mouse anti-human STAT5 and phosphotyrosine STAT5, AKT and phosphoserine AKT, ERK-1/2 and phosphotyrosine ERK-1/2, and monoclonal mouse anti-human JAK2 and phosphotyrosine JAK2 (Cell Signaling Technology); monoclonal mouse anti-human GM-CSFRα and GM-CSFRβ (BD Biosciences); monoclonal mouse anti-human β-actin (Sigma-Aldrich); or mouse anti-GFP (Millipore/Upstate) antibodies.
After binding with horseradish peroxidase-conjugated secondary antibodies, blots were visualized with an ECL detection system (GE Healthcare), and densitometry analysis was performed using the Epson Expression 1680 scanner (Epson). Densitometry results were normalized by dividing the numerical value of each sample signal by the numerical value of the signal from the corresponding loading control.
Immunohistochemistry
A standard immunohistochemistry method was used. Briefly, BM clots and smears were subjected to antigen retrieval performed with Diva Decloaker buffer in a Decloaking Chamber pressure cooker (Biocare Medical). After the slides were blocked with 3% hydrogen peroxide and Background Sniper protein block (Biocare Medical), they were incubated for 1 hour at room temperature with GM-CSFRα antibodies (1:100; Sigma-Aldrich) and counterstained with hematoxylin (Dako). Tris-buffered saline supplemented with 0.1% Tween were used for washing between the steps.
Immunoprecipitation
Whole-cell lysates, cytoplasmic or nuclear fractions, were incubated with 4 μg of polyclonal rabbit anti-human JAK2 or rabbit anti-GFP antibodies (Millipore/Upstate) for 16 hours at 4°C. Protein A agarose beads (Millipore/Upstate) were added for 2 hours at 4°C. As a negative control, whole-cell lysates, cytoplasmic fractions, or nuclear fractions were incubated either with rabbit serum and protein A agarose beads or with protein A agarose beads alone. After 3 washes with RIPA buffer, the beads were suspended in SDS sample buffer, boiled for 5 minutes, and removed by centrifugation, and the supernatant proteins were separated by SDS-PAGE, as described above.
Confocal Microscopy
CLL low-density cells were cytospun on poly-L-lysine–coated slides and fixed in 3.7% formaldehyde for 15 minutes at room temperature on a shaker. The slides were washed PBS, incubated with 1% Triton X-100 for 5 minutes and blocked with goat serum for 1 hour. After blocking, the slides were washed and incubated overnight with PE-conjugated mouse anti-human GM-CSFRα antibodies (BD Bioscience). The slides were then washed in PBS and incubated with the nuclear acid stain TOPRO-3 (Invitrogen). In other experiments, cells were incubated with mouse anti-S6 ribosomal protein (Calbiochem/Millipore) overnight, extensively washed in PBS, and then incubated with PE rabbit anti-mouse antibody (BD Biosciences) for 1 hour. The slides were then mounted with VECTASHIELD HardSet (Vector Laboratories) and viewed using an Olympus FluoView FV500 Confocal Laser Scanning Microscope (Olympus).
Isolation of Nuclear and Cytoplasmic Extracts
Nuclear and cytoplasmic extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific/Pierce) as previously described(22). To confirm that pure extracts were obtained, monoclonal mouse anti-human lamin B and mouse anti-human S6 ribosomal protein antibodies were used (Millipore/Calbiochem).
Generation of Luciferase Reporter Plasmids and Site-Specific Mutagenesis
A 4.0-kb fragment of the human GM-CSFRα promoter was generated by PCR using, as a template, genomic DNA isolated from PB low-density cells that were obtained from 2 patients with CLL. The DNA was purified via the Wizard DNA Purification Kit (Promega). Human GM-CSFRα promoter PCR primers were designed in accordance with the sequence of the GM-CSFRα-flanking region (Ensembl Resource, www.ensembl.org). The GM-CSFRα promoter PCR primers included a forward primer starting at bp-4012-CTTGGTGAACCGTCTGGTTGA (relative to the start of the first exon), a reverse primer starting at bp-23-CTTCTGAGTAGCTCCCTTCAG, and primers for several truncated constructs designed at bp-2517-GAATGGACTAAGACAGCTCCT and bp-496-TGTATGCCCATGTGCACTGTG. The amplified fragments were attached to BgIII- and KpnI-digestion sites of the pGL4-basic vector (Promega) containing a luciferase reporter gene. The pGL4 vectors harboring ligation products were introduced into One Shot TOP10 chemically competent Escherichia coli (Invitrogen), spread onto agar plates containing ampicillin (Invitrogen), and incubated overnight at 37°C. Single colonies were microaspirated and analyzed following restriction enzyme digestion. The sequences of all constructs were verified by automated sequencing (SeqWright).
MM1 Cell Transfection and Luciferase Assay
MM1 cells were transfected by electroporation with the Gene Pulser Xcell Electroporation System (Bio-Rad) as previously described (23). A total of 5 × 106 cells, suspended in 0.2 ml of serum-free OPTI-MEM medium (Invitrogen), were transfected with 2 μg of each of the specific reporter constructs. To assess luciferase activity, we used the Dual-Luciferase Reporter Assay System (Promega) and the SIRIUS luminometer V3.1 (Berthold Detection Systems) 24 hours after transfection. We determined the luciferase activity by calculating the constructs' luciferase activity relative to the activity of the Renilla reniformis luciferase produced by the pRL-SV40 control vector. The primers used in these experiments are listed in Supplemental Table 2.
RNA Purification and qRT-PCR
RNA was isolated using an RNeasy purification procedure (QIAGEN) and its quality and concentration were analyzed with a NanoDrop spectrophotometer (ND-1000; Thermo Scientific/NanoDrop Technologies). We used 500 ng of total RNA in 1-step qRT-PCR (Applied Biosystems). The sequence detection system ABI Prism 7700 (Applied Biosystems) used the TaqMan gene expression assay for ROR1, STAT3, Bcl2, Bcl-XL, c-Myc, p21 (WAF1), cyclin D1, and 18S according to the manufacturer's instructions. We ran the samples in triplicate, and used the comparative CT method for relative quantification (22).
ChIP Assay
We used a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) in accordance with the manufacturer's instructions. Briefly, cells were cross-linked with 1% formaldehyde for 10 minutes at room temperature and then harvested and incubated on ice for 10 minutes in lysis buffer. Nuclei were pelleted and digested by micrococcal nuclease. Following sonication and centrifugation, sheared chromatin was incubated with anti-STAT3 or rabbit serum (negative control) overnight at 4°C. Then, protein-G beads were added, and the chromatin was incubated for 2 hours in rotation. An aliquot of chromatin that was not incubated with an antibody was used as the “input” control sample. Antibody-bound protein/DNA complexes were eluted and subjected to PCR analysis. The primer sets used to amplify GM-CSFRα promoter sites are listed in Supplemental Table 2. The primers to amplify the human STAT3 promoter were forward, 5′-CCGAACGAGCTGGCCTTTCAT-3 and reverse, 5′-GGATTGGCTGAAGGGGCTGTA-3, which generated an 86-bp product; primers to amplify the WAF/p21 promoter were forward, 5′-TTGTGCCACTGCTGACTTTGTC-3 and reverse, 5′-CCTCACATCCTCCTTCTTCAGGCT-3, which generated a 303-bp product; and primers to amplify the c-Myc promoter were forward, 5′-TGAGTATAAAAGCCGGTTTTC-3 and reverse, 5′-AGTAATTCCAGCGAGAGGCAG-3, which generated a 63-bp product. The human RPL30 gene primers were provided by Cell Signaling Technology.
EMSA
Nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific/Pierce). Two μg of nuclear protein extracts were incubated with biotin-labeled GM-CSFRα promoter DNA probes in binding buffer for 30 minutes on ice. All probes were synthesized by Sigma-Genosys. The sequences of the probes used are listed in Supplemental Table 2. Following incubation, the samples were separated on a 5% polyacrylamide gel in Tris-borate EDTA, transferred onto a nylon membrane, and fixed on the membrane by UV cross-linking. The biotin-labeled probe was detected with streptavidin horseradish peroxidase (EMSA Gel-Shift Kit, Affymetrix/Panomics). A 7-fold excess of unlabeled cold probes combined with biotin-labeled probes was used for competition control. To determine the effect of antibodies on protein-DNA binding, 1 μg of monoclonal mouse anti-human STAT3 (BD Biosciences; Cell Signaling Technology) or mouse anti-human phosphoserine STAT3 (Cell Signaling Technology) antibody was incubated with the nuclear extracts for 30 minutes on ice prior to adding the biotin-labeled DNA probe. The isotypic controls consisted of mouse Ig G1 (BD Biosciences).
STAT3 and GM-CSFRα siRNA Transfection
MM1 and CLL cells were transfected by electroporation as described above. Knockdown of endogenous STAT3 was performed using the pre-designed siRNA and scrambled siRNA from Invitrogen/Ambion. The siRNA sequences used to target exons 14 and 15 of the human STAT3 gene were antisense 5′-GGGAAGCAUCACAAUUGGCtc-3′ and sense 5′-GCCAAUUGUGAUGCUUCCCtt-3′. We mixed 30 nM of siRNA with siPORT NeoFX Transfection Agent (Invitrogen/Ambion) and transfected them by electroporation into CLL cells. Transfected cells were maintained in co-culture with mesenchymal stroma cells, and qRT-PCR was performed 48 hours after transfection.
Lentiviral STAT3-shRNA Infection
HEK 293FT cells were co-transfected with GFP-lentivirus STAT3 shRNA or with GFP-lentivirus empty vector and the packaging vectors pCMVδR8.2 and pMDG (generously provided by Dr. G. Inghirami, Torino, Italy), using the superfect transfection reagent (QIAGEN). 293FT cell culture medium was filtered through a 45-μm syringe, the lentivirus was concentrated by filtration through an Amicon Ultra centrifugal filter device (Millipore), and the concentrated supernatant was used to infect CLL cells. CLL cells (5 × 106/ml) were incubated in 6-well plates (Becton Dickinson) in 2 ml of DMEM supplemented with 10% FBS and transfected with 100 μl of viral supernatant. Polybrene (10 ng/ml) was added to the viral supernatant at a ratio of 1:1000 (v/v). Transfection efficiency was measured after 48 hours and ranged between 45% and 60% (calculated on the basis of the ratio of propidium iodide (PI)-negative/GFP-positive cells). RNA was isolated and prepared for qRT-PCR analysis as previously described (22).
Results
GM-CSFRα, but not GM-CSFRβ, was Detected in CLL Cells
GM-CSF was found to upregulate the expression of the cell-surface receptor CD20 in CLL cells (20), and administering GM-CSF to patients with CLL appeared to improve the therapeutic efficacy of the anti-CD20 antibody rituximab (4). Therefore, we wondered whether CLL cells express the GM-CSFR and whether GM-CSF induces signal transduction in these cells. To explore whether GM-CSF activates GM-CSFR-induced signaling pathways in CLL cells (24), we incubated PB CLL cells with GM-CSF and used Western immunoblotting to assess the levels of unphosphorylated and phosphorylated STAT-3, STAT5, AKT (also known as protein kinase B), and ERK-1/2. As shown in Supplemental Figure 1A, GM-CSF did not induce phosphorylation of STAT3 or STAT5, nor did it change the phosphorylation levels of AKT or ERK, both of which were reported to be constitutively activated in CLL cells of some, but not all, CLL patients (25). Furthermore, unlike the process in myeloid cells, in which GM-CSF binds to the GM-CSFR and activates JAK2 (24), GM-CSF did not induce JAK2 phosphorylation in CLL cells (Supplemental Figure 1B) and GM-CSF or GM-CSFRα neutralizing antibodies did not affect CLL cell surface CD20 levels as assessed by flow cytometry (Supplemental Figure 1C).
Because activation of the GM-CSFR depends on the interaction between the GM-CSFRα and GM-CSFRβ subunits (24), we sought to determine whether both subunits are present in CLL cells. Using flow cytometry, we found that GM-CSFRα (CD116), but not GM-CSFRβ (CD131), is present on the surface of PB CD19+ CLL cells (Figure 1A, left panel). The myeloid cell lines OCI/AML3 and HMC860 were used as positive controls, and the T-cell Jurkat cell line was used as a negative control (Figure 1A, right panel). To confirm these data, we performed PCR and a Western blot analysis of cellular extracts of CLL cells from 6 and 7 patients and of normal B-cells from 2 healthy donors. GM-CSFRα transcripts were readily detected whereas weak signals of GM-CSFRβ were detected in all CLL samples by 50-cycle PCR. GM-CSFRα, but not GM-CSFRβ, was detected in cell lysates of all CLL PB samples, whereas neither GM-CSFRα nor GM-CSFRβ was detected in normal B-cells (Figure 1B). To confirm these results, we analyzed PB low-density cells from another CLL patient by flow cytometry. As shown in Figure 1C, 58.7% of the cells co-expressed CD19, CD5, and CD116, suggesting that CLL cells, typically co-expressing CD19 and CD5 antigens, also express GM-CSFRα. We repeated this analysis on PB low-density cells from 5 additional, randomly selected CLL patients. Co-expression of CD19, CD5, and CD116 was detected in 16.5%, 24.5%, 62.7%, 39.1%, and 28.9% of the 5 patients' CLL cells. Although CD116 expression was likely underestimated because of quenching (26), these data demonstrate that GM-CSFRα is expressed on the surface of CLL cells. Thus, although CLL cells express GM-CSFRα, GM-CSF does not activate the GM-CSFR, likely because the GM-CSFRβ subunit is not present in CLL cells.
Figure 1.
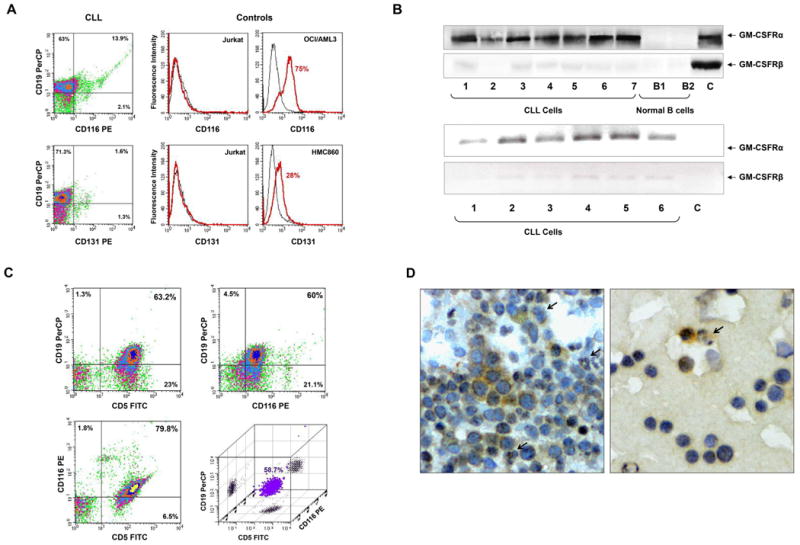
Chronic lymphocytic leukemia (CLL) cells express GM-CSFRα. (A) Flow cytometry analysis of peripheral blood (PB) CLL cells. Low-density cells from a patient with CLL were used. As depicted in the left panel, CLL CD19+ cells express CD116 but not CD131. PerCP is the isotype for CD19 and PE is the isotype controls for CD116 and CD131 are not shown. The positive and negative controls for CD116 and CD131 antibodies are depicted in the right panel. Jurkat cells were used as a negative control, and the myeloid lines OCI/AML3 and HMC860 were used as positive controls. (B) Upper panel: PCR analysis of low-density cells from different CLL patients and negative and normal B cells. As shown, GM-CSFRα was readily detected in all samples whereas very weak signals of GM-CSFRβ transcripts were detected by a 50 cycle touchdown PCR program. Lower panel: Western blot analysis of protein obtained from PB low-density cells of 7 patients with CLL and CD19+ B-lymphocytes from 2 healthy volunteers. As shown, GM-CSFRα, but not GM-CSFRβ, is detected in all CLL samples but not in normal B-cells. (C) Three-color flow cytometry analysis of PB low-density CLL cells. As shown, 63.2% of the cells co-expressed CD5 and CD19, 79.8% co-expressed CD5 and CD116, 60% co-expressed CD19 and CD116, and 58.7% co-expressed CD5, CD19 and CD116 (PerCP, peridinin-chlorophyll protein). (D) Immunohistochemistry staining with anti-GM-CSFRα antibodies of CLL BM clot (left panel) and smear (right panel). Several mononuclear cells, some of which exhibit typical CLL cell morphological features, stained positively for GM-CSFRα, whereas other lymphocytes did not. Both cytoplasmic and nuclear speckles are seen. Arrows depict positively stained granulocytes (positive control).
GM-CSFRα is Present Both in the Cytoplasm and Nucleus of CLL Cells
To confirm these data, we stained CLL BM smears with anti-GM-CSFRα antibodies. Similar to granulocytes, several mononuclear cells (some of which exhibited typical CLL cell morphology) expressed GM-CSFRα, and GM-CSFRα-positive speckles were detected both in the cytoplasm and nucleus of these cells (Figure 1D). To further delineate these findings, we performed confocal microscopy of PB CLL cells using GFP-conjugated mouse anti-human GM-CSFRα antibodies. Surprisingly, as in the CLL BM cells, GM-CSFRα was detected on the surface, to a lesser extent in the cytoplasm, and in the nucleus of CLL cells (Figure 2A). To confirm this observation, we prepared cytoplasmic and nuclear protein extracts of PB low-density CLL cells. S6 ribosomal protein was used as a positive control for the cytoplasmic fraction, and lamin B1 served as a positive control for the nuclear fraction. As shown in Figure 2C, GM-CSFRα was detected mainly in the nucleus of CLL cells obtained from 2 different patients.
Figure 2.

Detection of GM-CSFRα in the cytoplasm and nucleus of chronic lymphocytic leukemia (CLL) cells. (A, B) Confocal microscopic images of freshly isolated CLL cells that were cytospun and fixed on glass slides; the slides were stained with PE-conjugated anti-GM-CSFRα antibodies, as described in the Methods section. As shown (×400), GM-CSFRα is present in the cytosol and to a lesser extent in the nucleus of CLL cells. (C) Cytoplasmic and nuclear fractions were extracted from CLL cells of 2 patients and analyzed by Western immunoblotting using anti-GM-CSFRα antibodies. Adequate fractionation of cytoplasmic and nuclear extracts was confirmed using anti-S6 ribosomal protein and anti-lamin B1 antibodies. As shown, GM-CSFRα was detected in the nucleus of CLL cells.
STAT3 Activates the GM-CSFRα Promoter and Induces Production of GM-CSFRα Protein
Unlike normal B-lymphocytes (24), CLL cells produce GM-CSFRα, so we sought to determine what induces GM-CSFRα production in CLL cells. Recently, we found that CLL STAT3 is constitutively phosphorylated on serine 727 residues and activates a variety of genes (22). Typically, phosphorylated STAT3 binds to the γ-interferon activation sequence (GAS)-like element, also referred to as the sis-inducible element, located in the promoter region of various genes (27). Therefore, we performed a sequence analysis of the GM-CSFRα promoter and identified GAS-like elements, representing putative STAT3 binding sites (Supplemental Figure 2). We used MM1 as an in vitro model to test whether STAT3 induces GM-CSFRα (23). Unstimulated MM1 cells expressed low to undetectable levels of phosphotyrosine-STAT3, and exposure of MM1 cells to IL-6 induced STAT3 phosphorylation (28). We incubated MM1 cells with IL-6 and, using Western immunoblotting, established that IL-6 induced upregulation of both pSTAT3 and GM-CSFRα in a dose- and time-dependent manner (Figure 3A).
Figure 3.

IL-6-activated STAT3 activates the GM-CSFRα promoter and induces GM-CSFRα protein production in multiple myeloma cell line MM1 cells. (A) IL-6 phosphorylates STAT3 and upregulates GM-CSFRα protein levels in MM1 cells. Left panel: MM1 cells were incubated for 2 hours with increasing concentrations of IL-6. As determined by Western immunoblotting, IL-6 induced STAT3 tyrosine phosphorylation and upregulated GM-CSFRα protein levels in a dose-dependent manner. Right panel: MM1 cells were incubated with 20 ng/ml of IL-6 and harvested at different time points for further analysis. IL-6 induced STAT3 phosphorylation and upregulated GM-CSFRα protein levels in a time-dependent manner. (B) IL-6 induces GM-CSFRα promoter activity. A schematic diagram of the GM-CSFRα reporter fragments that were transfected into MM1 cells and maintained in the absence or presence of 20 ng/ml of IL-6 is presented on the left panel. The right panel depicts the mean ± standard deviation of the relative luciferase activity measured in 4 different experiments in which transfection efficiency of the truncated GM-CSFRα promoter constructs into MM1 cells was at least 45%. IL-6 induced luciferase activity in MM1 transfected with promoter fragments -4.0, -3.0, -2.5, and -0.5 Kb. Fragments -2.5 and 0.5 Kb yielded a similar IL-6-induced luciferase activity. IL-6 did not significantly increase luciferase activity in PGL4.17. (C) STAT3 binds to the promoter of GM-CSFRα and other STAT3-regulated genes. ChIP demonstrates that anti-STAT3 antibodies immunoprecipitated GM-CSFRα (left panel) and the STAT3-regulated genes STAT3, c-Myc, and WAF1/p21 (right panel). These genes were detected in MM1 cell nuclear extracts (Input) as well as in nuclear protein immunoprecipitated with anti-STAT3 antibodies in IL-6-stimulated MM1 cells, suggesting that STAT3 binds to known STAT3-regulated gene promoters and to the GM-CSFRα promoter. As shown in the left panel, binding of STAT3 to the GM-CSFRα promoter is detected by primers 1, 3, and 5, but not 2 and 4. The schematic diagram in the lower panel depicts the 3 active (1, 3, and 5) STAT3 binding sites. (D) EMSA results, using biotin-labeled and unlabeled DNA probes 1, 3, and 5 (Supplementary Table 2), show that IL-6-stimulated MM1 cell nuclear protein binds to the biotinylated DNA fragments 1, 3, and 5 and that the addition of the corresponding cold (unlabeled) DNA fragments or anti-STAT3 antibodies (but not IgG) attenuates the binding. The binding of IL-6-stimulated MM1 cell nuclear protein to biotinylated mutated DNA fragments 1, 3, and 5 (Table 2) is diminished or significantly reduced. (E) qRT-PCR demonstrates that STAT3-siRNA downregulates the mRNA levels of GM-CSFRα, and the STAT3-regulated genes STAT3, ROR1, c-Myc, cyclin D1, and p21. S18 mRNA (control) was not affected. (F) Western blot analysis shows that STAT3-siRNA, but not scrambled-siRNA or GAPDH, downregulates the protein levels of STAT3 and GM-CSFRα.
Then, using the MM1-inducible system, we cloned the proximal region of the human GM-CSFRα promoter (4.0 Kb). We generated a series of GM-CSFRα-truncated constructs (Figure 3B) and inserted them into a plasmid carrying a luciferase reporter. The location of the 5′ regions of these constructs is depicted in Supplemental Figure 2, and the primers used to amplify them are listed in Supplemental Table 2. All constructs were transfected into MM1 cells (transfection efficiency: 45%-50%; data not shown);following incubation of the constructs either with 20 ng/ml of IL-6 or with culture medium, we used a luciferase reporter assay and the Renilla reniformis system to assess their luciferase activity. Compared with results in control samples, exposure of MM1 cells to IL-6 significantly increased the luciferase activity of the truncated GM-CSFRα -4,000 (spanning from -4,012 to +23 bp), GM-CSFRα -3,000 (spanning from -2,965 to +23 bp), GM-CSFRα -2,500 (spanning from -2,517 to +23 bp), and to the same extent GM-CSFRα -500 (spanning from -496 to +23 bp). IL-6 did not affect the luciferase activity of PGL4.17 (Figure 3B). Among the serially truncated clones, GM-CSFRα -4,000 exerted the highest, and GM-CSFRα -2,500 and -500, the lowest IL-6-induced promoter activity (Figure 3B).
Because these data suggested that STAT3 activates the GM-CSFRα gene, we used ChIP to determine whether STAT3 indeed binds to GAS-like elements within the GM-CSFRα promoter. We obtained nuclear extracts of unstimulated and IL-6-stimulated MM1 cells and used ChIP and primers deduced from the luciferase activity data (Supplemental Table 2) to assess the binding of STAT3 to 5 putative STAT3-binding sites (Supplemental Figure 3). We found that primers 1, 3, and 5, but not primers 2 or 4, corresponding to the putative STAT3 binding sites depicted in Supplemental Figure 3, amplified DNA fragments that co-immunoprecipitated with anti-STAT3 antibodies (Figure 3C, left panel). These results suggest that STAT3 binds to sites 1, 3, and 5 of the GM-CSFRα promoter, shown at the bottom of Figure 3C. In addition, we found that anti-STAT3 antibodies co-immunoprecipitated DNA of the STAT3-regulated genes STAT3, c-Myc, and Waf1/P21, but not of the control gene RPL30 (Figure 3C, right panel), as previously reported (23). Then, to validate these findings, we performed EMSA. As shown in Figure 3D, MM1 nuclear protein bound the biotinylated GM-CSFRα promoter DNA fragments of regions 1, 3, and 5 (Supplemental Table 2), and access cold probes or anti-STAT3 antibodies attenuated this binding. In contrast, the binding of MM1 nuclear protein to the biotinylated mutant DNA fragments of regions 1, 3, or 5 (Supplemental Table 2) was diminished or significantly reduced, suggesting that activated STAT3 binds to the binding sites identified by our previous experiments.
Next, we sought to confirm that STAT3 activates the GM-CSFRα gene. We transfected MM1 cells with STAT3-siRNA, stimulated the cells with IL-6, and quantitated mRNA levels of GM-CSFRα, STAT3, and various STAT3-regulated genes by using relative qRT-PCR. As shown in Figure 3E, STAT3-siRNA downregulated mRNA levels of GM-CSFRα and the STAT3-regulated genes STAT3, Bcl-2, Bcl-XL, cyclin D1, c-Myc, and Waf1/p21. Furthermore, STAT3-siRNA, but not scrambled siRNA or GAPDH, downregulated STAT3 protein levels by 60% and GM-CSFRα protein levels by 70%suggesting that STAT3 activates the GM-CSFRα gene and induces production of GM-CSFRα protein by MM1 cells (Figure 3F).
We used IL-6 to induce STAT3 phosphorylation in MM1 cells. However, in CLL cells STAT3 is constitutively phosphorylated (22). We used ChIP to determine whether STAT3 activates GM-CSFRα in CLL cells. As shown in the left panel of Figure 4A, primers 1, 3, and 5, but not primers 2 or 4 (Supplemental Table 2), bound to CLL cell DNA fragments that were co-immunoprecipitated with anti-STAT3 antibodies (Supplemental Table 2; Supplemental Figure 3), suggesting that, as in MM1 cells, in CLL cells STAT3 bound the STAT3 binding sites 1, 3, and 5. In addition to co-immunoprecipitating DNA of GM-CSFRα, STAT3 co-immunoprecipitated DNA of the STAT3-regulated genes STAT3, c-Myc, p21/Waf1, ROR1, and VEGF, but not the control gene RPL30 (Figure 4A, right panel).
Figure 4.

STAT3 activates the GM-CSFRα promoter in chronic lymphocytic leukemia (CLL) cells. (A) STAT3 binds to the promoter of GM-CSFRα and other STAT3 regulated genes. ChIP demonstrates that anti-STAT3 antibodies immunoprecipitated GM-CSFRα (left panel) and the STAT3-regulated genes STAT3, c-Myc, WAF1/p21, ROR1, and VEGF (right panel) in CLL cells. These genes were detected in CLL nuclear extracts (Input) as well as in nuclear protein immunoprecipitated with anti-STAT3 antibodies, suggesting that STAT3 binds to known STAT3-regulated gene promoters and to the GM-CSFRα promoter. As shown in the left panel, binding of STAT3 to the GM-CSFRα promoter is detected by primers 1, 3, and 5, but not 2 and 4. (B) EMSA results, using biotin-labeled and unlabeled fragments 1, 3, and 5 and their corresponding mutant probes (Supplementary Table 2), show that CLL nuclear protein from 2 different patients binds to the biotinylated DNA fragments 1, 3, and 5 and that the addition of the corresponding cold (unlabeled) DNA fragments or anti-STAT3 antibodies (but not IgG) attenuates this binding. The binding of the labeled mutant DNA probes was diminished or significantly reduced compared with that of the unlabeled mutant DNA probes. (C) qRT-PCR demonstrates that lentiviral STAT3-shRNA downregulates mRNA levels of GM-CSFRα and the STAT3-regulated genes STAT3, Bcl-2, Bcl-XL, cyclin D1, c-Myc, and WAF1/p21. Expression of S18 mRNA (control) was not affected. (D) Western immunoblotting shows that STAT3-shRNA reduced GM-CSFRα protein levels. The empty vector reduced STAT3 protein levels by 10% and GM-CSFRα protein levels by 40%, whereas STAT3-shRNA downregulated STAT3 by 80% and GM-CSFRα protein levels by 90%.
To validate these findings, we performed EMSA. As shown in Figure 4B, the biotinylated DNA fragments of probes 1, 3, and 5 bound nuclear protein obtained from PB low-density CLL cells of 2 CLL patients, and excess cold probes or anti-STAT3 antibodies attenuated protein-DNA binding. Conversely, the binding of CLL nuclear protein to the mutated DNA biotinylated fragments (Supplemental Table 2) was diminished or significantly reduced, suggesting that, as in MM1 cells, activated STAT3 binds to the GM-CSFRα promoter binding sites 1, 3, and 5.
Finally, to confirm that STAT3 activates the GM-CSFRα promoter, PB low-density CLL cells were infected with lentiviral STAT3-shRNA (infection efficiency, 40%), and STAT3-regulated gene mRNA levels were quantitated by relative qRT-PCR. As shown in Figure 4C, infection with STAT3-shRNA reduced mRNA levels of GM-CSFRα and the STAT3-regulated genes STAT3, Bcl-2, Bcl-XL, cyclin D1, c-Myc, and Waf1/p21. Furthermore, unlike the empty vector that reduced STAT3 levels by 10% and GM-CSFRα levels by 40%, STAT3-shRNA downregulated the protein levels of STAT3 by 80% and of GM-CSFRα by 90% (Figure 4D).
The Full-Length GM-CSFRα Subunit is Present in the Cytosol and the Nucleus
The GM-CSFRα subunit is largely extracellular but contains a 54-amino acid intracytoplasmic tail (Figure 5A). We detected GM-CSFRα on the surface, in the cytosol, and in the nucleus of CLL cells. However, we have not established whether our GM-CSFRα antibodies detected the extracellular domain exclusively or also detected the intracellular domain. Removal of the intracytoplasmic tail of the GM-CSFRα subunit was reported to deactivate the GM-CSF-induced activity of GM-CSFR (29). To reveal the biological activity of GM-CSFRα in unstimulated cells lacking the GM-CSFRβ subunit, it was crucial to determine whether part of or the entire GM-CSFRα subunit was present in the cytosol and/or the nucleus. Therefore, GFP-tagged full-length GM-CSFRα (Supplemental Table 2) (amino acids 1–400) and its extracellular (amino acids 1–323) and intracellular (amino acids 347–400) regions (Figure 5A) were transfected into 293FT cells. The protein of transfected 293FT cells was immunoprecipitated with anti-GFP antibodies and, as shown in Figure 5B–D, full-lengthGM-CSFRα and its extracellular and intracellular domains were detected in total cell extract as well as in the cytosol and the nucleus. However, these anti-GM-CSFRα antibodies bound the extracellular, not the intracellular, GM-CSFRα domain (Figure 5E) and therefore detected both the full-length GM-CSFRα receptor and its extracellular domain in the cytosol and the nucleus (Figure 5F) of GM-CSFRα-transfected 293FT cells. Taken together, these data suggest that our anti-GM-CSFRα antibodies detected the extracellular domain of GM-CSFRα and that the entire GM-CSFRα subunit migrated to the cytosol and the nucleus.
Figure 5.

The full-length GM-CSFRα subunit is detected in the cytosol and the nucleus of GM-CSFRα-transfected 293TF cells. (A) Schematic diagram of GFP-tagged full-length GM-CSFRα and its truncated extracellular and intracellular fragments. The white box denotes the membrane domain. (B) The full-length GM-CSFRα subunit and its extracellular and intracellular fragments were successfully transfected into 293TF cells. Whole-cell protein extract of 293TF cells transfected with GFP-tagged full-length and its truncated extracellular and intracellular fragments were immunoprecipitated with rabbit anti-GFP antibodies; the GFP-tagged protein was detected with mouse anti-GFP antibodies. As shown, full-length-tagged GM-CSFRα and its tagged extracellular and intracellular regions were detected. Differences in signal intensity may represent differences in protein expression and/or transfection efficiency. I.P., immunoprecipitate; B, beads (control). (C) Similarly, confocal microscopy studies detected GFP-tagged full-length GM-CSFRα and its extracellular and intracellular fragments in the cytosol and the nucleus of the transfected 293FT cells. (D) The transfected full-length GM-CSFRα subunit and its extracellular and intracellular regions were detected in the cytosol and the nucleus of 293TF cells. Western immunoblot analysis detected GFP-tagged cytoplasmic (S6-positive, lamin B-negative) and nuclear (lamin B-positive, S6-negative) full-length, as well as extracellular and intracellular constructs of transfected GM-CSFRα. (E) GM-CSFRα antibodies detect the extracellular but not the intracellular region of the GM-CSFRα subunit. (F) Cytosolic (C) and nuclear (N) GFP-tagged full-length GM-CSFRα and its extracellular domain pulled down by GFP antibodies, but not by beads (B; control), bound GM-CSFRα antibodies.
Analysis of GM-CSFRα-Linked RNA Fragments and Proteins
The function of and biological effects of GM-CSF-stimulated GM-CSFR following ligand binding and dimerization of its GM-CSFRα and GM-CSFRβ subunits has been well established (24). However, little is known about the function of GM-CSFRα in the absence of the GM-CSFRβ subunit. To explore the role of GM-CSFRα, we transfected GFP or the GFP-tagged GM-CSFRα gene into 293FT cells, and, following overnight incubation, cell extract was immunoprecipitated with anti-GFP antibody-coated agarose beads. Both RNA and protein were extracted from the immunoprecipitate and analyzed.
The 1-way ANOVA program identified 7,220 RNA transcripts that co-immunoprecipitated with GFP-tagged GM-CSFRα and 1,593 RNA transcripts that co-immunoprecipitated with GFP-tagged GM-CSFRα, but not with GFP; the top 30 of these transcripts are depicted in the heat map presented in Figure 6. The GM-CSFRα-attached RNA transcripts at the top of the list are likely involved in active translation of processed GM-CSFRα mRNA. At the top of the list were also the SKI and MAFA oncogenes, 2 serine/threonine kinases, and 3 microRNAs (Table 1). Other top-list RNA transcripts included DEFT1P2 and DEFP1P, which encode proteins that belong to members of the death effector domains (DEDs) family, known to have a role in the control of programmed cell death and in the regulation of a variety of signal transduction pathways (30, 31). Pathways enriched with transcripts that co-immunoprecipitated with GFP-tagged GM-CSFRα but not with GFP included the JAK-STAT signaling and hematopoietic lineage pathways (Table 2).
Figure 6.

The top 30 RNA transcripts that co-immunoprecipitated with GFP-tagged GM-CSFRα. As described above, 293FT cells were transfected with a plasmid containing either GFP-tagged GM-CSFR-α or tagged GFP as controls. After immunoprecipitation with anti-GFP antibodies, RNA was extracted, sequenced, and aligned to hg.19 human genome. The 30 top RNA transcripts that differentially expressed between GFP-tagged GM-CSFRα and GFP are depicted.
Table 1. The top 10 RNA transcripts co-immunoprecipitated with GFP-tagged GM-CSFRα.
| Gene name | Fold change | Description |
|---|---|---|
| CSF2RA | 170.396 | Colony-stimulating factor 2 receptor, alpha, low-affinity (granulocyte-macrophage) |
| MIR3687 | 77.4649 | |
| MIR3648 | 67.7411 | |
| SKI | 7.39956 | v-SKI sarcoma viral oncogene homolog (avian) |
| TAF4 | 8.96483 | TAF4 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 135kDa |
| STK38 | 21.5572 | serine/threonine kinase 38 |
| MIR663 | 44.7745 | |
| SGK494 | 21.1261 | uncharacterized serine/threonine-protein kinase SgK494 |
| MAFA | 12.9538 | v-maf musculoaponeurotic fibrosarcoma oncogene homolog A (avian) |
| FLJ45445 | 17.3895 | hypothetical LOC399844 (unknown function) |
Table 2. Pathways enriched with RNA transcripts co-precipitated with GFP-tagged GM-CSFR-α.
| Pathway name | # of probe sets | P value |
|---|---|---|
| Hematopoietic cell lineage | 84 | 2.5 × 10-31 |
| JAK-STAT signaling pathways | 147 | 3.4 × 10-31 |
| Cytokine-Cytokine receptor interaction | 250 | 3.9 × 10-31 |
| Pathways in cancer | 328 | 1.3 × 10-29 |
| Maturity onset diabetes of the young | 25 | 10 × 10-17 |
| Vitamin B6 metabolism | 6 | 1.2 × 10-13 |
| Riboflavin metabolism | 12 | 6.6 × 10-13 |
| Nicotinate and nicotinamide metabolism | 26 | 3 × 10-12 |
Then, we used mass spectrometry to identify GM-CSFRα-interacting proteins. In addition to various keratins, heat shock proteins, and GM-CSFRα, Krüppel-associated protein (or KRAB zinc finger-associated protein 1[KAP1]), also known as transcription intermediary factor [TIF]-1β or tripartite motif-containing protein [TRIM] (28) (32) and interferon stimulated gene (ISG)-15 (also known as TRIM25) were pulled down together with GM-CSFRα (Supplemental Figure 4).
GM-CSFRα Provides CLL Cells with Survival Advantage
Because KAP1-siRNA was found to upregulate STAT3 transcription and induce accumulation of phosphoserine STAT3 in the nucleus (33) and because STAT3 is constitutively phosphorylated on serine 727 residues in CLL cells (22), we sought to explore how GM-CSFRα affects the transcription of STAT3. Using immunoprecipitation with anti-GM-CSFRα antibodies, we confirmed that KAP1 co-immunoprecipitated with GM-CSFRα (Figure 7A). Then, to determine how GM-CSFRα affects STAT3-DNA binding, we performed EMSA. As shown in Figure 7B, we found that transfection of CLL cells with GM-CSFRα-siRNA reduced CLL nuclear protein-DNA binding, whereas anti-STAT3 antibodies completely prevented the binding, as previously reported (22). Taken together, these data suggest that GM-CSFRα binds the negative regulator of STAT3 transcription KAP1 and as a result, enhances the transcriptional activity of STAT3.
Figure 7.

GM-CSFRα protects cells from apoptotic cell death. (A) KAP1 co-immunoprecipitates with anti-GM-CSFRα antibodies. Chronic lymphocytic leukemia (CLL) nuclear extracts from 3 CLL patients were immunoprecipitated (I.P.) with anti-GM-CSFRα antibodies using protein A–agarose beads. Incubation with beads only (B) was used as a negative control. The immune complex was separated by SDS-PAGE and analyzed by Western immunoblotting using the indicated antibodies. (B) GM-CSFRα-siRNA reduced the binding of CLL nuclear protein to the labeled STAT3 DNA binding-site. EMSA was conducted using a biotin-labeled DNA probe harboring a STAT3 binding-site. The binding was significantly attenuated by excess unbiotinylated (cold) probes or anti-STAT3 antibodies. The binding of nuclear extract from CLL cells transfected with GM-CSFRα-siRNA was significantly reduced as compared with its transfection control cells (GAPDH), and the binding was reversed by excess cold probes. (C) Upper panel: GM-CSFRα co-immunoprecipitates with ISG15. Nuclear extracts from cells of 3 CLL patients were immunoprecipitated (I.P.) with anti-ISG15 antibodies using protein A–agarose beads. Incubation with beads only (B) was used as a negative control. Lower panel: GM-CSFRα-siRNA downregulates IκB protein levels. Low-density peripheral blood (PB) CLL cells were transfected with GM-CSFRα-siRNA or GAPDH, and IκB protein levels were assessed by Western immunoblotting. (D) Transfection of CLL cells with GM-CSFRα-siRNA enhances NF-κB-DNA binding. EMSA results of CLL nuclear protein and a labeled NF-κB DNA-binding probe demonstrate a greater binding of protein from CLL cells transfected with GM-CSFRα-siRNA than from CLL cells transfected with GAPDH. Excess cold probes partially reversed the binding, proving its specificity. (E–G) GM-CSFRα-siRNA induced apoptosis in CLL cells. (E) CLL cells were co-transfected with GFP and GAPDH or GM-CSFRα-siRNA at a transfection efficiency of 33.5%, as assessed by flow cytometry. (F) Transfection of CLL cells with GM-CSFRα-siRNA but not with GAPDH downregulates GM-CSFRα mRNA levels as assessed by qRT-PCR of 4 different experiments (left panel) and GM-CSFRα protein levels (right panel). (G) A higher apoptosis rate was observed in CLL cells transfected with GM-CSFRα-siRNA than in cells transfected with GAPDH after incubation for 24 hours in 10% FCS. Data obtained from 2 different patients are depicted. (H) GM-CSFRα-siRNA protects multiple myeloma cell line MM1 cells from dexamethasone-induced apoptosis. MM1 cells were transfected with GFP or GFP-tagged GM-CSFRα at a transfection efficiency of 40% (a, b). The transfected cells were incubated for 48 hours with or without 20 μmol/l of dexamethasone. Apoptosis rates were assessed by flow cytometry using Annexin V and PI. This experiment was repeated 3 times. Rates of apoptosis are depicted as mean ± SD. To assess the statistical difference between the variables, we used Student's t-test.
The covalent binding of ISG15 to various proteins, known as ISGylation, negatively controls the NF-κB pathway (34); because NF-κB is constitutively activated in CLL (35) and because ISG15 binds to and co-immunoprecipitates with GM-CSFRα (Fig. 7C), we wondered how GM-CSFRα affects the activation of NF-κB in CLL cells. We transfected CLL cells with GM-CSFRα-siRNA and, by using Western immunoblotting, found that GM-CSFRα-siRNA downregulated the levels of IκB (Figure 7C). Because ubiquitination of IκB enables NF-κB to shuttle to the nucleus and bind to DNA, we performed EMSA. We found that transfection of CLL cells with GM-CSFRα-siRNA enhances NF-κB-DNA binding (Figure 7D), suggesting that, as in other proteins (34), GM-CSFRα ISGylation inhibits the NF-κB pathway.
As described above, we found that GM-CSFRα inversely affects 2 transcription factors known to be major regulators of cell survival (36) by enhancing the activity of STAT3 and contributing to the inhibition of NF-κB. To decipher the net effect of GM-CSFRα, we transfected PB low-density CLL cells with GM-CSFRα-siRNA and assessed the cellular apoptosis rate using PI and Annexin V. At a transfection rate of 33.5% to 55% (Figure 7E), GM-CSFRα-siRNA significantly downregulated GM-CSFRα mRNA and protein levels (Figure 7F) and induced CLL cell apoptosis (Figure 7G), suggesting that GM-CSFRα exerts an overall anti-apoptotic effect. CLL cells from four randomly selected patients were transfected with GM-CSFRα-siRNA and the rate of cellular apoptosis was assessed by Annexin V/PI staining using flow cytometry. The percent of apoptosis in GM-CSFRα-siRNA-transfected cells was higher than that of GAPDH-transfected cells by 20.4%, 44.6%, 18.7%, and 12.3%, respectively (P = 0.036 (paired t-test)). Figure 7F depicts results of Patients 1 and 2 results of patients 3 and 4 are not shown. To confirm that GM-CSFRα provides CLL cells with a survival advantage, we transfected MM1 cells with GFP or GM-CSFRα and incubated them for 48 hours with or without 20 μmol/l of dexamethasone, an agent known to induce apoptosis in MM1 cells (37). As shown in Figure 7H, dexamethasone induced apoptosis at a significantly higher rate in GFP-transfected cells than in GM-CSFRα-transfected cells, suggesting that GM-CSFRα protected MM1 cells from apoptosis.
Discussion
In this study we show that unlike in normal B lymphocytes in CLL cells constitutively activated STAT3 activates the GM-CSFRα promoter, and that GM-CSFRα, present on the cell surface, in the cytosol and in the nucleus provides the cells with survival advantage in a ligand-independent manner.
Several investigators have previously studied the expression GM-CSFR by normal and neoplastic B-lymphocytes. Using sensitive triple-layer immunophenotypic techniques, Till et al. (21) found that both the α- and β-chains of the GM-CSFR are expressed on hairy cells (HCs) and myelomatous plasma cells (PCs) but not on CLL or prolymphocytic leukemia (PLL) lymphocytes. In that study, the GM-CSFR was demonstrable on normal PCs in tonsils but not on either activated or resting tonsillar B-cells or on circulating normal B-lymphocytes. Our data confirm that GM-CSFR protein is not present in normal B-lymphocytes; however, we found that CLL cells express the α but not the β subunit of GM-CSFR. Like other investigators (38), we found that CLL cells express GM-CSFRα mRNA. We also detected GM-CSFRα, but not GM-CSFRβ, protein on the cell surface, in the cytosol, and in the nucleus of CLL cells. B1a cells were recently found to produce GM-CSF (5); however, GM-CSF does not affect any subtype of normal B-lymphocytes that lack the GM-CSFR. Although CLL cells are thought to be B1 cells, we found that GM-CSF did not elicit signal transduction in B-cell CLL cells, probably because CLL cells lack the β subunit of the GM-CSFR that associates with JAK2 to initiate signal transduction (17).
We detected the full-length GM-CSFRα in the cytosol and nucleus of CLL cells. Nuclear localization of cell-surface receptors has been reported by several investigators. Direct nuclear translocation of full-length growth factor receptors or fragments of them has been described in other cell types (39, 40), and importin-β and the nuclear pore protein Nup358 (40) were found to be involved in the nuclear transport process. Whether the karyopherin or another transport system is involved in shuttling GM-CSFRα to the nuclei of CLL cells, remains to be determined.
GM-CSFRα has been detected in various tissues, and ligation of GM-CSFRα in the absence of the β subunit (41) was reported to elicit effects in other nonhematopoietic cell lineages. Prostate carcinoma cells express GM-CSFRα (42), and GM-CSFRα is distributed throughout the CNS, predominantly in neuronal cells (43). A reduction in GM-CSFRα protein levels in the nuclei of the hippocampus, cortex, thalamus, and brainstem is thought to contribute to neurodegeneration in Alzheimer's disease (44). However, the factors responsible for GM-CSFRα expression in these cells have not been identified.
Unlike in normal B-lymphocytes, STAT3 is constitutively phosphorylated on serine 727 residues in CLL cells (22, 45) and is biologically active (22). Therefore, we sought to determine whether STAT3 affects the expression and protein production of GM-CSFRα in CLL cells. We performed a sequence analysis of the GM-CSFRα promoter and identified GAS-like elements, suggesting that STAT3 binds to the GM-CSFRα gene promoter in CLL cells. In MM1 cells, IL-6 induced both STAT3 and GM-CSFRα protein expression. Therefore, we used a luciferase assay to clone the GM-CSFRα promoter and used ChIP and EMSA to identify STAT3 binding sites in both MM1 and CLL cells. Transfection of MM1 and primary CLL cells with STAT3-siRNA or STAT3-shRNA downregulated GM-CSFRα mRNA and protein levels, confirming that STAT3 binds to the GM-CSFRα promoter and activates the GM-CSFRα gene in CLL cells.
In human embryos, GM-CSF was found to regulate cell viability through a mechanism independent of the β subunit (46). However the role of GM-CSFRα in CLL cells is unknown. GM-CSFRα does not have a DNA-binding site, and the function GM-CSFRα fulfills in the nucleus and/or cytosol is unknown. Several investigators demonstrated that mRNAs are co-regulated by one or more sequence-specific RNA-binding proteins that orchestrate their splicing, export, stability, localization, and translation and that posttranscriptional processes depend largely on the functions of RNA-binding proteins (reviewed in ref.(47)). RNA transcripts that co-immunoprecipitated with GM-CSFRα are those involved in GM-CSFRα translation of oncogenes (SKI and MAFA), of serine/threonine kinases, of microRNAs, and of members of the DED family (30, 31), suggesting that GM-CSFRα is involved in regulating cell-survival pathways.
Indeed, the proteins that co-immunoprecipitated with GM-CSFRα, KAP1 and ISG15, regulate cellular viability. KAP1 is thought to regulate the dynamic organization of chromatin structure by modifying epigenetic patterns and chromatin compaction (32); KAP1 is a transcriptional regulator of the IL-6/STAT3 signaling pathway, as KAP1 siRNA enhanced IL-6-induced STAT3-dependent transcription and gene expression (33). Furthermore, reduction of KAP1 expression resulted in the marked accumulation of phosphoserine STAT3 in the nucleus, a modification that regulates its transcriptional activation (33). We found that GM-CSFRα binds KAP1 and enhances the transcriptional activity of STAT3. In a previous report we showed that STAT3 provides CLL cells with survival advantage (22). Our current data suggest that GM-CSFRα enhances this effect.
IFNs promote the activation of a genetic program that controls the expression of hundreds of genes named ISGs. To the ISGs family belong all the components of the molecular machinery that modifies proteins by the addition of the ubiquitin-like protein ISG15. This process of covalent conjugation to different proteins is known as ISGylation. Expression of the ISGylation system suppressed NF-κB activation via TRAF6/TAK1 and reduced the level of polyubiquitinated TRAF6, suggesting that ISGylation negatively controls the NF-κB pathway (34). However, ISG15 upregulation was also associated with telomere shortening. Apparently, this regulation is unrelated to IFNs, TP53, and the DNA damage/senescence response. Overall, the ISGylation system and ISG15 in particular could provide some advantages to cancer cells, since a negative selection was rarely observed (48). We found that similar to other proteins (34), GM-CSFRα ISGylation inhibited the NF-κB pathway. The NF-κB pathway provides CLL cells with pro-survival signals (49). Nevertheless, overexpression of GM-CSFRα protected MM1 cells from dexamethasone-induced apoptosis, whereas transfection of CLL cells with GM-CSFRα-siRNA induced CLL cell apoptosis, suggesting that the STAT3-induced pro-survival effects overcome reduced ubiquitination of IκB. We have previously reported that in CLL cells unphophorylated STAT3 activates NF-κB in an IκB-independent manner (35). Therefore, as in neuronal (43) and embryonic (46) cells, GM-CSFRα provides CLL cells with a survival advantage.
Taken together, our data suggest that the α subunit of the GM-CSFR possesses GM-CSF-unrelated biological activities that protect CLL cells from apoptosis. Whether selective targeting of GM-CSFRα might be a useful therapeutic strategy in CLL remains to be determined.
Supplementary Material
Implications.
Constitutively activate STAT3 induces the expression of GM-CSFRα and protects CLL cells from apoptosis, suggesting that inhibition of STAT3 or GM-CSFRα may benefit patients with CLL.
Acknowledgments
We thank Jill Delsigne, Ph.D., and Diane S. Hackett for editing our manuscript.
Grant support: This study was supported by a grant from the CLL Global Research Foundation and the National Institutes of Health through MD Anderson's Cancer Center Support Grant CA016672.
Footnotes
Online Supplemental Material: See Supplementary data of patient characteristics (Table S1), primers used in this study ((Table S2), the effect of GM-CSF on CLL cell signaling ((Fig. S1), the sequence of the 5′-flanking region of the human GM-CSFRα (Fig. S2), and supplementary methods for the expression of human GM-CSFR-α in 293FT cells, epitp mapping of GM-CSFR-α antibodies, photoactivatable ribonucleoside enhanced crosslink and immunoprecipitation (PAR-CLIP), bioinformatics data analysis, mass spectrometry analysis, and apoptosis assay.
Disclosure of Potential Conflict of Interest: The authors have no potential conflict of interest.
Authors' Contributions: Conception and design: Z. Estrov, P. Li, D. Harris, A. Ferrajoli
Development of methodology: P. Li, D. Harris, Z. Liu, Y., Wang, Bueso-Ramos, C., I. Hazan-Halevy, S. Grgurevic
Acquisition of data (patients' samples and clinical data): M. Keating, W. Wierda, S. O'Brien, J. Burger, S. Faderl
Analysis of data and interpretation (e.g., statistical analysis, biostatistics, computational analysis): Z. Estrov, U. Rozovski, P. Li, D. Harris
Writing, review, and/or revision of the manuscript: Z. Estrov, P. Li, D. Harris, U. Rozoveski
Administrative, technical, or material support (i.e., obtaining data from database and organizing data): M. Keating, W. Wierda, S. O'Brien, S. Faderl
Study supervision: Z. Estrov
References
- 1.Tam CS, Keating MJ. Chemoimmunotherapy of chronic lymphocytic leukemia. Nat Rev Clin Oncol. 2010;7:521–32. doi: 10.1038/nrclinonc.2010.101. [DOI] [PubMed] [Google Scholar]
- 2.Cramer P, Hallek M. Prognostic factors in chronic lymphocytic leukemia-what do we need to know? Nat Rev Clin Oncol. 2011;8:38–47. doi: 10.1038/nrclinonc.2010.167. [DOI] [PubMed] [Google Scholar]
- 3.Cartron G, Zhao-Yang L, Baudard M, Kanouni T, Rouille V, Quittet P, et al. Granulocyte-macrophage colony-stimulating factor potentiates rituximab in patients with relapsed follicular lymphoma: results of a phase II study. J Clin Oncol. 2008;26:2725–31. doi: 10.1200/JCO.2007.13.7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrajoli A. Incorporating the use of GM-CSF in the treatment of chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:514–6. doi: 10.1080/10428190902763541. [DOI] [PubMed] [Google Scholar]
- 5.Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335:597–601. doi: 10.1126/science.1215173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guthridge MA, Stomski FC, Thomas D, Woodcock JM, Bagley CJ, Berndt MC, et al. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells. 1998;16:301–13. doi: 10.1002/stem.160301. [DOI] [PubMed] [Google Scholar]
- 7.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–8. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 8.Barouch DH, Santra S, Tenner-Racz K, Racz P, Kuroda MJ, Schmitz JE, et al. Potent CD4+ T cell responses elicited by a bicistronic HIV-1 DNA vaccine expressing gp120 and GM-CSF. J Immunol. 2002;168:562–8. doi: 10.4049/jimmunol.168.2.562. [DOI] [PubMed] [Google Scholar]
- 9.Walker F, Burgess AW. Specific binding of radioiodinated granulocyte-macrophage colony-stimulating factor to hemopoietic cells. Embo J. 1985;4:933–9. doi: 10.1002/j.1460-2075.1985.tb03721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park LS, Friend D, Gillis S, Urdal DL. Characterization of the cell surface receptor for human granulocyte/macrophage colony-stimulating factor. J Exp Med. 1986;164:251–62. doi: 10.1084/jem.164.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–44. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 12.Gearing DP, King JA, Gough NM, Nicola NA. Expression cloning of a receptor for human granulocyte-macrophage colony-stimulating factor. Embo J. 1989;8:3667–76. doi: 10.1002/j.1460-2075.1989.tb08541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashida K, Kitamura T, Gorman DM, Arai K, Yokota T, Miyajima A. Molecular cloning of a second subunit of the receptor for human granulocyte-macrophage colony-stimulating factor (GM-CSF): reconstitution of a high-affinity GM-CSF receptor. Proc Natl Acad Sci U S A. 1990;87:9655–9. doi: 10.1073/pnas.87.24.9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirza S, Walker A, Chen J, Murphy JM, Young IG. The Ig-like domain of human GM-CSF receptor alpha plays a critical role in cytokine binding and receptor activation. Biochem J. 2010;426:307–17. doi: 10.1042/BJ20091745. [DOI] [PubMed] [Google Scholar]
- 15.Sakamaki K, Miyajima I, Kitamura T, Miyajima A. Critical cytoplasmic domains of the common beta subunit of the human GM-CSF, IL-3 and IL-5 receptors for growth signal transduction and tyrosine phosphorylation. Embo J. 1992;11:3541–9. doi: 10.1002/j.1460-2075.1992.tb05437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muto A, Watanabe S, Itoh T, Miyajima A, Yokota T, Arai K. Roles of the cytoplasmic domains of the alpha and beta subunits of human granulocyte-macrophage colony-stimulating factor receptor. J Allergy Clin Immunol. 1995;96:1100–14. doi: 10.1016/s0091-6749(95)70195-8. [DOI] [PubMed] [Google Scholar]
- 17.Brizzi MF, Zini MG, Aronica MG, Blechman JM, Yarden Y, Pegoraro L. Convergence of signaling by interleukin-3, granulocyte-macrophage colony-stimulating factor, and mast cell growth factor on JAK2 tyrosine kinase. J Biol Chem. 1994;269:31680–4. [PubMed] [Google Scholar]
- 18.Selleri S, Deola S, Pos Z, Jin P, Worschech A, Slezak SL, et al. GM-CSF/IL-3/IL-5 receptor common beta chain (CD131) expression as a biomarker of antigen-stimulated CD8+ T cells. J Transl Med. 2008;6:17. doi: 10.1186/1479-5876-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farren TW, Giustiniani J, Liu FT, Tsitsikas DA, Macey MG, Cavenagh JD, et al. Differential and tumor-specific expression of CD160 in B-cell malignancies. Blood. 2011;118:2174–83. doi: 10.1182/blood-2011-02-334326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venugopal P, Sivaraman S, Huang XK, Nayini J, Gregory SA, Preisler HD. Effects of cytokines on CD20 antigen expression on tumor cells from patients with chronic lymphocytic leukemia. Leuk Res. 2000;24:411–5. doi: 10.1016/s0145-2126(99)00206-4. [DOI] [PubMed] [Google Scholar]
- 21.Till KJ, Burthem J, Lopez A, Cawley JC. Granulocyte-macrophage colony-stimulating factor receptor: stage-specific expression and function on late B cells. Blood. 1996;88:479–86. [PubMed] [Google Scholar]
- 22.Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–63. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Harris D, Liu Z, Liu J, Keating M, Estrov Z. Stat3 activates the receptor tyrosine kinase like orphan receptor-1 gene in chronic lymphocytic leukemia cells. PLoS One. 2010;5:e11859. doi: 10.1371/journal.pone.0011859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114:1289–98. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Efremov DG, Gobessi S, Longo PG. Signaling pathways activated by antigen-receptor engagement in chronic lymphocytic leukemia B-cells. Autoimmun Rev. 2007;7:102–8. doi: 10.1016/j.autrev.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 26.Wood B. 9-color and 10-color flow cytometry in the clinical laboratory. Arch Pathol Lab Med. 2006;130:680–90. doi: 10.5858/2006-130-680-CACFCI. [DOI] [PubMed] [Google Scholar]
- 27.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 28.Amit-Vazina M, Shishodia S, Harris D, Van Q, Wang M, Weber D, et al. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br J Cancer. 2005;93:70–80. doi: 10.1038/sj.bjc.6602637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polotskaya A, Zhao YM, Lilly MB, Kraft AS. Mapping the Intracytoplasmic Regions of the Alpha-Granulocyte-Macrophage Colony-Stimulating Factor-Receptor Necessary for Cell-Growth Regulation. Journal of Biological Chemistry. 1994;269:14607–13. [PubMed] [Google Scholar]
- 30.Alcivar A, Hu S, Tang J, Yang X. DEDD and DEDD2 associate with caspase-8/10 and signal cell death. Oncogene. 2003;22:291–7. doi: 10.1038/sj.onc.1206099. [DOI] [PubMed] [Google Scholar]
- 31.Valmiki MG, Ramos JW. Death effector domain-containing proteins. Cell Mol Life Sci. 2009;66:814–30. doi: 10.1007/s00018-008-8489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem. 2011;286:26267–76. doi: 10.1074/jbc.R111.252569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuruma R, Ohbayashi N, Kamitani S, Ikeda O, Sato N, Muromoto R, et al. Physical and functional interactions between STAT3 and KAP1. Oncogene. 2008;27:3054–9. doi: 10.1038/sj.onc.1210952. [DOI] [PubMed] [Google Scholar]
- 34.Minakawa M, Sone T, Takeuchi T, Yokosawa H. Regulation of the nuclear factor (NF)-kappaB pathway by ISGylation. Biol Pharm Bull. 2008;31:2223–7. doi: 10.1248/bpb.31.2223. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Hazan-Halevy I, Harris DM, Li P, Ferrajoli A, Faderl S, et al. STAT-3 activates NF-kappaB in chronic lymphocytic leukemia cells. Mol Cancer Res. 2011;9:507–15. doi: 10.1158/1541-7786.MCR-10-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 37.Agata N, Nogi H, Milhollen M, Kharbanda S, Kufe D. 2-(8-Hydroxy-6-methoxy-1-oxo-1H-2-benzopyran-3-yl) propionic acid, a small molecule isocoumarin, potentiates dexamethasone-induced apoptosis of human multiple myeloma cells. Cancer Res. 2004;64:8512–6. doi: 10.1158/0008-5472.CAN-04-2626. [DOI] [PubMed] [Google Scholar]
- 38.Falt S, Merup M, Gahrton G, Lambert B, Wennborg A. Identification of progression markers in B-CLL by gene expression profiling. Exp Hematol. 2005;33:883–93. doi: 10.1016/j.exphem.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–8. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 40.Reilly JF, Maher PA. Importin beta-mediated nuclear import of fibroblast growth factor receptor: role in cell proliferation. J Cell Biol. 2001;152:1307–12. doi: 10.1083/jcb.152.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding DX, Rivas CI, Heaney ML, Raines MA, Vera JC, Golde DW. The alpha subunit of the human granulocyte-macrophage colony-stimulating factor receptor signals for glucose transport via a phosphorylation-independent pathway. Proc Natl Acad Sci U S A. 1994;91:2537–41. doi: 10.1073/pnas.91.7.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rivas CI, Vera JC, Delgado-Lopez F, Heaney ML, Guaiquil VH, Zhang RH, et al. Expression of granulocyte-macrophage colony-stimulating factor receptors in human prostate cancer. Blood. 1998;91:1037–43. [PubMed] [Google Scholar]
- 43.Reed JA, Clegg DJ, Smith KB, Tolod-Richer EG, Matter EK, Picard LS, et al. GM-CSF action in the CNS decreases food intake and body weight. J Clin Invest. 2005;115:3035–44. doi: 10.1172/JCI25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ridwan S, Bauer H, Frauenknecht K, von Pein H, Sommer CJ. Distribution of granulocyte-monocyte colony-stimulating factor and its receptor alpha-subunit in the adult human brain with specific reference to Alzheimer's disease. J Neural Transm. 2012;119:1389–406. doi: 10.1007/s00702-012-0794-y. [DOI] [PubMed] [Google Scholar]
- 45.Frank DA, Mahajan S, Ritz J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest. 1997;100:3140–8. doi: 10.1172/JCI119869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sjoblom C, Wikland M, Robertson SA. Granulocyte-macrophage colony-stimulating factor (GM-CSF) acts independently of the beta common subunit of the GM-CSF receptor to prevent inner cell mass apoptosis in human embryos. Biology of Reproduction. 2002;67:1817–23. doi: 10.1095/biolreprod.101.001503. [DOI] [PubMed] [Google Scholar]
- 47.Keene JD. Minireview: global regulation and dynamics of ribonucleic Acid. Endocrinology. 2010;151:1391–7. doi: 10.1210/en.2009-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sgorbissa A, Brancolini C. IFNs, ISGylation and cancer: Cui prodest? Cytokine Growth Factor Rev. 2012;23:307–14. doi: 10.1016/j.cytogfr.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 49.Hewamana S, Alghazal S, Lin TT, Clement M, Jenkins C, Guzman ML, et al. The NF-kappaB subunit Rel A is associated with in vitro survival and clinical disease progression in chronic lymphocytic leukemia and represents a promising therapeutic target. Blood. 2008;111:4681–9. doi: 10.1182/blood-2007-11-125278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.