

Abstract
Fibroblast growth factor 23 (FGF23) is a hormone that inhibits renal phosphate reabsorption and 1,25-dihydroxyvitamin D biosynthesis. The FGF23 subtilisin-like proprotein convertase recognition sequence (176RHTR179↓) is protected by O-glycosylation through ppGalNAc-T3 (GALNT3) activity. Thus, inactivating GALNT3 mutations render FGF23 susceptible to proteolysis, thereby reducing circulating intact hormone levels and leading to hyperphosphatemic familial tumoral calcinosis. To further delineate the role of glycosylation in the Fgf23 function, we generated an inducible FGF23 transgenic mouse expressing human mutant FGF23 (R176Q and R179Q) found in patients with autosomal dominant hypophosphatemic rickets (ADHR) and bred this animal to Galnt3 knockout mice, a model of familial tumoral calcinosis. Due to the low intact Fgf23 level, Galnt3 knockout mice with wild-type Fgf23 alleles were hyperphosphatemic. In contrast, carriers of the mutant FGF23 transgene, regardless of Galnt3 mutation status, had significantly higher serum intact FGF23, resulting in severe hypophosphatemia. Importantly, serum phosphorus and FGF23 were comparable between transgenic mice with or without normal Galnt3 alleles. To determine whether the presence of the ADHR mutation could improve biochemical and skeletal abnormalities in Galnt3-null mice, these mice were also mated to Fgf23 knock-in mice, carrying heterozygous or homozygous R176Q ADHR Fgf23 mutations. The knock-in mice with functional Galnt3 had normal Fgf23 but were slightly hypophosphatemic. The stabilized Fgf23 ADHR allele reversed the Galnt3-null phenotype and normalized total Fgf23, serum phosphorus, and bone Fgf23 mRNA. However, the skeletal phenotype was unaffected. In summary, these data demonstrate that O-glycosylation by ppGaINAc-T3 is only necessary for proper secretion of intact Fgf23 and, once secreted, does not affect Fgf23 function. Furthermore, the more stable Fgf23 ADHR mutant protein could normalize serum phosphorus in Galnt3 knockout mice.
Fibroblast growth factor 23 (FGF23) is a protein hormone produced in bone, which inhibits reabsorption of phosphate through suppression of the sodium-phosphate cotransporters NaPi-IIa and NaPi-IIc in the kidney (1–3). The hormone also decreases renal production and increases catabolism of 1,25-dihydroxyvitamin D [1,25(OH)2D] (1–5). The reduced renal 1,25(OH)2D synthesis in turn suppresses intestinal absorption of phosphate and calcium.
The FGF23 protein harbors the subtilisin-like proprotein convertase (SPC) recognition sequence 176RHTR179↓, making it susceptible to proteolysis (4, 6, 7). In patients with autosomal dominant hypophosphatemic rickets (ADHR), either arginine in the SPC site is mutated to glutamine or tryptophan (6). As a result, ADHR mutant FGF23 is more stable and causes hypophosphatemia. In contrast, loss-of-function GALNT3 mutations found in patients with hyperphosphatemic familial tumoral calcinosis and its variant, hyperostosis-hyperphosphatemia syndrome render FGF23 less stable, resulting in reduced secretion of intact, bioactive FGF23 and consequent hyperphosphatemia with ectopic calcifications (8–10). GALNT3 encodes a Golgi-associated glycosyltransferase, UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 (ppGalNAc-T3). This enzyme O-glycosylates threonine 178 in the 176RHTR179↓ SPC recognition sequence and protects FGF23 from proteolysis (11). Thus, the absence of ppGalNAc-T3 results in cleavage of most FGF23 protein in vivo, leading to low or undetectable levels of circulating intact FGF23 and markedly elevated C-terminal fragments in both humans and Galnt3-null mice (12–16). Therefore, it is likely that the prevailing hyperphosphatemia due to loss of intact FGF23 further up-regulates this hormone, but with lack of glycosylation to stabilize FGF23, proteolytic fragments are secreted accounting for the increase in C-terminal FGF23 polypeptides.
In vivo and in vitro studies demonstrated that O-glycosylation by ppGalNAc-T3 is essential for secretion of intact, bioactive FGF23. However, the molecular interactions within the 176RHTR179↓ site and effects of post-translational glycosylation on FGF23 bioactivity are unknown. In this study, we used a new human mutant FGF23 transgenic mouse and a murine model of ADHR (ADHR Fgf23 knock-in mouse) (17), in concert with a model of tumoral calcinosis (Galnt3 knockout mouse) (16), to further delineate the role of glycosylation in FGF23 physiology. Furthermore, because the Galnt3 knockout mouse has abnormal biochemical and skeletal phenotypes (16, 18), we also sought to determine whether the presence of the ADHR mutation could correct these phenotypes.
Materials and Methods
Generation of inducible mutant FGF23 transgenic mouse
The mutant FGF23 transgene inducible by tetracycline was developed by combining core components of pCMV-Tet3G and pTRE3G-IRES from Tet-On 3G Vector Set (Bicistronic Version; Clontech Laboratories) into a single vector (Figure 1A). pCMV-Tet3G expresses Tet-On 3G, a tetracycline-controlled transactivator that exhibits high activity in the presence of the inducer doxycycline. To limit transgene expression to osteocytes, the CMV promoter in this vector was replaced with mouse Dmp1 promoter (a generous gift from Dr Teresita Bellido, Indiana University). pTRE3G-IRES is a Tet-inducible, mammalian expression vector designed to coexpress two genes of interest under the control of the Tet-responsive promoter PTRE3G. Human R176Q-R179Q FGF23 cDNA (generated from ColIa1-FGF23 R179Q from Dr Kenneth Jonsson, Uppsala University) and hrGFP II (Stratagene) were cloned into the first and second multiple cloning sites, respectively. Subsequently, the transactivator under the control of the Dmp1 promoter and inducible promoter controlling FGF23 and hrGFP II were inserted into CopyRight v2.0 pSMART BAC (Lucigen) in opposite directions.
Figure 1.

Generation and biochemical phenotype of inducible human FGF23 transgenic mouse. A, Generation of the inducible human FGF23 transgenic mouse. The human R176Q-R179Q FGF23 and hrGFP II genes were cloned into the multiple cloning sites of the pTRE3G-IRES vector, which coexpress the two genes when tetracycline-controlled transactivator Tet3G is present. The CMV promoter in pCMV-Tet3G was replaced with the mouse Dmp1 promoter (PDmp1) to limit the Tet3G expression to osteocytes. The main components of these two vectors were placed in a single vector to generate the transgenic mouse, which expresses mutant FGF23 and hrGFP II (the brighter variant of the humanized recombinant GFP) upon induction with doxycycline. SV40 PolyA signals were added at both ends. B, Serum biochemistries of uninduced transgenic animals. Number of animals = 7 or 8 per group. C) Serum biochemistries in transgenic animals following doxycycline induction. Number of animals = 8–10 per group. Dark gray bars, males; light gray, females. All values are presented as mean ± SEM. P values less than .05 by unpaired t-tests, compared with same-sex littermate controls (*). BUN, blood urea nitrogen.
Animals
Mutant FGF23 transgenic animals were generated by pronuclear injection of the transgene into fertilized C3H eggs at the Indiana University Transgenic and Knock-out Mouse Core. Five of 36 mice were positive for the transgene and were bred to establish independent lines. However, one of the five mice was infertile. Of the four remaining mouse lines, only one was hypophosphatemic upon 1-week induction of the transgene (data not shown). This line was used in further studies.
Experimental mice were generated by mating Galnt3 knockout mice, a model of familial tumoral calcinosis (16), to either human FGF23 transgenic mice or murine ADHR Fgf23-R176Q knock-in mice, a model of autosomal dominant hypophosphatemic rickets (17). To eliminate the effect of background strain, both tumoral calcinosis and ADHR mice were backcrossed to C57/BL6J for at least 10 generations. The FGF23 transgenic mice were in the original C3H strain. To ensure the equal inducibility, the transgene in mice was maintained at only one copy. Mice were fed a regular rodent diet, containing 1.01% calcium, 0.65% phosphorus, and 2.05 IU/g vitamin D3 (Teklad Global 18% Protein Extruded Rodent Diet, 2018SX; Harlan). The animals had access to the diets and acidified tap water ad libitum. To induce the FGF23 transgene, doxycycline (Clontech) was mixed in with drinking water (1 mg/ml) and given to the animals for 1 week prior to sacrifice. All mice were killed at 8 weeks of age. The study was approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
Biochemical measurements
Eight-week-old mice were anesthetized with ketamine/xylazine mix. Blood was then collected by cardiac puncture and stored at −80 C until analysis. Routine serum biochemistries (calcium, phosphorus, blood urine nitrogen, creatinine, and alkaline phosphatase) were measured, using an RX Daytona clinical chemistry analyzer (Randox Laboratories). Galnt3 knockout mice and Fgf23 knock-in mice express mouse Fgf23 only; however, the transgenic mice express human FGF23 in addition to endogenous mouse Fgf23. Thus, three different assays were used to differentiate species-specific intact and total FGF23/Fgf23 levels. Intact human FGF23 and mouse Fgf23 were measured using an Intact Fgf-23 ELISA kit, which detects only intact, bioactive FGF23/Fgf23 in both humans and mice (Kainos Laboratories). Species-specific total FGF23/Fgf23 was measured, using Mouse or Human FGF-23 (C-Terminal) ELISA kits (Immutopics International), which detect both intact FGF23/Fgf23 protein and C-terminal fragments.
Fgf23 mRNA quantification
After the blood draw, femurs were collected and stored in RNAlater RNA Stabilization Reagent (QIAGEN). Total RNA was extracted from the whole femurs, using TRIzol Plus RNA Purification System (Invitrogen) and used for first-strand cDNA synthesis, using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA was subsequently used for quantification of Fgf23 expression by probe-based quantitative PCR, using TaqMan Gene Expression Master Mix in the 7900HT Fast Real-Time PCR System (Applied Biosystems), as described elsewhere (19). TATA-box binding protein (Tbp) was used to normalize gene expression. Relative gene expression was determined by analyzing the data using the relative standard curve method.
Microcomputed tomography analysis
Femurs were collected and fixed in 10% neutral-buffered formalin for two days. Microcomputed tomography (Skyscan) of the distal femur metaphysis was performed using a 6-μm voxel size. Trabecular bone properties, including bone volume/tissue volume, trabecular number, and trabecular thickness were assessed on 1 mm of tissue (approximately 166 slices) in the distal metaphysis beginning at 0.5 mm from the growth plate.
Statistical analysis
Mean, SD, and standard error of the mean (SEM) were calculated for all outcome measures by genotype and sex. Unpaired t-tests were used to test differences between two genotype groups. ANOVA was used to test for overall differences among four or five genotypes within the same sex. P < .05 was considered significant for all analyses.
Results
Human FGF23 (R176Q-R179Q) transgene in Galnt3 knockout mice
O-glycosylation by ppGalNAc-T3 is essential for secretion of intact, bioactive FGF23. To further delineate the molecular interactions at the subtilisin convertase recognition site in FGF23, we generated inducible FGF23 transgenic mice, which carry two human FGF23 mutations (R176Q and R179Q) in the recognition site. The mutations stabilize FGF23, resulting in ADHR in humans (4, 6, 7, 20). At baseline, serum phosphorus was the same between wild-type and transgenic mice (Figure 1B), suggesting that there is no, if any, significant leaky expression from the transgene. Similarly, none of the biochemistries was affected by the presence of the transgene although there was a trend for increased alkaline phosphatase (t-test P = .09).
Upon doxycycline induction, transgenic mice were hypophosphatemic with 4.1 mg/dl lower phosphorus, compared with wild-type mice (Figure 1C). In addition, the transgenic mice had increased alkaline phosphatase and decreased body weight and blood urine nitrogen. Serum calcium and creatinine were also reduced, but only in male transgenic mice. Interestingly, the doxycycline administration itself changed serum parameters. Compared with uninduced wild-type males, induced males had higher serum alkaline phosphatase, calcium, and phosphorus (all t-test P < .001); however, creatinine and blood urea nitrogen were not affected by doxycycline (Figure 1B-C; data not shown).
The transgenic animals were mated to Galnt3 knockout mice, which lack the O-glycosylation needed to protect cleavage of intact FGF23 within the 176RHTR179↓ SPC site. Male Galnt3 knockout mice weighed slightly more than the other groups (ANOVA P = .044), but otherwise, there were no significant differences across groups (data not shown). In agreement with our previous results (16, 18), Galnt3-null mice had, on average, 1.9 mg/dl higher serum phosphorus (Figure 2). Serum creatinine was also slightly decreased, but only in male Galnt3 knockout mice. In contrast, after doxycycline administration, the human FGF23 transgenic animals carrying wild-type Galnt3 alleles were severely hypophosphatemic with a 3.0 mg/dl reduction. FGF23 transgenic mice with null Galnt3 alleles had a 1.9 mg/dl reduction of phosphorus. In addition to hypophosphatemia, all FGF23 transgene carriers had slightly lower serum calcium (on average, 0.7 mg/dl) and creatinine (0.04 mg/dl) with higher alkaline phosphatase (80 U/L). In light of the finding that iron deficiency increases Fgf23 expression in mice and the production of FGF23 fragments in normal mice and humans (17, 21), we tested whether FGF23 fragments altered iron and hematologic parameters. There were no major differences in iron, total iron binding capacity, or hemoglobin (Figure 2, data not shown)
Figure 2.

Serum biochemistries in human FGF23 transgenic Galnt3-null and control mice. Total human FGF23 and mouse Fgf23 concentrations represent intact FGF23/Fgf23 protein and C-terminal fragments of FGF23/Fgf23 measured by human and mouse C-terminal ELISA. Dark gray bars, males; light gray, females. All values are presented as mean ± SEM. Number of animals = 7–10 per group. ANOVA P values for same-sex groups are provided for measured parameters when P values are less than .05. N.S., not significant; TIBC, total iron binding capacity.
To understand the effects on phosphate metabolism, circulating intact human FGF23 or mouse Fgf23 was measured, using an assay that detects both human and mouse intact protein, and human FGF23 and mouse Fgf23 C-terminal fragments were measured, using species-specific assays. Consistent with the elevated serum phosphorus, Galnt3 knockout mice had only 50% of intact Fgf23 in normal mice (Figure 2). These mice also had approximately 10-fold higher total Fgf23 (intact plus C-terminal fragments) measured by mouse C-terminal assay, indicating that most of mouse Fgf23 in circulation is fragmented. In contrast, intact human FGF23 or mouse Fgf23 levels in the transgenic mice (upon induction with doxycycline) was 16 fold elevated, compared with wild-type mice. Total human FGF23 or mouse Fgf23 measured by species-specific assays revealed that human FGF23 expressed from the transgene is almost 24 times higher than wild type, whereas endogenous mouse Fgf23 was reduced to 31% of wild type in three transgenic groups although it remained similar between wild-type and double mutant females. Most importantly, intact and total FGF23/Fgf23 levels measured by three different assays were essentially the same in the transgenic animals regardless of the presence of normal Galnt3 alleles. Thus, overexpression of ADHR FGF23 completely abolished the need for O-glycosylation by ppGalNAC-T3 and allowed for secretion of intact mutant FGF23.
Murine Fgf23 ADHR knock-in allele in Galnt3 knockout mice
The transgenic study suggested that ADHR mutations in the SPC recognition site of human FGF23 allows almost complete FGF23 secretion despite the absence of normal Galnt3 alleles. Thus, we asked whether endogenous Fgf23 when mutated could correct the skeletal and biochemical phenotype of Galnt3 knockout mice. For this purpose, a murine ADHR allele (knock-in R176Q Fgf23 ADHR mutation) that results in stabilized Fgf23 protein in vivo (17) was placed on the Galnt3-null background, and mice heterozygous or homozygous for the ADHR mutation and null for Galnt3 were examined with appropriate control genotypes. Mouse genotypes did not significantly affect body weights of the animals (data not shown). The Galnt3-null mouse has high bone volume/tissue volume, increased trabecular number, and cortical thickening at 24 weeks of age (18); however, at 8 weeks of age the Galnt3-null mice did not have severe bone disease and the genetic crosses were all similar to control (Supplemental Table 1).
Analysis of biochemistries demonstrated that in Galnt3 knockout mice serum calcium was mildly elevated (0.4 mg/dl) and alkaline phosphatase was 22% lower (Figure 3). Male Galnt3 knockout mice with homozygous ADHR mutations also had a 9% reduction in alkaline phosphatase. Regardless of sex, Galnt3 knockout mice (with wild-type Fgf23 alleles) were hyperphosphatemic (on average, 1.9 mg/dl), whereas mice carrying a heterozygous ADHR mutation (with wild-type Galnt3 alleles) were hypophosphatemic (average −1.1 mg/dl). Of note, the mice lacking Galnt3 but carrying one or two ADHR mutant alleles had normal serum phosphorus.
Figure 3.

Serum biochemistries and femur Fgf23 expression in ADHR-Galnt3-null and control mice. Total Fgf23 represents intact Fgf23 protein and C-terminal fragments of Fgf23 measured by C-terminal ELISA. Fgf23 mRNA expression was normalized to TATA-box binding protein (Tbp) and is presented by setting wild-type males as 1.0. Dark gray bars, males; light gray, females. All values are presented as mean ± SEM. Number of animals = 10–15 per group. ANOVA P values for same-sex groups are provided for measured parameters when P values are less than .05. N.S., not significant; +, wild-type allele; −, null allele; a, ADHR knock-in allele.
Galnt3 knockout mice with wild-type Fgf23 alleles had less than half of the intact Fgf23 concentrations compared with normal mice (Figure 3). However, total Fgf23 (intact plus C-terminal fragments) were, on average, 17-fold higher than that of normal mice. Similarly, Fgf23 gene expression in bone was 3-fold higher in Galnt3 knockout mice than in controls. Interestingly, although slightly hypophosphatemic, ADHR mutant mice with wild-type Galnt3 alleles had normal intact Fgf23 levels as well as normal total serum Fgf23 and Fgf23 mRNA expression. Furthermore, intact Fgf23 in the mice carrying the ADHR mutations (without normal Galnt3 alleles) were as low as in Galnt3 knockout mice, despite their normal phosphorus levels. With the addition of the ADHR mutations to the Galnt3-null background, total Fgf23 concentrations were almost completely normalized, albeit still slightly higher than wild type. The ratio of intact to total Fgf23 revealed that only 1% of Fgf23 produced in Galnt3 mutant mice was secreted as intact Fgf23. Presence of the ADHR mutation significantly improved secretion of intact Fgf23, regardless of the Galnt3 genotype, but did not completely correct the intact/total Fgf23 ratios. Therefore, in the context of normal physiological regulation of Fgf23 expression, the presence of a single ADHR knock-in mutation mostly abolished the need for ppGalNAc-T3-mediated glycosylation, normalizing the elevated serum phosphate and reducing total FGF23 observed in Galnt3 knockout mice.
Discussion
Both in vitro and in vivo studies (11–15) demonstrated that secretion of intact FGF23 requires O-glycosylation of threonine 178 by ppGalNAc-T3. However, it is unknown how the ADHR mutation and ppGalNAc-T3 interact at the 176RHTR179↓ SPC recognition sequence and whether the glycosylation is critical for FGF23 activity at target organs. To address these questions, we generated mice expressing the human ADHR mutations in the absence of ppGalNAc-T3. Loss of normal Galnt3 alleles renders Fgf23 susceptible to proteolysis, leading to lower intact Fgf23 concentrations and consequent hyperphosphatemia. In contrast, because the ADHR mutation disrupts the SPC recognition sequence, intact Fgf23 escapes degradation by proteolysis (4, 7, 20). In agreement with this finding, transgenic mice expressing human ADHR mutations had comparable intact and total concentrations of human FGF23 and mouse Fgf23, regardless of Galnt3 genotypes. Collectively, these data suggest that in the absence of the SPC recognition sequence, O-glycosylation of the threonine is no longer required for secretion of intact Fgf23. In addition to the similar intact FGF23 and Fgf23 levels, the FGF23 transgenic mice had the similar phosphorus levels in the presence or absence of normal Galnt3 alleles. This suggests that once secreted, the glycosylation by ppGalNAc-T3 does not significantly affect the function of FGF23, at least in the ADHR mutant form.
In murine Fgf23 ADHR knock-in mice, placement of an ADHR mutation in the recognition sequence also allowed intact Fgf23 to be released into circulation in Galnt3 knockout mice. The protective effect of ADHR mutation is consistent with the finding in the transgenic mouse study. However, compared with ADHR knock-in mice carrying wild-type Galnt3 alleles, intact Fgf23 was slightly lower and total Fgf23 production was higher in the knock-in mice with Galnt3-null background. As a result, proportion of intact Fgf23 secreted did not reach the level of ADHR mutant carriers with normal ppGalNAc-T3 function. This suggests that the ADHR mutation abolishing the SPC site may interfere with O-glycosylation process and may not completely protect intact Fgf23 proteins from proteolysis. Given that an in vitro study (12) demonstrated that not all ADHR mutant proteins are fully glycosylated, it is possible that these underglycosylated mutant proteins are still susceptible to proteolytic degradation, albeit far less than wild-type Fgf23 protein. Transgenic mice had human FGF23 with both arginines in the SPC site mutated, whereas the knock-in mice had endogenous murine Fgf23 gene with one arginine mutated. Thus, we cannot rule out the possibility that the species or the number of mutations affected the stability of mutant Fgf23 in the absence of normal Galnt3 alleles.
Unlike human FGF23 transgenic mice, phosphorus and Fgf23 each influences the other via a feedback loop in murine Fgf23 ADHR knock-in mice (Figure 4). Interestingly, ADHR mutants with normal Galnt3 alleles were mildly hypophosphatemic even with the same intact Fgf23 level as in wild-type mice, while ADHR mutants without ppGalNAc-T3 had lower Fgf23, yet maintained normal serum phosphorus. This suggests that the mutant form of Fgf23 may have a slightly higher potency at the target receptor complex. Speculatively, the complete reversal of the phosphate metabolism in the ADHR-Galnt3 double mutants also suggests that patients with tumoral calcinosis due to GALNT3 and FGF23 inactivating mutations (both with the same ELISA profile of low intact and high C-terminal FGF23) would benefit from the delivery of ADHR-mutant FGF23 because this form of the hormone may be more stable and potentially more effective in vivo. Although the intact FGF23 ELISA recognizes ADHR FGF23 in humans (21, 22) and Fgf23 in mice (17), it is also possible that due to the subtle structural differences the assay does not target ADHR-mutated FGF23 as efficiently as wild type, resulting in slightly lower baseline serum readings.
Figure 4.
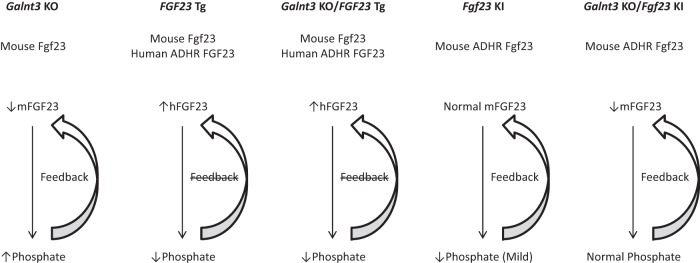
Feedback between phosphorus and FGF23/Fgf23 in mouse models. Because expression of human FGF23 transgene is controlled by doxycycline levels, feedback loops between phosphorus and FGF23 in these mice are disrupted. In contrast, Galnt3 knockout mice and mouse Fgf23 ADHR knock-in mice retain the feedback loops. KO, knockout; Tg, transgenic; KI, knock-in.
The primary comparisons in the transgenic mouse study were made within induced and uninduced groups separately. Interestingly, secondary comparisons between uninduced and induced wild-type males revealed that doxycycline treatment itself affects serum biochemistries. Serum alkaline phosphatase, calcium and phosphorus were all elevated in the induced mice, whereas renal function makers – creatinine and blood urea nitrogen – were unaffected. Other investigators also found that the antibiotic treatment also had direct effect on urinary sodium and calcium excretions (23). The mechanism for these biochemical changes is unknown. However, these observations suggest that effect of doxycycline on mineral metabolism must be taken into account when this common inducing agent is used.
In summary, the addition of an ADHR mutation (in the form of transgene or knock-in) in the human FGF23 or mouse Fgf23 SPC site on the Galnt3-null background essentially eliminated the need for FGF23/Fgf23 O-glycosylation by ppGalNAc-T3 and reversed the hyperphosphatemic phenotype of the tumoral calcinosis model. Furthermore, once secreted, the glycosylation of mature FGF23/Fgf23 was no longer required for biological activity. Additional studies are needed to understand this regulatory mechanism for competition between protection by glycosylation and proteolysis by subtilisin-like proprotein convertases. However, these data suggest that O-glycosylation is only necessary to protect FGF23/Fgf23 from these convertases and therefore acts as an important intracellular regulatory step to control FGF23/Fgf23 production.
Acknowledgments
We thank Anthony Acton for performing routine biochemical measurements and Drew Brown for microcomputed tomography analysis.
This study was supported by National Institutes of Health Grants R01 AR042228 (to M.J.E.), DK063934, DK095784 (to K.E.W.), and S10-RR023710 (for microcomputed tomography) and KL2 career development award (to S.I.) from the Indiana Clinical and Translational Sciences Institute funded in part by the National Institutes of Health Grant KL2 RR025760.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ADHR
- autosomal dominant hypophosphatemic rickets
- FGF23
- fibroblast growth factor 23
- ppGalNAc-T3
- UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3
- 176RHTR179↓
- convertase recognition sequence
- SPC
- subtilisin-like proprotein convertase.
References
- 1. Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–435 [DOI] [PubMed] [Google Scholar]
- 2. Larsson T, Marsell R, Schipani E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145(7):3087–3094 [DOI] [PubMed] [Google Scholar]
- 3. Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145(11):5269–5279 [DOI] [PubMed] [Google Scholar]
- 4. Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimada T, Yamazaki Y, Takahashi M, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289(5):F1088–F1095 [DOI] [PubMed] [Google Scholar]
- 6. ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(3):345–348 [DOI] [PubMed] [Google Scholar]
- 7. White KE, Carn G, Lorenz-Depiereux B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60(6):2079–2086 [DOI] [PubMed] [Google Scholar]
- 8. Ichikawa S, Baujat G, Seyahi A, et al. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010;152A(4):896–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab. 2005;90(4):2420–2423 [DOI] [PubMed] [Google Scholar]
- 10. Topaz O, Shurman DL, Bergman R, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36(6):579–581 [DOI] [PubMed] [Google Scholar]
- 11. Kato K, Jeanneau C, Tarp MA, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281(27):18370–18377 [DOI] [PubMed] [Google Scholar]
- 12. Frishberg Y, Ito N, Rinat C, et al. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22(2):235–242 [DOI] [PubMed] [Google Scholar]
- 13. Garringer HJ, Mortazavi SMJ, Esteghamat F, et al. Two novel GALNT3 mutations in familial tumoral calcinosis. Am J Med Genet A. 2007;143(20):2390–2396 [DOI] [PubMed] [Google Scholar]
- 14. Ichikawa S, Guigonis V, Imel EA, et al. Novel GALNT3 mutations causing hyperostosis-hyperphosphatemia syndrome result in low intact fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2007;92(5):1943–1947 [DOI] [PubMed] [Google Scholar]
- 15. Ichikawa S, Imel EA, Sorenson AH, et al. Tumoral calcinosis presenting with eyelid calcifications due to novel missense mutations in the glycosyl transferase domain of the GALNT3 gene. J Clin Endocrinol Metab. 2006;91(11):4472–4475 [DOI] [PubMed] [Google Scholar]
- 16. Ichikawa S, Sorenson AH, Austin AM, et al. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150(6):2543–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farrow EG, Yu X, Summers LJ, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. 2011;108(46):E1146–E1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ichikawa S, Austin AM, Gray AK, Allen MR, Econs MJ. Dietary phosphate restriction normalizes biochemical and skeletal abnormalities in a murine model of tumoral calcinosis. Endocrinology. 2011;152(12):4504–4513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ichikawa S, Austin AM, Gray AK, Econs MJ. A Phex mutation in a murine model of X-linked hypophosphatemia alters phosphate responsiveness of bone cells. J Bone Miner Res. 2012;27(2):453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimada T, Muto T, Urakawa I, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143(8):3179–3182 [DOI] [PubMed] [Google Scholar]
- 21. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011;96(11):3541–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2007;22(4):520–526 [DOI] [PubMed] [Google Scholar]
- 23. Myakala K, Motta S, Murer H, et al. Renal-specific and inducible depletion of NaPi-IIc/Slc34a3, the cotransporter mutated in HHRH, does not affect phosphate or calcium homeostasis in mice. Am J Physiol Renal Physiol. 2014;306(8):F833–F843 [DOI] [PubMed] [Google Scholar]