

Abstract
Background:
We report a case of a neonate with proximal spinal muscular atrophy (SMA) type 1 (also known as Werdnig-Hoffmann disease or severe infantile acute SMA) associated with a Blake's pouch cyst; a malformation that is currently classified within the spectrum of Dandy-Walker complex. The association of the two conditions has not been previously reported in the English literature. A comprehensive review of the pertinent literature is presented.
Case Description:
A male neonate was noted to have paucity of movement of the four limbs with difficulty of breathing and poor feeding soon after birth. Respiratory distress with tachypnea, necessitated endotracheal intubation and mechanical ventilation. Pregnancy was uneventful except for decreased fetal movements reported by the mother during the third trimester. Neurological examination revealed generalized hypotonia with decreased muscle power of all limbs, nonelicitable deep tendon jerks, and occasional tongue fasciculations. Molecular genetic evaluation revealed a homozygous deletion of both exons 7 and 8 of the survival motor neuron 1 (SMN1) gene, and exon 5 of the neuronal apoptosis inhibitory protein (NAIP) gene on the long arm of chromosome 5 consistent with Werdnig-Hoffmann disease (SMA type 1). At the age of 5 months, a full anterior fontanelle and abnormal increase of the occipito-frontal circumference were noted. Computed tomographic (CT) scan and magnetic resonance imaging (MRI) of the brain revealed a tetraventricular hydrocephalus and features of Blake's pouch cyst of the fourth ventricle.
Conclusions:
This case represents a previously unreported association of Blake's pouch cyst and SMA type 1.
Keywords: Blake's Pouch Cyst, Dandy–Walker complex, spinal muscular atrophy, Werdnig-Hoffmann disease
INTRODUCTION
The spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of disorders characterized by degeneration and loss of anterior horn cells of the spinal cord leading to muscle weakness and atrophy.[84] Proximal SMA (types I-IV) accounts for 80-90% of all SMA cases and is primarily caused by recessive mutations in the survival motor neuron 1 (SMN1) gene located in the chromosome region 5q11.2-5q13.3, with homozygous absence of exon 7 in more than 95% of cases.[48,59] Blake's pouch cyst, in contrast, is defined as a failure of regression of Blake's pouch (the rudimentary fourth ventricular tela choroidea) secondary to nonperforation of the foramen of Magendi resulting in a posterior ballooning into the cisterna magna.[16,75] Failure of perforation of the foramen of Magendi results in enlargement of the fourth ventricle and the supratentorial ventricular system until the foramina of Luschka open and establish equilibrium of cerebrospinal fluid (CSF) outflow from the ventricles into the cisterns.[16] Blake pouch cyst is one of the anomalies within the Dandy-Walker complex (DWC), which is a continuum of congenital anomalies comprising Dandy-Walker malformation (DWM), Dandy-Walker variant (DWV), mega cisterna magna (MCM) in addition to Blake's pouch cyst.[20,78] To the best of our knowledge, the coexistence of SMA type 1 and Blake's pouch cyst (BPC) has not been previously reported in English literature.
CASE REPORT
A male neonate who is the first product of a nonconsanguineous marriage born at term with a body weight of 3.6 kg to a healthy young mother. Pregnancy was uneventful except for decreased fetal movements reported by the mother during the third trimester. Family history was negative for both parents.
Soon after birth, he was noticed to have paucity of movement of the four limbs with difficulty of breathing and poor feeding. He then developed respiratory distress with tachypnea, increased work of breathing, and oxygen desaturation that necessitated endotracheal intubation and mechanical ventilation. Clinically, his body weight, height and occipito-frontal circumference were all above 25th percentile; neurological examination revealed generalized hypotonia with decreased muscle power of all limbs, nonelicitable deep tendon jerks and occasional tongue fasciculations. No other clinical abnormalities were detected. Extensive metabolic workup and a TORCH (Toxoplasmosis, Rubella, Cytomegalovirus, Herpes simplex, HIV) screen revealed no abnormalities. Cranial and abdominal ultrasound examinations as well as cardiac echocardiogram were normal. Molecular genetic evaluation revealed a homozygous deletion of both exons 7 and 8 of the SMN1 gene, and exon 5 of the neuronal apoptosis inhibitory protein (NAIP) gene on the long arm of chromosome 5 consistent with Werdnig-Hoffmann disease (SMA type 1).
At the age of 5 months, a full anterior fontanelle and abnormal increase of the occipito-frontal circumference were noted. Computed tomographic (CT) scan and magnetic resonance imaging (MRI) of the brain revealed a tetraventricular hydrocephalus and features of BPC of the fourth ventricle. Endoscopic third ventriculostomy or ventriculoperitoneal shunt insertion were both refused by the parents after detailed counseling. A follow up MRI done 3 months later showed progressive hydrocephalus [Figure 1].
Figure 1.
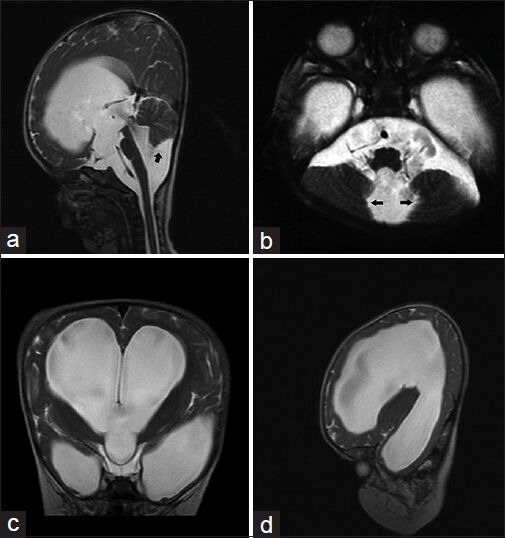
(a) Sagittal T2-weighted MR image demonstrating severe hydrocephalus with bulging third ventricular floor, open aqueduct and dilated fourth ventricle. Cerebellar vermis is compressed, relatively well-developed and is nonrotated. A thin line between dilated fourth ventricle and cisterna magna (Black arrow) indicates a Blake's pouch cyst. A high torcular Herophili with upward displacement of the tentorium is seen (b) Axial T2-weighted MR image with cystic dilatation of the fourth ventricle and compressed medial cerebellar hemispheres (Black arrows) (c) Coronal and (d) Sagittal T2-weighted MR images further demonstrate features of severe hydrocephalus
The parents refused any surgical maneuver for CSF diversion including ventriculoperitoneal shunting or endoscopic third ventriculostomy. Now, for 11 months since admission to neonatal intensive care unit (NICU), many trials of extubation failed and he had to be re-intubated every time, due to increased work of breathing and desaturation. Over his NICU course, he developed frequent respiratory tract infections that were timely treated. Currently, the patient is kept on mechanical ventilation and is receiving only supportive care.
DISCUSSION
We report a case of proximal SMA type 1 (Werdnig–Hoffmann disease or severe infantile acute SMA) associated with a Blake's pouch cyst; a malformation that is currently classified within the spectrum of DWC. An exhaustive search of the Medline failed to retrieve any previously reported association of the two conditions in the English literature. The patient had a homozygous deletion of both exons 7 and 8 of the SMN1 gene, and exon 5 of the NAIP gene on the long arm of chromosome 5. In addition to the posterior fossa anomaly and hydrocephalus in our patient, both clinical and genetic findings are consistent with the diagnosis of proximal SMA type 1. In a seemingly similar case, Panas et al. reported a combination of distal SMA with DWC and anterior polar cataracts in two brothers aged 23 and 25 years.[57] However, distal SMA and proximal SMAs represent completely different entities, making this case the first reported in the literature.
The spinal muscular atrophies
The SMAs are a genetically and clinically heterogeneous group of disorders characterized by degeneration and loss of anterior horn cells of the spinal cord leading to muscle weakness and atrophy.[84] Proximal SMA (types I-IV) accounts for 80-90% of all SMA cases and is primarily caused by recessive mutations in the SMN1 gene located in the chromosome region 5q11.2- 5q13.3, with homozygous absence of exon 7 in more than 95% of cases.[48,59] Alias et al. found homozygous absence of SMN1 exons 7 and 8 in 671 (90%) of 745 SMA patients. SMN gene is present in two highly homologous copies, SMN1 and SMN2, but only deletions of the SMN1 gene (exons 7 and 8 or exon 7) are responsible for clinical manifestations of SMA.[5] Extended deletions that include the NAIP gene may correlate with the severtity of SMA.[48]
Depending on the age of onset, the maximum muscular activity achieved, and survivorship, proximal SMA types are classified as type I (Phenotype MIM number 253300), severe infantile acute SMA, or Werdnig–Hoffman disease; type II (Phenotype MIM number 253550), or infantile chronic SMA; type III (Phenotype MIM number 253400), juvenile SMA, or Wohlfart–Kugelberg–Welander disease; and type IV (Phenotype MIM number 271150), or adult-onset SMA.[54]
Non-SMN1 SMAs include nonproximal SMA, bulbar palsy, spinobulbar muscular atrophy (SBMA), and infantile SMA variants also known as “SMA plus”.[57,84] These variants are characterized by SMA with additional or atypical features. They include SMA with respiratory distress, which can be caused by recessive mutations in Immunoglobulin μ-binding protein 2 (IGHMBP2); infantile lethal X-linked SMA with arthrogryposis and congenital fractures (SMAX2), caused by mutations in ubiquitin-activating enzyme E1 (UBE1); SMA1 with arthrogryposis and bone fractures, and SMA with pontocerebellar hypoplasia (SMA-PCH), also known as PCH type 1.[59]
Dandy-Walker complex and Blake's pouch cyst
The posterior fossa anomaly and the associated hydrocephalus in the patient of this report represent a BPC that is currently classified within the spectrum of DWC. DWC is a continuum of congenital anomalies comprising DWM, DWV, Blake's pouch cyst, and MCM.[20,78] These anomalies are characterized by varying degrees of malformation of the medullary vela, the cerebellar vermis and hemispheres, the fourth ventricle choroid plexus, the posterior fossa subarachnoid cisterns, and the enveloping meningeal structures.[16] Persistent Blake's pouch and MCM are thought to represent less severe abnormalities within the Dandy–Walker continuum.[60]
The original description of DWM dates back to the year 1914 when Dandy and Blackfan described a huge cystic dilatation of the fourth ventricle with anterior displacement of the cerebellar vermis that was attributed to primary atresia of the foramina of the fourth ventricle.[22] Over the following years many cases were reported, expanding the limits of the malformation to include findings of one particular case or another, and in the meanwhile creating a great deal of confusion about the definition and limits of the syndrome.[42] Taggart and Walker in 1942 further defined the condition.[70] Subsequently, Benda in 1954 introduced the now widely accepted name of DWM and was the first to introduce the currently held opinion that atresia of the cerebellar foramina is not an essential feature of the malformation.[12]
Tortori-Donati et al. added persistent BPC as an independent entity within the DWC.[75] BPC is thought to result from failure of regression of Blake's pouch (the rudimentary fourth ventricular tela choroidea) secondary to nonperforation of the foramen of Magendi.[75] Failure of perforation of the foramen of Magendi results in enlargement of the fourth ventricle and the supratentorial ventricular system until the foramina of Luschka open and establish equilibrium of CSF outflow from the ventricles into the cisterns.[16] However, as the larger foramen of Magendi is permanently missing, the ventricles will stay enlarged.[20] The cerebellar hemispheres and vermis would rather be compressed (to a certain degree) than underdeveloped and would therefore re-expand in case of ventricular shunting.[9]
Typical radiological features of BPC are (i) tetraventricular hydrocephalus, (ii) infra- or retrocerebellar localization of the cyst, (iii) a relatively well-developed, nonrotated cerebellar vermis (as opposed to a DW), (iv) a cystic dilation of the fourth ventricle without cisternal communication, and (v) some degree of compression on the medial cerebellar hemispheres. Ideally, one may see the fourth ventricular choroid plexus continuing in the roof of the cyst on sagittal MR images.[20] These features were all noted in our patient's MR images [Figure 1]. Classically, the choroid plexus of the fourth ventricle is displaced into the superior cyst wall in Blake's pouch, absent in DWM, and normal in arachnoid cyst.[52] As it is evident in our patient [Figure 1], BPC may push the developing tentorium into an abnormal, relatively high position.[25] BPC is capable of producing a broad spectrum of findings ranging from being asymptomatic to a full-blown hydrocephalus.[20]
DWC has been associated with a long list of chromosomal and phenotypic abnormalities of both neural and mesenchymal elements [Table 1]. Heterozygous deletion of ZIC1 and ZIC4 on chromosome 3q24 was the first molecularly defined cause of classic DWM.[31] A second DWM-linked locus on 6p25.3 was associated with deletions or duplications encompassing FOXC1 causing cerebellar and posterior fossa malformations including cerebellar vermis hypoplasia (CVH), MCM and DWM. Foxc1-null mice have embryonic abnormalities of the rhombic lip due to loss of mesenchyme-secreted signaling molecules with subsequent loss of Atoh1 expression in vermis. Specific loss of FOXC1 and general defects in mesenchymal signaling may result in cerebellar malformations.[3]
Table 1.
Clinical and chromosomal abnormalities associated with Dandy-Walker complex
Spinal muscular atrophy with pontocerebellar hypoplasia
SMA-PCH (PCH-1) is an important differential diagnosis of the case reported herein, and can be excluded based on both genetic and radiological grounds; SMA-PCH is a non-SMN1 condition on contrary to our patient with homozygous deletion of both exons 7 and 8 of the SMN1 gene on chromosome 5. MRI of our patient reveals a normal appearance of the brainstem and ventral pons in addition to other findings consistent with BPC [Figure 1]. PCH-1 is an autosomal-recessive disease of prenatal or infantile onset characterized by PCH and ventral horn cell degeneration. Clinically, polyhydramnios is usually present during pregnancy, and affected individuals present with bulbar dysfunction that leads to neonatal respiratory insufficiency, feeding difficulty, and congenital contractures; death often occurs before 1 year of age.[46] An extended clinical spectrum of PCH 1 with later onset of hypotonia, varying degrees of cerebellar or pontine hypoplasia and atrophy, peripheral nerve involvement, and longer survival has been reported.[61] All PCH syndromes [types 1-6] include a small cerebellum and brainstem with progressive microcephaly being a common finding.[59]
It is of relevance to this discussion to review the molecular genetic abnormality underlying PCH-1; namely the Vaccinia-related kinase 1 (VRK1) gene abnormality owing to its role in the pathogenesis of SMA and its probable contribution to the evolution of some cerebellar and posterior fossa congenital anomalies [Figure 2]. Renbaum et al. discovered a premature stop codon in the VRK1 gene that encodes a serine-threonine kinase and is located on chromosome 14q32 in affected siblings with PCH-1 in one family. VRK1 may be specifically important for spinal motor neuron survival or that it may also play a role in tRNA processing. In either case, identification of a VRK1 mutation as a cause of SMA-PCH points to new roles for this protein and suggests VRK2 [chromosome 2p16] and VRK3 [chromosome 19q13] as candidate genes for related phenotypes, including other PCHs and other SMAs.[59]
Figure 2.
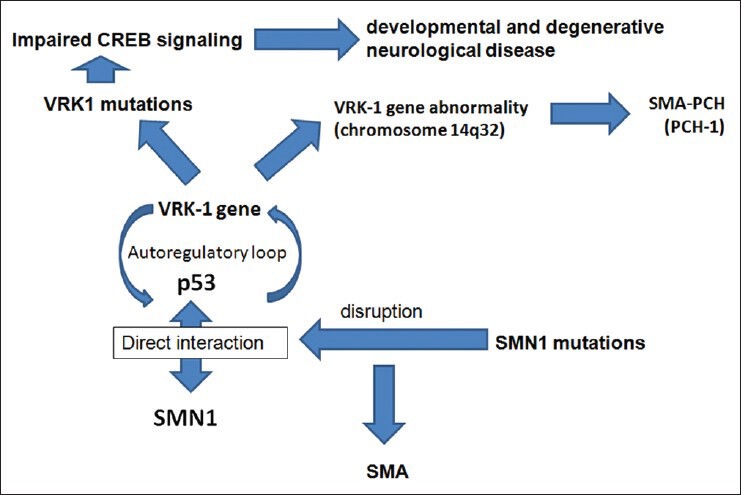
Schematic representation of the genetic events and interactions between VRK-1, SMA, and PCH-1
The existence of an autoregulatory loop between VRK1 and p53 may be of relevance to both development and maintenance of the nervous system. p53 regulates cell division and death during nervous system development and in response to neuronal insult or injury during life. Recessive mutations in Ataxia telangiectasia mutated (ATM), which phosphorylate p53 in response to DNA damage cause ataxia telangiectasia, in which loss of cerebellar neurons and ataxia are prominent features.[59] Moreover, p53 interacts directly with SMN1, an association disrupted by SMN1 mutations associated with SMA,[81] and might be of relevance to SMA pathogenesis.[76] In addition, VRK1 may be involved in cell cycle regulation, organization of chromatin, and organization of the nuclear envelope.[46] The role of VRK1 in cyclic AMP (cAMP) response element-binding (CREB) activation suggests that VRK1 mutations may lead to impaired CREB signaling, which can result in both developmental and degenerative neurological disease; Coffin–Lowry syndrome is caused by mutations in RSK2, another CREB kinase, Rubinstein–Taybi syndrome can be caused by mutations in CREBBP (a CREB binding protein). Interference with CREB-dependent transcription is a feature of polyglutamine stretches, common in spinocerebellar ataxias. CREB also binds to the SMN promoter and increases SMN expression, so its deficiency could promote an SMA phenotype.[59]
As it is well known that mutations in multiple genes can cause various central nervous system malformations,[46] we think that future reports and studies may reveal important molecular genetic links between DWC (specifically BPC) and Werdnig-Hoffmann disease.
CONCLUSION
This case represents a previously unreported association of BPC and SMA type 1 that further expands the current literature, and potentially directs future investigation of probable molecular genetic links between these conditions.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2014/5/5/282/139390
Contributor Information
Sherien A. Shohoud, Email: sherienshohoud@hotmail.com.
Waleed A. Azab, Email: waleedazab@hotmail.com.
Tarek M. Alsheikh, Email: tmalsheikh@gmail.com.
Rania M. Hegazy, Email: raniahegazy@hotmail.com.
REFERENCES
- 1.Al-Achkar W, Wafa A, Jarjour RA. A new case of de novo translocation (12;17;18) (q212;q22;q21.1) and cranio-cerebello-cardiac (3C) syndrome. Am J Med Genet A. 2011;155A:648–51. doi: 10.1002/ajmg.a.33742. [DOI] [PubMed] [Google Scholar]
- 2.Aluclu MU, Bahceci S, Bahceci M. A rare embryological malformation of brain-Dandy-Walker syndrome-and its association with Kallmann's syndrome. Neuro Endocrinol Lett. 2007;28:255–8. [PubMed] [Google Scholar]
- 3.Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41:1037–42. doi: 10.1038/ng.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexiou GA, Prodromou N. Dandy-Walker malformation in Crouzon syndrome. J Child Neurol. 2010;25:653. doi: 10.1177/0883073809349323. [DOI] [PubMed] [Google Scholar]
- 5.Alias L, Bernal S, Fuentes-Prior P, Barcelo M, Also E, Martinez-Hernandez R, et al. Mutation update of spinal muscular atrophy in Spain: Molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet. 2009;125:29–39. doi: 10.1007/s00439-008-0598-1. [DOI] [PubMed] [Google Scholar]
- 6.Al-Mendalawi MD. Dandy Walker malformation and hypertrophic cardiomyopathy. Unusual fatal association. Neurosciences (Riyadh) 2010;15:55. [PubMed] [Google Scholar]
- 7.Altman NR, Naidich TP, Braffman BH. Posterior fossa malformations. AJNR Am J Neuroradiol. 1992;13:691–724. [PMC free article] [PubMed] [Google Scholar]
- 8.Asai A, Hoffman HJ, Hendrick EB, Humphreys RP. Dandy-Walker syndrome: Experience at the Hospital for Sick Children, Toronto. Pediatr Neurosci. 1989;15:66–73. doi: 10.1159/000120445. [DOI] [PubMed] [Google Scholar]
- 9.Barkovich AJ. Congenital malformations of the brain and skul. In: Barkovich AJ, editor. Pediatric Neuroimaging. 2nd ed. New York: Raven; 1994. pp. 177–275. [Google Scholar]
- 10.Baxi LV, Walsh CA. Cerebral venous sinus thrombosis, Dandy-Walker malformation and polymicrogyria in Turner syndrome: An unreported association. Prenat Diagn. 2009;29:899–900. doi: 10.1002/pd.2308. [DOI] [PubMed] [Google Scholar]
- 11.Bekiesiñska-Figatowska M, Rokicki D, Walecki J, Gremida M. Menkes’ disease with a Dandy-Walker variant: Case report. Neuroradiology. 2001;43:948–50. doi: 10.1007/s002340100576. [DOI] [PubMed] [Google Scholar]
- 12.Benda CE. The Dandy-Walker syndrome of the so-called atresia of the foramen of Magendie. J Neuropathol Exp Neurol. 1954;13:14–29. doi: 10.1093/jnen/13.1.14. [DOI] [PubMed] [Google Scholar]
- 13.Ben Hamouda H, Sfar MN, Braham R, Ben Salah M, Ayadi A, Soua H, et al. Association of severe autosomal recessive osteopetrosis and Dandy-Walker syndrome with agenesis of the corpus callosum. Acta Orthop Belg. 2001;67:528–32. [PubMed] [Google Scholar]
- 14.Blank MC, Grinberg I, Aryee E, Laliberte C, Chizhikov VV, Henkelman RM, et al. Multiple developmental programs are altered by loss of Zic1 and Zic 4 to cause Dandy-Walker malformation cerebellar pathogenesis. Development. 2011;138:1207–16. doi: 10.1242/dev.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cakmak A, Zeyrek D, Cekin A, Karazeybek H. Dandy-Walker syndrome together with occipital encephalocele. Minerva Pediatr. 2008;60:465–8. [PubMed] [Google Scholar]
- 16.Calabrò F, Arcuri T, Jinkins JR. Blake's pouch cyst: An entity within the Dandy-Walker continuum. Neuroradiology. 2000;42:290–5. doi: 10.1007/s002340050888. [DOI] [PubMed] [Google Scholar]
- 17.Chen CP, Tzen CY, Chang TY, Lin CJ, Wang W, Lee CC. Prenatal diagnosis of partial trisomy 3p and partial monosomy 11q in a fetus with a Dandy-Walker variant and trigonocephaly. Prenat Diagn. 2002;22:1112–3. doi: 10.1002/pd.471. [DOI] [PubMed] [Google Scholar]
- 18.Chen CP, Chen M, Su YN, Tsai FJ, Chern SR, Hsu CY, et al. Prenatal diagnosis and molecular cytogenetic characterization of de novo partial trisomy 7p (7p15.3 → pter) and partial monosomy 13q (13q33.3 → qter) associated with Dandy-Walker malformation, abnormal skull development and microcephaly Taiwan. J Obstet Gynecol. 2010;49:320–6. doi: 10.1016/S1028-4559(10)60068-X. [DOI] [PubMed] [Google Scholar]
- 19.Coban D, Akin MA, Kurtoglu S, Oktem S, Yikilmaz A. Dandy-Walker malformation: A rare association with hypoparathyroidism. Pediatr Neurol. 2010;43:439–41. doi: 10.1016/j.pediatrneurol.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Cornips EM, Overvliet GM, Weber JW, Postma AA, Hoeberigs CM, Baldewijns MM, et al. The clinical spectrum of Blake's pouch cyst: Report of six illustrative cases. Childs Nerv Syst. 2010;26:1057–64. doi: 10.1007/s00381-010-1085-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Agostino AN, Kernohan JW, Brown JR. Dandy-Walker syndrome. J Neuropathol Exp Neurol. 1963;22:450–70. doi: 10.1097/00005072-196307000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Dandy WE, Blackfan KD. Internal hydrocephalus. Am J Dis Child. 1914;8:406–82. [Google Scholar]
- 23.Donahue ML, Ryan RM. Interstitial deletion of 8q21-->22 associated with minor anomalies, congenital heart defect, and Dandy-Walker variant. Am J Med Genet. 1995;56:97–100. doi: 10.1002/ajmg.1320560122. [DOI] [PubMed] [Google Scholar]
- 24.Ecker JL, Shipp TD, Bromley B, Benacerraf B. The sonographic diagnosis of Dandy-Walker and Dandy-Walker variant: Associated findings and outcomes. Prenat Diagn. 2000;20:328–32. [PubMed] [Google Scholar]
- 25.Epelman M, Daneman A, Blaser SI, Ortiz-Neira C, Konen O, Jarrín J, et al. Differential diagnosis of intracranial cystic lesions at head US: Correlation with CT and MR imaging. Radiographics. 2006;26:173–96. doi: 10.1148/rg.261055033. [DOI] [PubMed] [Google Scholar]
- 26.Estroff JA, Scott MR, Benacerraf BR. Dandy-Walker variant: Prenatal sonographic features and clinical outcome. Radiology. 1992;185:755–8. doi: 10.1148/radiology.185.3.1438757. [DOI] [PubMed] [Google Scholar]
- 27.Furtado LV, Bayrak-Toydemir P, Hulinsky B, Damjanovich K, Carey JC, Rope AF. A novel X-linked multiple congenital anomaly syndrome associated with an EBP mutation. Am J Med Genet A. 2010;152A:2838–44. doi: 10.1002/ajmg.a.33674. [DOI] [PubMed] [Google Scholar]
- 28.Gibson JB. Congenital hydrocephalus due to atresia of the foramen of Magendie. J Neuropathol Exp Neurol. 1955;14:244–62. doi: 10.1097/00005072-195507000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Golden JA, Rorke LB, Bruce DA. Dandy-Walker syndrome and associated anomalies. Pediatr Neurosci. 1987;13:38–44. doi: 10.1159/000120299. [DOI] [PubMed] [Google Scholar]
- 30.Gonsette R, Potvliege R, Andre-Galisaux G, Stenuit J. La mega grande citerne: Etude clinique, radiologique et anatomopathologique. Acta Neurol Psychiatr Belg. 1968;68:559–70. [PubMed] [Google Scholar]
- 31.Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. 2004;36:1053–5. doi: 10.1038/ng1420. [DOI] [PubMed] [Google Scholar]
- 32.Guschmann M, Horn D, Entezami M, Urban M, Hänal S, Kunze J, et al. Mesomelic campomelia, polydactyly and Dandy-Walker cyst in siblings. Prenat Diagn. 2001;21:378–82. doi: 10.1002/pd.70. [DOI] [PubMed] [Google Scholar]
- 33.Gverić-Ahmetasević S, Colić A, Gverić T, Gasparović VE, Pavlisa G, Ozretić D. Coexistance of cerebral sinovenous thrombosis and Dandy Walker malformation in newborn. Coll Antropol. 2011;(35 Suppl 1):303–7. [PubMed] [Google Scholar]
- 34.Hart MN, Malamud N, Ellis WG. The Dandy-Walker syndrome: A clinicopathological study based on 28 cases. Neurology. 1972;22:771–80. doi: 10.1212/wnl.22.8.771. [DOI] [PubMed] [Google Scholar]
- 35.Harwood-Nash DC, Fitz CR. Vol. 3. St Louis: Mosby; 1976. Neuroradiology in infants and children; pp. 1014–9. [Google Scholar]
- 36.Hindryckx A, De Catte L, Van Esch H, Fryns JP, Moerman P, Devlieger R. First trimester prenatal diagnosis of 13q-syndrome presenting with increased nuchal translucency, Dandy-Walker malformation and small parietal encephalocoele. Prenat Diagn. 2008;28:445–6. doi: 10.1002/pd.1989. [DOI] [PubMed] [Google Scholar]
- 37.Hirsch JF, Pierre-Kahn A, Renier D, Sainte-Rose C, Hoppe-Hirsch E. The Dandy-Walker malformation. A review of 40 cases. J Neurosurg. 1984;61:515–22. doi: 10.3171/jns.1984.61.3.0515. [DOI] [PubMed] [Google Scholar]
- 38.Imai T, Hattori H, Miyazaki M, Higuchi Y, Adachi S, Nakahata T. Dandy-Walker variant in Coffin-Siris syndrome. Am J Med Genet. 2001;100:152–5. doi: 10.1002/ajmg.1231. [DOI] [PubMed] [Google Scholar]
- 39.Jalali A, Aldinger KA, Chary A, McLone DG, Bowman RM, Le LC, et al. Linkage to chromosome 2q36.1 in autosomal dominant Dandy-Walker malformation with occipital cephalocele and evidence for genetic heterogeneity. Hum Genet. 2008;123:237–45. doi: 10.1007/s00439-008-0467-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kasliwal MK, Suri A, Sharma BS. Dandy Walker malformation associated with syringomyelia. Clin Neurol Neurosurg. 2008;110:317–9. doi: 10.1016/j.clineuro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 41.Kim YJ, Won YD, Kim KT, Chang ED, Huh PW. Parenchymal neurocutaneous melanosis in association with intraventricular dermoid and Dandy-Walker variant: A case report. Korean J Radiol. 2006;7:145–8. doi: 10.3348/kjr.2006.7.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kollias SS, Ball WS, Jr, Prenger EC. Cystic malformations of the posterior fossa: Differential diagnosis clarified through embryologic analysis. Radiographics. 1993;13:1211–31. doi: 10.1148/radiographics.13.6.8031352. [DOI] [PubMed] [Google Scholar]
- 43.Lim BC, Park WY, Seo EJ, Kim KJ, Hwang YS, Chae JH. De novo interstitial deletion of 3q22.3-q25.2 encompassing FOXL2, ATR, ZIC1, and ZIC4 in a patient with blepharophimosis/ptosis/epicanthus inversus syndrome, Dandy-Walker malformation, and global developmental delay. J Child Neurol. 2011;26:615–8. doi: 10.1177/0883073810384996. [DOI] [PubMed] [Google Scholar]
- 44.Lin AE, Doshi N, Flom L, Tenenholz B, Filkins KL. Beemer-Langer syndrome with manifestations of orofaciodigital syndrome. Am J Med Genet. 1991;39:247–51. doi: 10.1002/ajmg.1320390303. [DOI] [PubMed] [Google Scholar]
- 45.Lopez-Gutierrez JC. PHACES syndrome and ectopia cordis. Interact Cardiovasc Thorac Surg. 2011;12:642–4. doi: 10.1510/icvts.2010.258442. [DOI] [PubMed] [Google Scholar]
- 46.Maricich SM, Aqeeb KA, Moayedi Y, Mathes EL, Patel MS, Chitayat D, et al. Pontocerebellar hypoplasia: Review of classification and genetics, and exclusion of several genes known to be important for cerebellar development. J Child Neurol. 2011;26:288–94. doi: 10.1177/0883073810380047. [DOI] [PubMed] [Google Scholar]
- 47.Marnet D, Vinchon M, Mostofi K, Catteau B, Kerdraon O, Dhellemmes P. Neurocutaneous melanosis and the Dandy-Walker complex: An uncommon but not so insignificant association. Childs Nerv Syst. 2009;25:1533–9. doi: 10.1007/s00381-009-0976-6. [DOI] [PubMed] [Google Scholar]
- 48.Miskovic M, Lalic T, Radivojevic D, Cirkovic S, Vlahovic G, Zamurovic D, et al. Lower incidence of deletions in the survival of motor neuron gene and the neuronal apoptosis inhibitory protein gene in children with spinal muscular atrophy from Serbia. Tohoku J Exp Med. 2011;225:153–9. doi: 10.1620/tjem.225.153. [DOI] [PubMed] [Google Scholar]
- 49.Moerman P, Vandenberghe K, Fryns JP, Haspeslagh M, Lauweryns JM. A new lethal chondrodysplasia with spondylocostal dysostosis, multiple internal anomalies and Dandy-Walker cyst. Clin Genet. 1985;27:160–4. doi: 10.1111/j.1399-0004.1985.tb00204.x. [DOI] [PubMed] [Google Scholar]
- 50.Mortier GR, Messiaen L, Espeel M, Smets KJ, Vanzieleghem BD, Roels F, et al. Chondrodysplasia punctata with multiple congenital anomalies: A new syndrome? Pediatr Radiol. 1998;28:790–3. doi: 10.1007/s002470050466. [DOI] [PubMed] [Google Scholar]
- 51.Namavar Y, Barth PG, Kasher PR, van Ruissen F, Brockmann K, Bernert G, et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134:143–56. doi: 10.1093/brain/awq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nelson M, Maher K, Gilles F. A different approach to cysts of the posterior fossa. Pediatr Radiol. 2004;34:720–32. doi: 10.1007/s00247-004-1253-1. [DOI] [PubMed] [Google Scholar]
- 53.Notaridis G, Ebbing K, Giannakopoulos P, Bouras C, Kovari E. Neuropathological analysis of an asymptomatic adult case with Dandy-Walker variant. Neuropathol Appl Neurobiol. 2006;32:344–50. doi: 10.1111/j.1365-2990.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 54.Bethesda: National Center for Biotechnology Information, U.S. National Library of Medicine; [Last accessed on 2013 Nov 08]. Online Mendelian Inheritance in Man (OMIM®) [database online] Available from: http://omim.org/entry/253300 . [Google Scholar]
- 55.Osenbach RK, Menezes AH. Diagnosis and management of the Dandy-Walker malformation: 30 years of experience. Pediatr Neurosurg. 1992;18:179–89. doi: 10.1159/000120660. [DOI] [PubMed] [Google Scholar]
- 56.Ozdemir O, Polat A, Cinbis M, Kurt F, Kucuktasci K, Kiroglu Y. Dandy-Walker's variant and tetralogy of Fallot with atrial septal defect and patent ductus arteriosus and primary hypothyroidy-a new association. Indian J Pediatr. 2009;76:433–5. doi: 10.1007/s12098-009-0130-1. [DOI] [PubMed] [Google Scholar]
- 57.Panas M, Spengos K, Tsivgoulis G, Kalfakis N, Sfagos C, Vassilopoulos D, et al. Spinal muscular atrophy, Dandy-Walker complex, and cataracts in two siblings: A new entity? J Neurol Neurosurg Psychiatry. 2005;76:1183–4. doi: 10.1136/jnnp.2004.055855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raybaud C. Cystic malformations of the posterior fossa abnormalities associated with development of the roof of the fourth ventricle and adjoint meningeal structures. J Neuroradiol. 1982;9:103–33. [PubMed] [Google Scholar]
- 59.Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85:281–9. doi: 10.1016/j.ajhg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson AJ, Goldstein R. The cisterna magna septa: Vestigial remnants of Blake's pouch and a potential new marker for normal development of the rhombencephalon. J Ultrasound Med. 2007;26:83–95. doi: 10.7863/jum.2007.26.1.83. [DOI] [PubMed] [Google Scholar]
- 61.Rudnik-Schoneborn S, Sztriha L, Aithala GR, Houge G, Laegreid LM, Seeger J, et al. Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. Am J Med Genet A. 2003;117A:10–7. doi: 10.1002/ajmg.a.10863. [DOI] [PubMed] [Google Scholar]
- 62.Sasaki-Adams D, Elbabaa SK, Jewells V, Carter L, Campbell JW, Ritter AM. The Dandy-Walker variant: A case series of 24 pediatric patients and evaluation of associated anomalies, incidence of hydrocephalus, and developmental outcomes. J Neurosurg Pediatr. 2008;2:194–9. doi: 10.3171/PED/2008/2/9/194. [DOI] [PubMed] [Google Scholar]
- 63.Seidahmed MZ, Alkuraya FS, Shaheed M, Al Zahrani M, Al Manea W, Mansour F, et al. Ritscher-Schinzel (cranio-cerebello-cardiac, 3C) syndrome report of four new cases with renal involvement. Am J Med Genet A. 2011;155A:1393–7. doi: 10.1002/ajmg.a.33966. [DOI] [PubMed] [Google Scholar]
- 64.Sener RN. Rhombencephalosynapsis associated with Dandy-Walker malformation. J Neuroimaging. 2007;17:355–7. doi: 10.1111/j.1552-6569.2007.00066.x. [DOI] [PubMed] [Google Scholar]
- 65.Shenoy RD, Kamath N. Dandy-Walker malformation, occipital meningoencephalocele, meso-axial polydactyly and bifid hallux. Clin Dysmorphol. 2010;19:166–8. doi: 10.1097/MCD.0b013e32833a77f1. [DOI] [PubMed] [Google Scholar]
- 66.Silengo M, Gianino P, Longo P, Battistoni G, Defilippi C. Dandy-Walker complex in a child with Jeune's asphyxiating thoracic dystrophy. Pediatr Radiol. 2000;30:430. doi: 10.1007/s002470050779. [DOI] [PubMed] [Google Scholar]
- 67.Stevens CA, Lachman RS. New lethal skeletal dysplasia with Dandy-Walker malformation, congenital heart defects, abnormal thumbs, hypoplastic genitalia, and distinctive facies. Am J Med Genet A. 2010;152A:1915–8. doi: 10.1002/ajmg.a.33488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stoll C, Huber C, Alembik Y, Terrade E, Maitrot D. Dandy- Walker variant malformation, spastic paraplegia, and mental retardation in two sibs. Am J Med Genet. 1990;37:124–7. doi: 10.1002/ajmg.1320370129. [DOI] [PubMed] [Google Scholar]
- 69.Sudha T, Dawson AJ, Prasad AN, Konkin D, de Groot GW, Prasad C. De novo interstitial long arm deletion of chromosome 3 with facial dysmorphism, Dandy-Walker variant malformation and hydrocephalus. Clin Dysmorphol. 2001;10:193–6. doi: 10.1097/00019605-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 70.Taggart TK, Walker AE. Congenital atresia of the foramina of Luschka and Magendie. Arch Neurol Psychiatry. 1942;48:583–612. [Google Scholar]
- 71.Talamonti G, Picano M, Debernardi A, Bolzon M, Teruzzi M, D’Aliberti G. Giant occipital meningocele in an 8-year-old child with Dandy-Walker malformation. Childs Nerv Syst. 2011;27:167–74. doi: 10.1007/s00381-010-1154-6. [DOI] [PubMed] [Google Scholar]
- 72.Todt I, Mazereeuw-Hautier J, Binder B, Willems PJ. Dandy-Walker malformation in patients with KID syndrome associated with a heterozygote mutation (p. Asp50Asn) in the GJB2 gene encoding connexin 26. Clin Genet. 2009;76:404–8. doi: 10.1111/j.1399-0004.2009.01211.x. [DOI] [PubMed] [Google Scholar]
- 73.Tohyama J, Kato M, Kawasaki S, Harada N, Kawara H, Matsui T, et al. Dandy-Walker malformation associated with heterozygous ZIC1 and ZIC4 deletion: Report of a new patient. Am J Med Genet A. 2011;155A:130–3. doi: 10.1002/ajmg.a.33652. [DOI] [PubMed] [Google Scholar]
- 74.Tonni G, Azzoni D, Ambrosetti F, de Felice C, Ventura A. Cerebro- fronto- facial syndrome (Dandy-Walker Variant and frontofacial dysmorphisms): Report of the first case identified by increased nuchal translucency beyond 13(+6) weeks. Congenit Anom (Kyoto) 2007;47:68–71. doi: 10.1111/j.1741-4520.2007.00146.x. [DOI] [PubMed] [Google Scholar]
- 75.Tortori-Donati P, Fondelli MP, Rossi A, Carini S. Cystic malformations of the posterior cranial fossa originating from a defect of the posterior membranous area. Mega cisterna magna and persisting Blake's pouch: Two separate entities. Childs Nerv Syst. 1996;12:303–8. doi: 10.1007/BF00301017. [DOI] [PubMed] [Google Scholar]
- 76.Tsai MS, Chiu YT, Wang SH, Hsieh-Li HM, Li H. Abolishing Trp53-dependent apoptosis does not benefit spinal muscular atrophy model mice. Eur J Hum Genet. 2006;14:372–5. doi: 10.1038/sj.ejhg.5201556. [DOI] [PubMed] [Google Scholar]
- 77.Walbert T, Sloan AE, Cohen ML, Koubeissi MZ. Symptomatic neurocutaneous melanosis and Dandy-Walker malformation in an adult. J Clin Oncol. 2009;27:2886–7. doi: 10.1200/JCO.2008.21.5830. [DOI] [PubMed] [Google Scholar]
- 78.Warf BC, Dewan M, Mugamba J. Management of Dandy-Walker complex-associated infant hydrocephalus by combined endoscopic third ventriculostomy and choroid plexus cauterization. J Neurosurg Pediatr. 2011;8:377–83. doi: 10.3171/2011.7.PEDS1198. [DOI] [PubMed] [Google Scholar]
- 79.Weimer J, Cohen M, Wiedemann U, Heinrich U, Jonat W, Arnold N. Proof of partial imbalances 6q and 11q due to maternal complex balanced translocation analyzed by microdissection of multicolor labeled chromosomes (FISH-MD) in a patient with Dandy-Walker variant. Cytogenet Genome Res. 2006;114:235–9. doi: 10.1159/000094206. [DOI] [PubMed] [Google Scholar]
- 80.Wilson ME, Lindsay DJ, Levi CS, Ackerman TE, Gordon WL. US case of the day. Dandy-Walker variant with agenesis of the corpus callosum. Radiographics. 1994;14:678–81. doi: 10.1148/radiographics.14.3.8066281. [DOI] [PubMed] [Google Scholar]
- 81.Young PJ, Day PM, Zhou J, Androphy EJ, Morris GE, Lorson CL. A direct interaction between the survival motor neuron protein and p53 and its relationship to spinal muscular atrophy. J Biol Chem. 2002;277:2852–9. doi: 10.1074/jbc.M108769200. [DOI] [PubMed] [Google Scholar]
- 82.Zangwill KM, Boal DK, Ladda RL. Dandy-Walker malformation in Ellis-van Creveld syndrome. Am J Med Genet. 1988;31:123–9. doi: 10.1002/ajmg.1320310114. [DOI] [PubMed] [Google Scholar]
- 83.Zhang XB, Li CX. A case of keratitis ichthyosis deafness (KID) syndrome associated with Dandy-Walker. J Eur Acad Dermatol Venereol. 2007;21:706–7. doi: 10.1111/j.1468-3083.2006.02001.x. [DOI] [PubMed] [Google Scholar]
- 84.Zerres K, Rudnik-Schöneborn S. Spinal muscular atrophies. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Principles and Practice of Medical Genetics. 5th ed. Vol. 3. New York: Churchill Livingstone; 2006. pp. 3001–23. [Google Scholar]