

Abstract
TGF-β1, -β2, and -β3 are small, secreted signaling proteins. They share 71–80% sequence identity and signal through the same receptors, yet the isoform-specific null mice have distinctive phenotypes and are inviable. The replacement of the coding sequence of TGF-β1 with TGF-β3 and TGF-β3 with TGF-β1 led to only partial rescue of the mutant phenotypes, suggesting that intrinsic differences between them contribute to the requirement of each in vivo. Here, we investigated whether the previously reported differences in the flexibility of the interfacial helix and arrangement of monomers was responsible for the differences in activity by generating two chimeric proteins in which residues 54–75 in the homodimer interface were swapped. Structural analysis of these using NMR and functional analysis using a dermal fibroblast migration assay showed that swapping the interfacial region swapped both the conformational preferences and activity. Conformational and activity differences were also observed between TGF-β3 and a variant with four helix-stabilizing residues from TGF-β1, suggesting that the observed changes were due to increased helical stability and the altered conformation, as proposed. Surface plasmon resonance analysis showed that TGF-β1, TGF-β3, and variants bound the type II signaling receptor, TβRII, nearly identically, but had small differences in the dissociation rate constant for recruitment of the type I signaling receptor, TβRI. However, the latter did not correlate with conformational preference or activity. Hence, the difference in activity arises from differences in their conformations, not their manner of receptor binding, suggesting that a matrix protein that differentially binds them might determine their distinct activities.
Transforming growth factor beta isoforms, TGF-β1, -β2, and -β3, are small (25 kDa) secreted homodimeric signaling proteins. They coordinate wound healing, modulate immune cell function, maintain the extracellular matrix, and regulate epithelial and endothelial cell growth and differentiation.1 Their importance is demonstrated by the many human diseases that result from disruption or dysregulation of the TGF-β signaling pathway, including developmental disorders, such as Marfan’s disease,2 and adult diseases such as cancer3 and fibrosis.4
TGF-β isoforms signal through two surface receptors, known as the TGF-β type I and type II receptors (TβRI and TβRII, respectively). TGF-βs assemble TβRI and TβRII into a TβRI2-TβRII2 heterotetramer in a sequential manner, first by binding TβRII followed by recruitment of TβRI.5 The stepwise assembly of TβRI and TβRII into a TβRI2-TβRII2 heterotetramer is driven by binding of TβRI to a composite TGF-β/TβRII interface.6,7 The assembly of the TβRI2-TβRII2 heterotetramer triggers a transphosphorylation cascade whereby the constitutively active TβRII kinase phosphorylates TβRI in a negative regulatory domain known as a GS box. This activates the TβRI kinase, which phosphorylates cytoplasmic effectors,8 including the canonical nuclear-translocating Smad proteins,9 but others as well.10 The TGF-β type III receptor (TβRIII), a cell surface proteoglycan with a short noncatalytic cytoplasmic domain, binds all three TGF-β isoforms and promotes the formation of the signaling complex with TβRI and TβRII.11 The ability of TβRIII to potentiate assembly of the signaling complex is especially important for TGF-β2 since TGF-β2 binds TβRII weakly, and most cells are poorly responsive to TGF-β2 in the absence of TβRIII.12
The TGF-β isoforms are encoded by distinct genes and are highly conserved relative to one another and across species, with the human TGF-β isoforms sharing between 71–79% identity (Supporting Information, Figure S1A). The three-dimensional structures of the TGF-βs are similar, consisting of two cystine-knotted monomers tethered together by a disulfide bond.13−16 The monomers are described as adopting the shape of an extended hand, and the homodimer is formed by packing the palm of one hand into the heel of the other (Supporting Information, Figure S1B).
TGF-β isoforms are indistinguishable in most cell-based reporter gene and growth inhibition assays.17 The primary exception is TGF-β2, which is 100–1000-fold less potent than TGF-β1 and TGF-β3 in cell lines that lack TβRIII.12,18,19 The TGF-β isoforms are nonetheless each required in vivo as the isoform-specific null mice are inviable: TGF-β1 null mice have an autoimmune-like inflammatory disease and die within a few days of weaning,20,21 TGF-β2 null mice exhibit developmental defects of the heart, spinal column, urogenital tract, eye, and inner ear and die just before birth,22 and TGF-β3 null mice are defective in lung development, have cleft palate, and die about a day after birth.23 The phenotypic differences in the null mice correlate with differences in tissue expression patterns. TGF-β1, for example, is widely expressed in both developing embryos and adults, consistent with the widespread multifocal inflammatory disease characteristic of the TGF-β1 null mice, while TGF-β2 and TGF-β3 are abundantly expressed in the developing heart and lungs, consistent with major developmental defects of these organs in the corresponding null mice.
Though the three-dimensional structures of the isoforms are similar, intrinsic differences among them might also contribute to the requirement of each in vivo. This was first suggested when it was shown that TGF-β1 and TGF-β3 have opposing effects in skin wound healing, with topical application of purified TGF-β3 preventing and TGF-β1 promoting scarring.24 The mechanism in wound healing has recently been shown to be due to differences in chemoregulated cell migration,25 with TGF-β3 promoting epidermal cell migration, and TGF-β1 neither promoting nor inhibiting migration.26 The differences between isoforms have been further suggested by tissue explant studies in which cleft palates, excised from developing TGF-β3 null embryos, developed normally when exogenous purified TGF-β3 was added to the culture medium, but not when TGF-β1 or TGF-β2 was added27−29 and by gene replacement studies where “knock-in” of the coding sequence for the mature domain of TGF-β1 into the TGF-β3 locus, and vice versa, led to partial, but not complete, rescue of the mutant phenotype.30,31 These results indicate that there are indeed intrinsic differences between isoforms that contribute to their distinct functions in vivo.
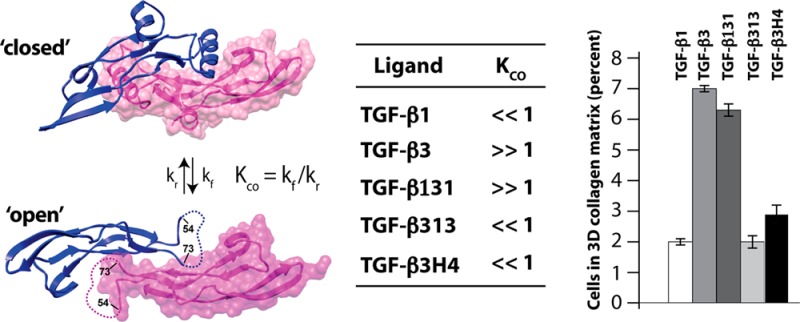
The solution structure of TGF-β113 and the crystal structure of TGF-β315 are similar: the palm α-helix, α3, packs into the heel of the other monomer to form a compact “closed” dimer (Supporting Information, Figure S1B). TGF-β1 and TGF-β3 nevertheless appear to differ in solution, as NMR secondary shifts and {1H}-15N heteronuclear NOEs indicate that α3 is well-ordered and rigid in TGF-β1,13 but not in TGF-β332,33 (Supporting Information, Figure S2). The disorder in the region corresponding to α3 in TGF-β3 is also suggested by the crystal structure of the TβRII/TGF-β3 complex, where the two subunits are rotated away from one another by 101° in a noncanonical “open” conformation and there is no electron density for α3 (Figure 1A).34 Though it is conceivable that the open form of TGF-β3 might be a consequence of either differences in the solution conditions under which the NMR studies were performed (TGF-β1 in 5% D2O at pH 4.2 and TGF-β3 in 6% dioxane-d8, 2% methanol-d3, 87% H2O, and 5% D2O at pH 2.9) or the low pH at which the TGF-β3/TβRII complex was crystallized (pH 4.5), this is unlikely as TGF-β3 alone was recently crystallized in the open form at neutral pH.35 The α-helix 3 region cannot be structurally ordered in the open conformation since it would sterically overlap with residues in the heel of the other monomer (Figure 1A). The overall conclusion based on a synthesis of these observations is that in solution TGF-β3 undergoes a conformational equilibrium between the “closed” and “open” states, with the open state predominating (KCO ≫ 1; Figure 1A). TGF-β1 may also undergo a similar equilibrium, but with the closed state predominating (KCO ≪ 1).
Figure 1.

Closed and open forms of TGF-β3 and construct design for this study. (A) Closed and open conformations of TGF-β3. Closed form is from the crystal structure of unbound TGF-β3 (PDB code 1TGJ), while the open form is from the crystal structure of the TGF-β3/TβRII complex (PDB code 1KTZ; TβRII not displayed). Open form differs from the closed by a 101° degree rotation of the monomers away from one another; the open form also has no electron density for the α-helix 3 and connecting loops (residues 55–72, dashed line). Disorder of α-helix 3 in the open form is likely caused by steric overlap between residues near the C-terminal end of α-helix 3 and the heel of the opposing monomer; this is shown by the structure below the “closed” form in which TGF-β3 monomers from the closed form (with an intact α-helix 3) have been overlaid onto the partial monomer structures of the open form. Steric clashes between α3 and the heel of one monomer and the other are highlighted in green. (B) Amino acid sequence alignment of TGF-β1 and -β3 and the sequences of TGF-β131, TGF-β313 and TGF-β3H4. Secondary structural elements correspond to those from the crystal structure of TGF-β3 (PDB code 1TGJ). Residues from TGF-β1 are highlighted with a gray background, while those from TGF-β3 with a blue background; the same color scheme is used for TGF-β131, TGF-β313, and TGF-β3H4. (C, D) Interaction of α-helix 3 of one monomer with the heel of the other for the closed form of TGF-β1 and -β3 (PDB codes 1KLC and 1TGJ, respectively). Heel residues conserved between TGF-β1 and -β3 (L28, I22, M104, V106, and A41) are highlighted in light and dark pink. Helical residues that differ between TGF-β1 and -β3 are highlighted in red.
The difference in KCO between TGF-β1 and TGF-β3 might be due to differences in packing interactions in the dimer interface, with more favorable interactions favoring the closed form. The differences in KCO might alternatively be due to differences in the intrinsic stability of α-helix 3, with a more stable helix more strongly resisting the transition from the closed to open form (thus favoring the closed form). The objective of this study was to investigate the underlying basis for the differences in KCO between TGF-β1 and TGF-β3 and whether this might underlie their differences in biological activity.
Experimental Procedures
Plasmid Constructs
The coding sequences for the mature TGF-β3, TGF-β131, and TGF-β313 signaling ligands (each 336 nucleotides in length) were synthesized (Genscript). The synthetic genes were inserted between the NdeI and HindIII sites downstream of the T7 promoter in plasmid pET32a (Novagen). The TGF-β3 A54L, H58Y, G63A, and T67Q mutant, termed TGF-β3H4, was constructed by performing site-directed mutagenesis (QuikChange Site-Directed Mutagenesis kit, Stratagene) of the T7-based TGF-β3 construct described above. The coding sequence for the mature domain of TGF-β131 was also used to replace the corresponding sequence for the mature domain of TGF-β1 in the CHO cell expression vector pcDNA-GS-TGF-β1 (kindly provided by Dr. Peter Sun, NIAID, Rockville, MD).36 The coding sequence for TGF-β131 in this expression vector (designated pcDNA-GS-TGF-β131) is preceded by the rat serum albumin leader peptide and the TGF-β1 pro-domain, both of which are required for the proper maturation and secretion of the mature dimer into the culture medium.
Protein Preparation
TGF-β3, TGF-β313, and TGF-β3H4 were expressed in bacteria, refolded, and purified using the procedure previously described for TGF-β3.37 TβRII-ED (residues 15–130) and TβRI-ED (residues 7–91) were expressed in bacteria, refolded, and purified as previously described.38,39 TGF-β1 and TGF-β131 were expressed and purified from conditioned medium produced by an overexpressing stably transfected CHO cell line. The cell line used to produce TGF-β1 has been previously described36 and was kindly provided from Dr. Peter Sun (NIAID, Rockville, MD). The TGF-β131 cell line was generated by transfecting the pcDNA-GS-TGF-β131 construct into CHO-lec3.2.8.1 cells using Lipofectamine 2000 (Invitrogen) following the same protocol for TGF-β1.36 The highest expressing stable transfected clone was selected by ELISA and chosen for large-scale protein production as described.36 TGF-β1 and TGF-β131 were purified as described,36 but with the modification that ion-exchange chromatography was used in the last step of the purification in place of size exclusion chromatography. The ion-exchange chromatography was performed by dialyzing the TGF-β1 or TGF-β131 preparation into 0.1 M acetic acid and loading onto a Source 15S cation exchange column (GE Healthcare) previously equilibrated in ligand buffer (20 mM sodium acetate, 30% isopropanol, pH 4.0). The column was then washed with three column volumes of ligand buffer and eluted with 200–600 mM linear NaCl gradient in ligand buffer over 10 column volumes. The corresponding dimer peak was pooled and dialyzed against 4 L of 100 mM acetic acid (4 h/cycle, 3 cycles).
NMR Sample Preparation
Uniformly isotopically labeled TGF-β313 and TGF-β3H4 used in the NMR studies were obtained by growing transformed Escherichia coli BL21(DE3) cells (EMD Biosciences) on M9 minimal medium enriched with 0.1% (w/v) 15NHCl4 or 0.1% (w/v) 15NHCl4 and 0.3% (w/v) uniformly 13C-labeled d-glucose (Cambridge Isotope Laboratories) following the procedure described by Marley.40 Selectively isotopically labeled forms of TGF-β131 were obtained by culturing CHO-lec3.2.8.1 cells stably transfected with the pcDNA-GS-TGF-β131 construct in SFM4CHO serum free medium (Hyclone) custom formulated so that it lacked leucine, tyrosine, glycine, valine, and cysteine. The labeled amino acids (Cambridge Isotope Laboratories) were added to the medium according to the concentrations in its original formulation. Four different selective labeling schemes, each with one 15N-labeled amino acid and one 13CO labeled amino acid, were used as shown in Table 2. NMR samples of TGF-β313, TGF-β131, and TGF-β3H4 were prepared by buffer exchanging the purified protein into 87% H2O, 5% D2O, 6% dioxane-d8 and 2% methanol-d3 at pH 2.9. The buffer exchanged protein samples, which had a concentration of 0.25–0.5 mM and a volume of 325 μL, were transferred to clean Shigemi thinwall microcells (Shigemi).
Table 2. Selective Labeling Schemes for TGF-β131.
| scheme | 13CO labeled | 15N labeled | unique dipeptide |
|---|---|---|---|
| 1 | Gly | Tyr | G38-Y39 |
| 2 | Cys | Leu | C44-L45 |
| 3 | Val | Leu | V61-L62 |
| 4 | Gly | Leu | G63-L64 |
NMR Spectroscopy
NMR data were collected at 40 °C using a Bruker 700 MHz spectrometer equipped with a 5 mm 1H{13C,15N} z-gradient “TCI” cryogenically cooled probe (Bruker Biospin). Backbone resonances of TGF-β313 and TGF-β3H4 were assigned by recording and analyzing HNCACB,41 CBCA(CO)NH,42 HNCO,43 HCACO,44 and HBHACONH42 triple resonance data sets. NMR data were processed and analyzed using the nmrPipe45 and Sparky46 software packages, respectively. Backbone amide 15N T1, 15N T2, and {1H}-15N NOE relaxation parameters were measured in an interleaved manner at 40 °C at a 15N frequency of 70.95 MHz using 1H-detected pulse schemes previously described.47 The T1 and T2 data sets were each collected using 8–10 delay times, varying between 16–3200 ms and 16–192 ms, respectively. The T1 and T2 relaxation times were obtained by fitting relative peak intensities as a function of T1 or T2 delay time to a two-parameter decaying exponential. NOE values were obtained by taking the ratio of peak intensities from experiments performed with and without 1H presaturation and by applying a correction factor to account for the incomplete recovery of both 15N and 1H magnetization.48
Circular Dichroism Spectroscopy
CD measurements were performed at 25 °C in the far UV (200–250 nm) on a JASCO J-815 spectropolarimeter using quartz cells with a path length of 1 mm. CD spectra were measured with 0.25 mg mL–1 TGF-β samples in 10 mM H3PO4 (pH 2.9) and were processed by subtracting the buffer contribution. Experimental data were expressed as mean residue ellipticity (degree × cm2/dmol).
Surface Plasmon Resonance (SPR) Binding Assays
SPR binding studies were performed with a BIAcore 3000 instrument (GE Healthcare) and were analyzed using the software package Scrubber2 (Biologic Software). TGF-βs were biotinylated and captured on carboxymethyl dextran (CM5) chips. This was accomplished by adding a 1.25-fold excess of the purified TβRI and TβRII ectodomains in 0.1 M NaHCO3 at pH 7.5 to TGF-β1, TGF-β3, TGF-β131, TGF-β313, and TGF-β3H4 and then by adding 10 molar equivalents of sulfo-NHS-LC-LC-Biotin (Pierce). Singly biotinylated TGF-βs were separated from receptors and doubly and multiply biotinylated forms by applying them to a Source S cation exchange column (GE Healthcare) in the presence of 30% isopropanol at pH 4.0 and eluting with a linear 0–0.5 M NaCl gradient. Biotinylated TGF-βs were captured by injecting them over a CM5 sensor chip (GE Healthcare, Piscataway, NJ) to which 5000 RU streptavidin had been covalently attached to all four flow cells using an amine coupling kit (GE Healthcare). Surface densities of captured TGF-βs were kept at 50–300 RU to minimize rebinding artifacts. Binding assays were performed by injecting 2-fold serial dilutions of the receptors in duplicate or triplicate in HBS-EP buffer (GE Healthcare) at a flow rate of 50 or 100 μL min–1 for kinetic experiments. Surfaces were regenerated by a brief injection of 4 M guanidine hydrochloride (10 s contact time at a flow rate of 100 μL min–1). Baseline correction was performed by double referencing.49 Kinetic analyses were performed by global fitting with a simple 1:1 model. Standard errors were obtained from the variation in the fitted parameters.
Sandwich Assay for Quantifying Chemoregulated Cell Migration
The chemoregulated migration assay was performed as previously described.25 This was accomplished by monitoring the migration of dermal fibroblasts from the center of native collagen gels where the TGF-βs had been added at concentrations of 1, 10, 100, and 1000 pg mL–1. The migration of the fibroblasts was then quantified by counting the cells along the length of the collagen-containing cylinder using a digitally stepped microscope. The measurements for each TGF-β at each concentration were performed in triplicate. The experiment was repeated three times, and the reported values are the mean from a single representative experiment; reported errors correspond to the deviation among replicates.
Results
Design and Production of TGF-β313 and TGF-β131
The hypothesis of this study is that the difference in KCO is mainly determined by intrinsic differences in the stability of α-helix 3 and that the isoform-specific biological activities arise from differences in KCO. This hypothesis is based on the differences in the calculated helical propensity of α-helix 3, which is nearly 10-fold higher for TGF-β1 compared to TGF-β3 (Table 1). These calculations, which were performed using the program Agadir,50,51 show that much but not all of the difference in the helical stability is due to substitution of an α-helix-stabilizing alanine at position 63 in TGF-β1 with an α-helix-destabilizing glycine in TGF-β3 (Figure 1B). The alternative explanation, that packing interactions between monomers differ, seems unlikely since all of the residues in the “heel” region against which α3 is packed are conserved between TGF-β1 and TGF-β3 (Figure 1C,D). The residues within α-helix 3 that interact with residues in the heel region are also the same, except residue 58, which is a histidine in TGF-β3 but a tyrosine in TGF-β1 (Figure 1C,D).
Table 1. Sequences and Predicted Helix Propensity for Peptides of α-Helix 3a.
| constructs | sequences | helical content (%) |
|---|---|---|
| TGF-β1 | QYSKVLALYNQH | 4.8 |
| TGF-β1 Q57T | TYSKVLALYNQH | 5.1 |
| TGF-β1 Y58H | QHSKVLALYNQH | 2.6 |
| TGF-β1 K60T | QYSTVLALYNQH | 5.2 |
| TGF-β1 A63G | QYSKVLGLYNQH | 0.9 |
| TGF-β1 Q67T | QYSKVLALYNTH | 3.8 |
| TGF-β1 H68L | QYSKVLALYNQL | 4.4 |
| TGF-β1 Y58H, A63G, Q67T | QHSKVLGLYNTH | 0.4 |
| TGF-β3 | THSTVLGLYNTL | 0.5 |
| TGF-β3 T57Q | QHSTVLGLYNTL | 0.5 |
| TGF-β3 H58Y | TYSTVLGLYNTL | 0.7 |
| TGF-β3 T60K | THSKVLGLYNTL | 0.5 |
| TGF-β3 G63A | THSTVLALYNTL | 2.5 |
| TGF-β3 T67Q | THSTVLGLYNQL | 0.7 |
| TGF-β3 L68H | THSTVLGLYNTH | 0.5 |
| TGF-β3 H58Y, G63A, T67Q | TYSTVLALYNQL | 5.0 |
To investigate our hypothesis, two TGF-β chimeras were generated in which residues 54–75 were swapped between TGF-β1 and TGF-β3 (Figure 1B). The swapped region corresponds to the region shown to be flexible in TGF-β332,33,52 and includes α-helix 3 (residues 57–68) as well as several flanking residues. There are nine amino acid differences in the swapped region, including L54, Q57, Y58, K60, A63, Q67, H68, G71, and A75 in TGF-β1 and A54, T57, H58, T60, G63, T67, L68, E71, and S75 in TGF-β3. The expectation was that the TGF-β1 variant with the α3 region from TGF-β3 (designated TGF-β131) would be altered such that the open form was favored (KCO ≫ 1), while the TGF-β3 variant with the α3 region from TGF-β1 (designated TGF-β313) would favor the closed form (KCO ≪ 1). To generate the TGF-β131 and TGF-β313 chimeric proteins, the coding sequences of both were synthesized and inserted into a T7-based expression vector. TGF-β313 was obtained by expressing it in E. coli and refolding and purifying it as described for TGF-β3.53 TGF-β131 was also expressed at a level comparable to that of TGF-β313 in E. coli, but attempts to refold the denatured monomers into native disulfide-linked dimers were unsuccessful. The parent protein, TGF-β1, is similarly behaved, and therefore TGF-β131 was produced using the same CHO cell expression system previously used to produce TGF-β1.36 TGF-β313 and TGF-β131, as well as another variant described below designated TGF-β3H4, were found to run as a single band at 25 kDa on SDS gels under nonreducing conditions, but as a single band at 12.5 kDa under reducing conditions (Supporting Information, Figure S3).
Backbone Amide Assignments
The 1H–15N HSQC spectrum of TGF-β3 includes many intense signals in the random coil region that arise from residue 54–75, which are disordered.32 The 1H–15N HSQC spectrum of TGF-β313, when recorded under identical conditions, 87% H2O, 5% D2O, 6% dioxane-d8, and 2% methanol-d3 at pH 2.9 and 40 °C, has significantly improved spectral dispersion and much more uniform signal intensities. These improved spectral features enabled full assignment (102/102 expected non-proline residues) of the backbone resonances of TGF-β313 using a single 0.4 mM 15N, 13C sample and standard triple-resonance experiments implemented on a 700 MHz spectrometer (Figure 2A).
Figure 2.

Assigned 1H–15N HSQC spectra of TGF-β313 and TGF-β131. (A) Assigned HSQC of TGF-β313 with peaks labeled by their one letter amino acid code and residue number. (B) 1H–15N HSQC of 15N-tyrosine selectively labeled TGF-β131 overlaid with the HN plane of HNCO data for Scheme 1 (cyan) labeled TGF-β131. (C) 1H–15N HSQC of 15N-leucine selectively labeled TGF-β131 (black contours) overlaid with the HN plane of an HNCO for Scheme 2 (green contours), Scheme 3 (magenta contours) and Scheme 4 (cyan contours) labeled TGF-β131. Peak labeled with a green asterisk (*) was observed in the HN planes of all HNCO data.
TGF-β131 was expressed at a high level and could be readily purified from the conditioned medium produced by the stably transfected CHO cells, but it proved impractical to prepare an 15N or 15N,13C uniformly labeled sample for assignment purposes owing to the high cost of the culture medium. Therefore, an alternative labeling strategy was adopted in which residues in unique i–i+1 dipeptides were selectively labeled with the corresponding 13CO-labeled amino acid at position i and the corresponding 15N-labeled amino acid at position i+1. This strategy is designed to enable definitive assignment of the amide of residue i+1 by recording the 1H–15N plane of an HNCO. This strategy was previously used to successfully assign a number of residues in CHO-cell derived TGF-β1,54,55 but has the disadvantage that unambiguous assignment of each amide requires preparation of a separate sample. This limited the number of amides that could be definitively assigned; accordingly two judiciously chosen residues inside and outside the swapped region (L62 and L64 and Y39 and L45, respectively) were assigned. The residues inside the swapped region corresponded to those that had {1H}-15N NOEs greater than 0.75 (i.e., were rigid) in TGF-β1, but significantly less than 0.75 in TGF-β3 (i.e., were flexible), while those outside the swapped region were chosen in regions that had {1H}-15N NOEs greater than 0.75 (i.e., were rigid) in both TGF-β1 and TGF-β3. The labeling schemes designed to assign Y39, L45, L62, and L64 are listed in Table 2.
The 1H–15N HSQC of the Scheme 1 sample (13CO-Gly, 15N-Tyr) exhibited 7–8 strong signals, while the 1H–15N HSQC spectra of the Scheme 2, 3, and 4 samples (all of which were labeled with 15N-Leu, but with a different 13CO-labeled amino acid) each exhibited 11–12 strong signals. These spectra, shown in black contours in Figure 2B,C, are largely consistent with expectations based on the amino acid content of TGF-β131, which contains 7 tyrosine and 10 leucine residues. The corresponding 1H–15N planes of the HNCO spectra, in contrast, exhibit either a single signal in the Scheme 1 sample (Figure 2A, cyan contours) or one signal with a unique chemical shift and one signal that has the same chemical shift in the Scheme 2, 3, and 4 samples (Figure 2B, green, magenta, and cyan contours, respectively). The single signal of the Scheme 1 sample and the unique signals of the Scheme 2, 3, and 4 samples are presumed to correspond to Y39, L45, L62, and L64, respectively. The same signal present in each of the Scheme 2, 3, and 4 samples has a very narrow line width and is presumed to be L2, which lies near the N-terminus and is highly flexible.
Flexibility and Conformational Preference Switched for TGF-β313 and TGF-β131
The backbone amide {1H}-15N NOEs were measured for both TGF-β313 and TGF-β131 at 40 °C on a Bruker 700 MHz spectrometer (Figure 3A, B). TGF-β313 residues 59, 60, 63, 66, 67, and 68 in α-helix 3 all have high NOE values (>0.75) (Figure 3A). This suggests that the α-helix 3 of TGF-β313 is structurally ordered, which is similar to TGF-β1, not TGF-β3 (Supporting Information, Figure S2). TGF-β131 residues 62 and 64 in α-helix 3 region have low NOE values (0.35 and 0.47 respectively), while residues Y39 and L45 have high NOE values (>0.75), indicating that the α-helix 3 region of TGF-β131 is flexible. Thus, substitution of residues 54–75 from TGF-β1 into TGF-β3 caused α3 to become rigid, while substitution of residues 54–75 from TGF-β3 into TGF-β1 caused α3 to become flexible.
Figure 3.

NMR relaxation and secondary structure analysis of TGF-β313 and TGF-β131. (A) {1H}-15N NOEs of TGF-β313. {1H}-15N NOE values for residues 57, 58, 61 62, 64, and 65 in α-helix 3 region of TGF-β313 could not be accurately measured due to peak overlap. (B) {1H}-15N NOEs of residues Y39, L45, L62, and L64 in TGF-β131. (C) Secondary structure probabilities of TGF-β313 calculated from NMR secondary shifts by the program PECAN;56 positive and negative values indicate α-helical and β-strand probabilities, respectively. Secondary structure diagram shown along the top corresponds to that of TGF-β1 (PDB 1KLC).
The secondary structure of TGF-β313 was assessed by analyzing the secondary shifts using the program PECAN, which provide a sensitive and accurate indicator of secondary structure propensities.56 This showed that α3 is highly probable in TGF-β313 (Figure 3C). To directly confirm that α3 was present in TGF-β313, we recorded a three-dimensional 15N-edited NOESY spectrum and identified numerous dαN(i,i+3) and αN(i,i+4) NOEs which are diagnostic for well-ordered α-helices (Supporting Information, Figure S4). Thus, α3 is structurally ordered in TGF-β313, which is similar to TGF-β1, not TGF-β3.
TGF-β313, in addition to forming a stable α-helix 3, is also expected to adopt the closed state, with α3 from one monomer packing against the heel of the other monomer. To investigate whether the stable α3 in TGF-β313 also promoted the “closing” of the dimer, the assigned chemical shifts common to TGF-β3 and TGF-β313 were compared (Figure 4A). This showed that there were three regions with relative large chemical shift perturbations centered about Ile22, Leu28, and Tyr50. These residues were found to be either in the heel region or in the prehelix loop, which in the closed form of TGF-β3, directly interact with α3 (Figure 4B). This indicates that the conformation of TGF-β313 differs from TGF-β3, most likely because it adopts a closed conformation similar to TGF-β1.
Figure 4.

Chemical shift perturbation between TGF-β3 and TGF-β313 and TGF-β3 and TGF-β3H4. (A, C) Chemical shift perturbation between TGF-β3 and TGF-β313 and TGF-β3 and TGF-β3H4, respectively. Plotted values correspond to the average chemical shift perturbation of 1H and 15N chemical shifts given by Δav = [(Δδ2NH + Δδ2N/25)/2]1/2 between corresponding residues. (B, D) Residues with the largest shift perturbations between TGF-β3 and TGF-β313 and TGF-β3 and TGF-β3H4, respectively, mapped onto the crystal structure of TGF-β3 (PDB code 1TGJ).
The backbone 15N T1 and T2 relaxation times for residues in regions of regular secondary structure of TGF-β313 were found to have mean values of 1116 ± 66 and 60.0 ± 7.0 ms, respectively, when measured at a 15N frequency of 70.95 MHz. These relaxation times correspond to an isotropic rotational correlation time (τc) of 12.7 ns, which is very close to the 12.2 ns rotational correlation time of TGF-β1 deduced from backbone 15N T1 and T2 relaxation times measured at 50.68 MHz (Supporting Information, Tables S1 and S2). The correspondence of the correlation times for TGF-β313 with those of TGF-β1 would be unexpected if the monomers were not rigidly packed against one another as in TGF-β1, thereby providing further evidence that TGF-β313 predominantly adopts the closed form with the two monomers rigidly packed against one another.
Flexibility and Conformational Preference Also Switched for Helix-Stabilized TGF-β3 Variant, TGF-β3H4
The data presented above clearly demonstrate that swapping the region between residues 54–75 swaps the flexibility and the position of the KCO equilibrium. The effect of swapping residues 54–75 on KCO could be through an increase in the intrinsic stability of α3, or alternatively through alterations in interactions between residues in α-helix 3 and residues in the heel region of the other monomer. Though it was previously hypothesized that helical stability was the major contributor to differences in KCO between TGF-β1 and TGF-β3, this was not directly evaluated through the studies of the two chimeras, TGF-β313 and TGF-β131. To evaluate this more directly, a helix-stabilized variant of TGF-β3 was designed that had just four amino acid substitutions relative to TGF-β3. The substitutions include H58Y, G63A and T67Q within α3 and A54L outside α3. The H58, G63, and T67 residues of TGF-β3 were selected for substitution since Agadir calculations showed that these residues contribute the greatest to the helical stability of α3 in TGF-β1 (Table 1). The Agadir calculations further showed that simultaneous substitution of all three residues increases the helical stability of α3 in TGF-β3 such that it is comparable to that of TGF-β1 (Table 1). Two of the substituted residues within α3 are expected to have no direct contact with the opposing monomer A63 and Q67, while the third substituted residue, H58 packs against the other monomer with surface area of 117 Å2. The A54L substitution was selected because L54 packs against A63 in TGF-β1 and may therefore indirectly stabilize the helix.
The helix-stabilized TGF-β3 variant, TGF-β3H4, was expressed, refolded, and purified using the same procedure as TGF-β313. The 1H–15N HSQC spectrum of TGF-β3H4 showed highly dispersed peaks with uniform intensities. This indicated that TGF-β3H4 likely adopts an ordered structure, similar to TGF-β1 and TGF-β313. To examine this more directly, a 15N,13C labeled TGF-β3H4 sample was prepared, and its backbone resonances were assigned using standard triple-resonance methodology (Figure 5A). To examine the backbone flexibility, a 15N-labeled TGF-β3H4 sample was prepared, and the backbone {1H}-15N NOEs were measured (Figure 5B). The {1H}-15N NOE measurements showed that TGF-β3H4 had a similar overall pattern as TGF-β313, including T60, V61, A63, and Q67 within α3, which all had high (>0.75) {1H}-15N NOE values. The analysis of secondary structure propensities using the program PECAN further showed there is a high probability for helix formation in the α3 region (Figure 5C). Thus, TGF-β3H4 has a structurally ordered α-helix 3 region, similar to TGF-β1 and TGF-β313. TGF-β3H4 also appears to adopt the closed conformation as a comparison of the assigned chemical shifts of TGF-β3H4 with those of TGF-β3 identified the same three regions in the comparison of TGF-β313 and TGF-β3 (Figure 4C,D).
Figure 5.

Resonance assignment, {1H}-15N NOE measurements, and secondary structure analysis of TGF-β3H4. (A) Assigned 1H–15N HSQC of TGF-β3H4. Assigned peaks are indicated by their one letter amino acid code and residue number. (B) {1H}-15N NOEs for TGF-β3H4. (C) Secondary structure probabilities of TGF-β3H4 calculated from NMR secondary shifts by the program PECAN;56 positive and negative values indicate α-helical and β-strand probabilities, respectively. Secondary structure diagram shown along the top corresponds to that of TGF-β1 (PDB 1KLC).
Circular Dichroism Measurements
The TGF-β isoforms have been previously analyzed by circular dichroism (CD) under acidic conditions.57 These studies showed that the measured helical content of TGF-β1 matched that calculated from its three-dimensional structure, while that of TGF-β3 was much lower than expected (at the time the only available structure was the crystal structure of TGF-β3, which as noted, adopts the closed form15). Thus, it should be possible to complement the NMR measurements described above with CD measurements. TGF-β1, TGF-β3, TGF-β313, TGF-β131, and TGF-β3H4 were therefore analyzed by CD under conditions comparable to that previously used in Pellaud’s study (10 mM phosphoric acid, pH 2.9) (Figure 6). The CD spectrum of TGF-β1 shows more negative ellipticity at 220 nm and more positive elliptictiy at 200 nm compared to TGF-β3, consistent with the NMR analysis that shows that TGF-β1 has greater helical content. The CD spectra of TGF-β313 and TGF-β3H4 were furthermore shown to be similar to that of TGF-β1, while the TGF-β131 spectrum was shown to be similar to that of TGF-β3. These results confirm the prior conclusions drawn from the NMR studies: TGF-β1, TGF-β313, and TGF-β3H4 include a stable α3 and are predominantly closed, while TGF-β3 and TGF-β131 include a disordered α3 and are predominantly open. The similarity of the CD spectra for the proteins in the two groups further suggest that the position of the KCO equilibrium is also qualitatively similar; i.e., KCO for TGF-β313 and TGF-β3H4 is similar to that of TGF-β1, and KCO of TGF-β131 is similar to that TGF-β3.
Figure 6.
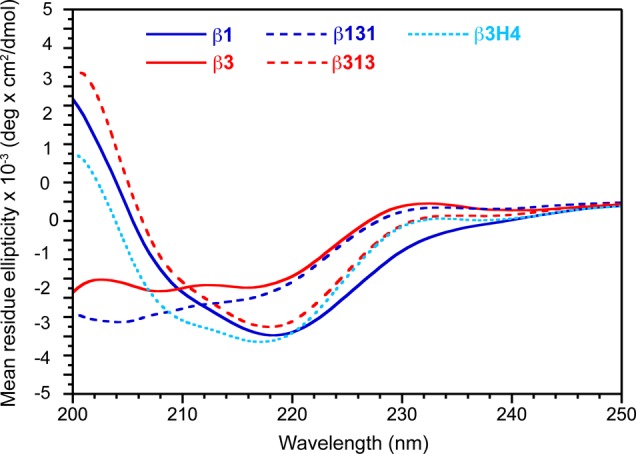
CD spectra of TGF-β1, TGF-β3, TGF-β131, TGF-β313, and TGF-β3H4 in 10 mM H3PO4 at pH 2.9 and 25 °C.
Biological Activities
TGF-β1 and TGF-β3 are indistinguishable in most commonly used cell-culture assays to assess TGF-β activity, such as growth inhibition or Smad phosphorylation assays. TGF-β1 and TGF-β3 have however been shown to be distinguishable in a chemoregulated migration assay in three-dimensional collagen matrices, which is thought to underlie their distinct functions in dermal wound healing.25 The assay is performed using a “sandwich” format in which dermal fibroblasts are plated at the interface between an upper and lower matrix compartment in the presence of an isotropic distribution of test mitogen. The migration of the fibroblasts in response to the different conditions is examined by quantifying their disposition within the collagen matrix. TGF-β3 is active in the sandwich assay, eliciting directional migration in response to cytokine distribution, while TGF-β1 and TGF-β2 neither promote nor inhibit directional migration. This assay was performed using TGF-β131, TGF-β313, and TGF-β3H4, together with TGF-β1 and TGF-β3 as controls (Figure 7). The assay was repeated three times, and errors were calculated by statistical analysis.25 TGF-β131 gained function in this assay so that it induced migration similar to TGF-β3, while TGF-β313 lost function and thus does not induce migration, similar to TGF-β1. TGF-β3H4 had significantly diminished activity compared to TGF-β3 and is similar, but not identical, to TGF-β1 (Figure 7). These data show that the biological activity correlates with KCO: TGF-β3 and TGF-β131 both favor the open form (KCO ≫ 1) and potently induce migration, while TGF-β1, TGF-β313, and TGF-β3H4 favor the closed form (KCO ≪ 1) and have little to no ability to induce migration.
Figure 7.

Chemoregulated motogenic response of dermal fibroblasts to TGF-β ligands in the sandwich migration assay. Measurements for each TGF-β at each concentration were performed in triplicate. The experiment was repeated three times, and the reported values are the mean from a single representative experiment; reported errors correspond to the standard deviation among replicates. Baseline migration controls in the absence of ligands ranged from 1.3 to 2.0% and is represented by the dashed horizontal lines.
Receptor Binding Properties
TGF-β1 and TGF-β3 had previously been characterized in terms of binding TβRII and recruiting TβRI using SPR-based methods with immobilized biotinylated TGF-βs.37,58 These measurements, while demonstrating very overall similar affinities and kinetics, had shown small differences in the dissociation rate for TβRI binding by the TGF-β:TβRII complex. TGF-β1 and TGF-β3 have a similar association rate for TβRI recruitment, but TGF-β3 has slower dissociation rate and hence a slightly greater affinity.58 To determine whether these small differences in TβRI binding were caused by differences in KCO, SPR kinetic analysis was performed with biotinylated TGF-β1, TGF-β3, TGF-β313, TGF-β131, and TGF-β3H4. To minimize artifacts that might arise from biotinylation, TGF-βs were each prebound to the purified TβRI and TβRII ectodomains and then biotinylated. The biotinylated TGF-βs were then isolated using high resolution ion-exchange chromatography under denaturing conditions, which not only enabled complete removal of the receptors, but also separation of singly, doubly, and multiply biotinylated forms. The singly biotinylated TGF-βs were then captured onto a high density streptavidin surface. The sensorgrams, along with the fitted kinetic traces, are shown in Figure 8. The tabulated rate and dissociation constants are presented in Table 3.
Figure 8.

Quantitative SPR measurements of receptor binding properties with biotinylated TGF-βs. (A) Sensorgrams obtained as TβRII alone was injected (left) or TβRI was injected in the presence of 4 μM TβRII (right) over a TGF-β1 surface. The traces shown (black) correspond to triplicate measurements of a 2-fold serial dilution of the receptor over the concentration range shown. The orange curves correspond to global fits of each data set to a 1:1 binding model. (B–E) Same SPR experiments as in A, except over TGF-β3, TGF-β131, TGF-β313, or TGF-β3H4 surfaces, respectively.
Table 3. Binding Constants for TGF-β Ligands to the Signaling Receptors.
| surface | analyte | buffer suppl | kon (M–1 s–1) | koff (s–1) | Kd (μM) | Rmax (RU) |
|---|---|---|---|---|---|---|
| TGF-β1 | TβRII | none | 7.2(±0.4) × 105 | 0.121(±0.007) | 0.17 ± 0.01 | 53.6 ± 4.3 |
| TGF-β3 | TβRII | none | 1.2(±0.1) × 106 | 0.199(±0.009) | 0.17 ± 0.01 | 79.2 ± 4.0 |
| TGF-β131 | TβRII | none | 5.5(±0.1) × 105 | 0.124(±0.002) | 0.22 ± 0.01 | 41.3 ± 1.0 |
| TGF-β313 | TβRII | none | 1.4(±0.1) × 106 | 0.171(±0.003) | 0.13 ± 0.01 | 64.5 ± 3.2 |
| TGF-β3H4 | TβRII | none | 5.1(±0.2) × 105 | 0.083(±0.002) | 0.16 ± 0.01 | 67.0 ± 3.2 |
| TGF-β1 | TβRI | 4 μM TβRII | 3.3(±0.1) × 104 | 7.6(±0.4)x × | 0.24 ± 0.01 | 27.6 ± 1.4 |
| TGF-β3 | TβRI | 4 μM TβRII | 3.5(±0.1) × 104 | 1.2(±0.1) × 10–3 | 0.034 ± 0.002 | 59.3 ± 3.0 |
| TGF-β131 | TβRI | 4 μM TβRII | 5.2(±0.1) × 104 | 2.6(±0.1) × 10–3 | 0.051 ± 0.001 | 27.4 ± 1.4 |
| TGF-β313 | TβRI | 4 μM TβRII | 5.4(±0.1) × 104 | 5.1(±0.2) × 10–2 | 0.95 ± 0.04 | 56.5 ± 2.9 |
| TGF-β3H4 | TβRI | 4 μM TβRII | 4.0(±0.1) × 104 | 1.9(±0.4) × 10–3 | 0.047 ± 0.003 | 56.4 ± 3.0 |
TGF-β313 and TGF-β131 are shown to retain the same kinetics and affinity for binding TβRII as TGF-β3 and TGF-β1. The small difference in TβRI recruitment by TGF-β1 and TGF-β3 noted in the prior study is reversed: TGF-β131 has a characteristically slow off-rate, which is similar to TGF-β3, and TGF-β313 has a faster off-rate, similar to TGF-β1. TGF-β3H4 binds TβRII with the same kinetics and affinity as TGF-β3, but its kinetics and affinity for recruiting TβRI are characteristic of TGF-β3, not TGF-β1 (Figure 8E, Table 3). Thus, even though TGF-β3H4 has a stable α-helix 3 and adopts the closed form, this does not affect the kinetics of TβRI binding. Together, these results show that the small difference in TβRI recruitment is not correlated with the differences in KCO.
Discussion
The unique phenotypes of the TGF-β isoform-specific null mice are largely explained by differences in the expression patterns of the isoforms, indicating that the diversification of the TGF-β superfamily to include three closely related TGF-β isoforms has largely been driven by advantages that arise from differences in their spatial-temporal expression patterns.20−23 The expansion of functionality by duplicating genes to fine-tune spatial-temporal expression, without altering the properties of the ligands so that they can be distinguished by the signaling receptors or other binding proteins, does impose the fundamental limitation that the isoforms can only achieve their distinct functions without overlap in their expression patterns. Thus, there is some advantage for ligands of the superfamily to be distinguished, either by the signaling receptors or by other binding proteins that differentially regulate access to the signaling receptors.
The objective of this study was to investigate whether the previously reported differences in the equilibrium between the closed and open monomer arrangements, KCO, was caused by differences in helical stability and whether the differences in KCO might contribute to the unique requirement of TGF-β1 and TGF-β3 in vivo that has been previously suggested based on gene replacement30,31 and other studies.24,26−29 The construction of the chimeric proteins, in which the unstable palm helix in TGF-β3 was swapped into TGF-β1 and the stable palm helix in TGF-β1 was swapped into TGF-β3, and analysis of these by NMR and CD, showed that swapping the helical regions swapped their preferred conformation. TGF-β313 was shown to adopt the closed conformation characteristic of TGF-β1 (Figures 3–4, 6), while TGF-β131 was shown to adopt the open conformation characteristic of TGF-β3 (Figures 3–6). The migration of dermal fibroblasts through a matrix of native collagen was shown to correlate with the preferred conformation of the ligand, with the predominantly closed ligands lacking detectable chemoregulated migration activity, and the open ligands possessing potent chemoregulated migration activity (Figure 7).
The primary limitation of the chimeras is that it is not possible to determine whether the altered properties—specifically the shifts in the KCO equilibrium or changes in migration—were a direct consequence of changes in the helical stability or altered conformation, or whether the inclusion of sequence from the other isoform affected the equilibrium and migration through other mechanisms. This limitation was addressed by generating another TGF-β3 variant, TGF-β3H4, in which only four residues were substituted. The substitution of these four residues was predicted to increase the helical stability to a similar extent as the TGF-β313 chimera, but because only four residues were substituted rather than nine, this decreased the likelihood that these residues would alter either the conformation or migration through other mechanisms. Thus, the finding that TGF-β3H4 adopted the closed conformation and lost most of its activity in the migration assay provides strong, though not conclusive evidence, that the shift in the KCO equilibrium toward the closed state is caused by an increase in helical stability and that the activity in the migration assay is specifically due to the open conformation. The finding that the migration activity of TGF-β3H4 was not entirely abolished might be because KCO is not as far shifted to the closed form as that of the TGF-β313. This is not apparent from either the TGF-β3H4 α3 secondary structure propensities or magnitude of the TGF-β3H4–TGF-β3 shift perturbations, which were indistinguishable from those of TGF-β313, but is suggested by the {1H}-15N NOE data, which showed somewhat increased flexibility in the prehelix loop (Figure 5b).
To investigate the underlying mechanistic basis by which the alterations in the sequence and conformation of the TGF-βs might alter activity, the binding properties of TGF-β313, TGF-β131, and TGF-β3H4 for the signaling receptors, TβRI and TβRII, were investigated using SPR. The previous SPR studies performed in our laboratory showed that TGF-β1 and TGF-β3 bound TβRII with the same overall affinity and kinetics, but had small differences in their ability to cooperate with TβRII to bind and recruit TβRI.58 This, together with the accompanying structures of TGF-β1 and TGF-β3 bound to the TβRI and TβRII ectodomains (Supporting Information, Figure S3), suggested that these differences were due to residues 51, 57, and 60 within the TβRI binding interface, which differ between TGF-β1 and TGF-β3.58
The SPR binding studies reported here showed that TGF-β131 had a somewhat higher affinity for TβRI due to a somewhat slower disassociation rate, while TGF-β313 has a somewhat lower affinity for TβRI due to a somewhat faster disassociation rate (Table 3). This change in TβRI binding by TGF-β131 and TGF-β313 is consistent with the swap of residues 57 and 60, although it is impossible to rule out that the altered conformation might have also had an effect since the change in TβRI binding also correlates with the change of conformation. The SPR binding studies further showed that the ability of TGF-β3H4 to bind and recruit TβRI was indistinguishable from that of TGF-β3. The absence of an effect on TβRI binding is consistent with the substitution of none of the residues known to affect TβRI binding, but it is inconsistent with the altered conformation, which was closed for TGF-β3H4, but open for TGF-β3. The overall conclusion, therefore, is that the small differences in the rate at which TβRI disassociates is determined by whether the TGF-βs include the residues previously shown to influence TβRI binding, not whether they adopt the open or closed state.
This finding is perhaps not surprising as the TβRI- and TβRII-bound forms of TGF-β1 and TGF-β3 have been shown to both be closed,6,58 and the rate of dissociation is presumably determined by the rate at which all of the stabilizing interactions, including those between residues 51, 57, and 60 of TGF-β and TβRI mentioned above, are simultaneously disrupted. The finding that the TβRI association kinetics do not correlate with the conformation of the TGF-βs is somewhat surprising given that some of the TGF-βs are predominantly open and must undergo a transition to the closed form upon binding TβRI (since the bound forms for TGF-β1 and TGF-β3 have been shown to be closed6,58). The fact that the association rates do not correlate suggests that the kinetics of the open and closing transition are likely fast compared to other steps required for TβRI recruitment, which might well be the case given that the TβRII N-terminus has previously been shown to undergo a disorder-to-order transition as it binds TβRI.6,34,58
This leads us to conclude that the differential effect of the TGF-βs in the migration assay is due to differential interactions with other TGF-β binding proteins that might be present. These differential interactions could be mediated by the overall conformational differences between the TGF-βs, or alternatively through direct recognition of the substituted amino acid residues. There is at present insufficient data to distinguish these mechanisms, but it is nevertheless tempting to speculate that it is the former, not the latter, as the opening and closing of TGF-β results in a very large overall change in conformation that has a high potential to be differentially recognized, either structurally or as a result of differences in charge or hydrophobicity.
There are several TGF-β binding proteins known, including membrane-bound nonsignaling receptors, such the type III receptor, or soluble proteins, such as the shed form of the type III receptor, α2-macroglobulin, or decorin. These proteins could be derived from the relatively low levels of calf serum (1%) that were added to the collagen gels or proteins that are secreted or shed by the dermal fibroblasts. The majority of TGF-β binding proteins preferentially bind one isoform over the others. TGF-β2 binds the soluble type III receptor, for example, with an affinity that is roughly 3-fold greater than TGF-β1 and 4-fold greater than TGF-β3.49 Both α2-macroglobulin and decorin preferentially bind TGF-β1 and TGF-β2.59,60 Hence, α2-macroglobulin and decorin might bind TGF-β3 more transiently compared to TGF-β1, which in turn could increase the rate at which TGF-β3 diffuses through the collagen gels compared to TGF-β1. Collagen isoforms have not been reported to bind TGF-βs, but if they do have a slight propensity to interact, this could lead to the observed differences in the migration assay, as TGF-βs that are closed or open might have a differing propensity to interact with collagen thus affecting their diffusion through the collagen matrix. Abundant decorin and collagen is present in the epidermis; thus it seems plausible that the TGF-βs are differentially interacting with these molecules, as opposed to the soluble type III receptor or α2-macroglubin, which are mainly present in serum.
The results present here have shown that residues within helix 3 determine whether TGF-βs adopt the open or closed state and that the open or closed state is likely responsible for the differential activity of TGF-β1 and TGF-β3, as least as it relates to signaling activity in the epidermis. The fact that intrinsic differences among the isoforms do contribute to their unique activities most likely reflects an added adaptation that has enabled the TGF-βs to take upon their unique and distinctive functions in vivo.
Acknowledgments
The authors would like to acknowledge Dr. Peter Sun for the TGF-β1 cell line and plasmid, Dr. Kathy Flanders for guidance and suggestions regarding the cell-based migration assays, and Dr. Eileen Lafer who helped with the design of some of the SPR experiments.
Glossary
Abbreviations
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser enhancement
- TGF-β
transforming growth factor-β
- TGF-β313
TGF-β3 in which residues 54–75 were swapped with the corresponding residues from TGF-β1
- TGF-β131
TGF-β1 in which residues 54–75 were swapped with the corresponding residues from TGF-β3
- TGF-β3H4
TGF-β3 bearing A54L, H58Y, G63A, and T67Q single amino acid substitutions
- TβRI 7–91
residues 7–91 of the extracellular domain of the TGF-β type I receptor
- TβRII ED
residues 15–130 of the extracellular domain of the TGF-β type II receptor
- SPR
surface plasmon resonance
- CD
circular dichroism
Supporting Information Available
Supplementary Figures S1–S5 and Tables S1–S2. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Novo Nordisk Research Center China, Beijing, China.
The authors declare no competing financial interest.
Dedication
This paper is dedicated to Seth L. Schor who had a lifelong enthusiasm for science and passed away just prior to the submission of this paper.
This research was supported by grants from the NIH (GM58670 awarded to A.P.H., CA172886 awarded to A.P.H and L-Z.S., and CA54174 awarded to the U. Texas HSC San Antonio Cancer Therapy and Research Center), the Robert A. Welch Foundation (AQ-1842 awarded to A.P.H.), and the Cancer Prevention and Research Institute in Texas (RP120867 awarded to A.P.H).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Massague J. (1998) TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Loeys B. L.; Mortier G.; Dietz H. C. (2013) Bone lessons from Marfan syndrome and related disorders: fibrillin, TGF-B and BMP at the balance of too long and too short. Pediatr. Endocrinol. Rev. 10(Suppl. 2), 417–423. [PubMed] [Google Scholar]
- Massague J. (2008) TGFbeta in Cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A.; Dobaczewski M.; Frangogiannis N. G. (2011) TGF-beta signaling in fibrosis. Growth Factors 29, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana J. L.; Attisano L.; Carcamo J.; Zentella A.; Doody J.; Laiho M.; Wang X. F.; Massague J. (1992) TGF beta signals through a heteromeric protein kinase receptor complex. Cell 71, 1003–1014. [DOI] [PubMed] [Google Scholar]
- Groppe J.; Hinck C. S.; Samavarchi-Tehrani P.; Zubieta C.; Schuermann J. P.; Taylor A. B.; Schwarz P. M.; Wrana J. L.; Hinck A. P. (2008) Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 29, 157–168. [DOI] [PubMed] [Google Scholar]
- Massague J. (2008) A very private TGF-beta receptor embrace. Mol. Cell 29, 149–150. [DOI] [PubMed] [Google Scholar]
- Wrana J. L.; Attisano L.; Wieser R.; Ventura F.; Massague J. (1994) Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Massague J. (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700. [DOI] [PubMed] [Google Scholar]
- Moustakas A.; Heldin C. H. (2005) Non-Smad TGF-beta signals. J. Cell Sci. 118, 3573–3584. [DOI] [PubMed] [Google Scholar]
- Lopez-Casillas F.; Wrana J. L.; Massague J. (1993) Betaglycan presents ligand to the TGF beta signaling receptor. Cell 73, 1435–1444. [DOI] [PubMed] [Google Scholar]
- Cheifetz S.; Hernandez H.; Laiho M.; ten Dijke P.; Iwata K. K.; Massague J. (1990) Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J. Biol. Chem. 265, 20533–20538. [PubMed] [Google Scholar]
- Hinck A. P.; Archer S. J.; Qian S. W.; Roberts A. B.; Sporn M. B.; Weatherbee J. A.; Tsang M. L.; Lucas R.; Zhang B. L.; Wenker J.; Torchia D. A. (1996) Transforming growth factor beta 1: three-dimensional structure in solution and comparison with the X-ray structure of transforming growth factor beta 2. Biochemistry 35, 8517–8534. [DOI] [PubMed] [Google Scholar]
- Daopin S.; Li M.; Davies D. R. (1993) Crystal structure of TGF-beta 2 refined at 1.8 A resolution. Proteins 17, 176–192. [DOI] [PubMed] [Google Scholar]
- Mittl P. R.; Priestle J. P.; Cox D. A.; McMaster G.; Cerletti N.; Grutter M. G. (1996) The crystal structure of TGF-beta 3 and comparison to TGF-beta 2: implications for receptor binding. Protein Sci. 5, 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlunegger M. P.; Grutter M. G. (1992) An unusual feature revealed by the crystal structure at 2.2 A resolution of human transforming growth factor-beta 2. Nature 358, 430–434. [DOI] [PubMed] [Google Scholar]
- Nilsen-Hamilton M. (1990) Transforming growth factor-beta and its actions on cellular growth and differentiation. Curr. Top Dev. Biol. 24, 95–136. [PubMed] [Google Scholar]
- Qian S. W.; Burmester J. K.; Merwin J. R.; Madri J. A.; Sporn M. B.; Roberts A. B. (1992) Identification of a structural domain that distinguishes the actions of the type 1 and 2 isoforms of transforming growth factor beta on endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 89, 6290–6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baardsnes J.; Hinck C. S.; Hinck A. P.; O’Connor-McCourt M. D. (2009) TbetaR-II discriminates the high- and low-affinity TGF-beta isoforms via two hydrogen-bonded ion pairs. Biochemistry 48, 2146–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A. B.; Huh C. G.; Becker D.; Geiser A.; Lyght M.; Flanders K. C.; Roberts A. B.; Sporn M. B.; Ward J. M.; Karlsson S. (1993) Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U. S. A. 90, 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull M. M.; Ormsby I.; Kier A. B.; Pawlowski S.; Diebold R. J.; Yin M.; Allen R.; Sidman C.; Proetzel G.; Calvin D.; et al. (1992) Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford L. P.; Ormsby I.; Gittenberger-de Groot A. C.; Sariola H.; Friedman R.; Boivin G. P.; Cardell E. L.; Doetschman T. (1997) TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G.; Pawlowski S. A.; Wiles M. V.; Yin M.; Boivin G. P.; Howles P. N.; Ding J.; Ferguson M. W.; Doetschman T. (1995) Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M.; Foreman D. M.; Ferguson M. W. (1995) Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 108(Pt 3), 985–1002. [DOI] [PubMed] [Google Scholar]
- Schor S. L.; Ellis I. R.; Harada K.; Motegi K.; Anderson A. R.; Chaplain M. A.; Keatch R. P.; Schor A. M. (2006) A novel ’sandwich’ assay for quantifying chemo-regulated cell migration within 3-dimensional matrices: wound healing cytokines exhibit distinct motogenic activities compared to the transmembrane assay. Cell Motil. Cytoskeleton 63, 287–300. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay B.; Fan J.; Guan S.; Li Y.; Chen M.; Woodley D. T.; Li W. (2006) A ″traffic control″ role for TGFbeta3: orchestrating dermal and epidermal cell motility during wound healing. J. Cell Biol. 172, 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gato A.; Martinez M. L.; Tudela C.; Alonso I.; Moro J. A.; Formoso M. A.; Ferguson M. W.; Martinez-Alvarez C. (2002) TGF-beta(3)-induced chondroitin sulphate proteoglycan mediates palatal shelf adhesion. Dev. Biol. 250, 393–405. [PubMed] [Google Scholar]
- Taya Y.; O’Kane S.; Ferguson M. W. (1999) Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 126, 3869–3879. [DOI] [PubMed] [Google Scholar]
- Tudela C.; Formoso M. A.; Martinez T.; Perez R.; Aparicio M.; Maestro C.; Del Rio A.; Martinez E.; Ferguson M.; Martinez-Alvarez C. (2002) TGF-beta3 is required for the adhesion and intercalation of medial edge epithelial cells during palate fusion. Int. J. Dev Biol. 46, 333–336. [PubMed] [Google Scholar]
- Yang L. T.; Kaartinen V. (2007) Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev. Biol. 312, 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. E.; Wankhade U. D.; Konkel J. E.; Cherukuri K.; Nagineni C. N.; Flanders K. C.; Arany P. R.; Chen W.; Rane S. G.; Kulkarni A. B. (2013) Transforming growth factor-beta3 (TGF-beta3) knock-in ameliorates inflammation due to TGF-beta1 deficiency while promoting glucose tolerance. J. Biol. Chem. 288, 32074–32092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocharov E. V.; Blommers M. J.; Kuhla J.; Arvinte T.; Burgi R.; Arseniev A. S. (2000) Sequence-specific 1H and 15N assignment and secondary structure of transforming growth factor beta3. J. Biomol. NMR 16, 179–180. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Korzhnev D. M.; Blommers M. J.; Arvinte T.; Orekhov V. Y.; Billeter M.; Arseniev A. S. (2002) Dynamics-modulated biological activity of transforming growth factor beta3. J. Biol. Chem. 277, 46273–46279. [DOI] [PubMed] [Google Scholar]
- Hart P. J.; Deep S.; Taylor A. B.; Shu Z.; Hinck C. S.; Hinck A. P. (2002) Crystal structure of the human TbetaR2 ectodomain--TGF-beta3 complex. Nat. Struct. Biol. 9, 203–208. [DOI] [PubMed] [Google Scholar]
- Grutter C.; Wilkinson T.; Turner R.; Podichetty S.; Finch D.; McCourt M.; Loning S.; Jermutus L.; Grutter M. G. (2008) A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions. Proc. Natl. Acad. Sci. U.S.A. 105, 20251–20256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z.; Sun P. D. (2004) Overexpression of human transforming growth factor-beta1 using a recombinant CHO cell expression system. Protein Expression Purif. 37, 265–272. [DOI] [PubMed] [Google Scholar]
- Huang T.; David L.; Mendoza V.; Yang Y.; Villarreal M.; De K.; Sun L.; Fang X.; Lopez-Casillas F.; Wrana J. L.; Hinck A. P. (2011) TGF-beta signalling is mediated by two autonomously functioning TbetaRI:TbetaRII pairs. EMBO J. 30, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga J. E.; Groppe J. C.; Cui Y.; Hinck C. S.; Contreras-Shannon V.; Pakhomova O. N.; Yang J.; Tang Y.; Mendoza V.; Lopez-Casillas F.; Sun L.; Hinck A. P. (2005) Assembly of TbetaRI:TbetaRII:TGFbeta ternary complex in vitro with receptor extracellular domains is cooperative and isoform-dependent. J. Mol. Biol. 354, 1052–1068. [DOI] [PubMed] [Google Scholar]
- Hinck A. P.; Walker K. P. 3rd; Martin N. R.; Deep S.; Hinck C. S.; Freedberg D. I. (2000) Sequential resonance assignments of the extracellular ligand binding domain of the human TGF-beta type II receptor. J. Biomol. NMR 18, 369–370. [DOI] [PubMed] [Google Scholar]
- Marley J.; Lu M.; Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75. [DOI] [PubMed] [Google Scholar]
- Wittekind M.; Mueller L. (1993) Hncacb, a High-Sensitivity 3d Nmr Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha-Carbon and Beta-Carbon Resonances in Proteins. J. Magn. Reson. Ser. B 101, 201–205. [Google Scholar]
- Grzesiek S.; Bax A. (1993) Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR 3, 185–204. [DOI] [PubMed] [Google Scholar]
- Grzesiek S.; Dobeli H.; Gentz R.; Garotta G.; Labhardt A. M.; Bax A. (1992) 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-gamma. Biochemistry 31, 8180–8190. [DOI] [PubMed] [Google Scholar]
- Kay L. E.; Ikura M.; Tschudin R.; Bax A. (1990) 3-Dimensional Triple-Resonance Nmr-Spectroscopy of Isotopically Enriched Proteins. J. Magn. Reson. 89, 496–514. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G.. SPARKY 3; University of California: San Francisco, 2008. [Google Scholar]
- Kay L. E.; Torchia D. A.; Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979. [DOI] [PubMed] [Google Scholar]
- Freedberg D. I.; Ishima R.; Jacob J.; Wang Y. X.; Kustanovich I.; Louis J. M.; Torchia D. A. (2002) Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 11, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza V.; Vilchis-Landeros M. M.; Mendoza-Hernandez G.; Huang T.; Villarreal M. M.; Hinck A. P.; Lopez-Casillas F.; Montiel J. L. (2009) Betaglycan has two independent domains required for high affinity TGF-beta binding: proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry 48, 11755–11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz V.; Serrano L. (1994) Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1, 399–409. [DOI] [PubMed] [Google Scholar]
- Munoz V.; Serrano L. (1995) Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol. 245, 275–296. [DOI] [PubMed] [Google Scholar]
- Deep S.; Walker K. P. 3rd; Shu Z.; Hinck A. P. (2003) Solution structure and backbone dynamics of the TGFbeta type II receptor extracellular domain. Biochemistry 42, 10126–10139. [DOI] [PubMed] [Google Scholar]
- Cerletti N. (2000) Process for the production of biologically active dimeric protein, U.S. Patent 6057430.
- Archer S. J.; Bax A.; Roberts A. B.; Sporn M. B.; Ogawa Y.; Piez K. A.; Weatherbee J. A.; Tsang M. L.; Lucas R.; Zheng B. L.; et al. (1993) Transforming growth factor beta 1: secondary structure as determined by heteronuclear magnetic resonance spectroscopy. Biochemistry 32, 1164–1171. [DOI] [PubMed] [Google Scholar]
- Archer S. J.; Bax A.; Roberts A. B.; Sporn M. B.; Ogawa Y.; Piez K. A.; Weatherbee J. A.; Tsang M. L.; Lucas R.; Zheng B. L.; et al. (1993) Transforming growth factor beta 1: NMR signal assignments of the recombinant protein expressed and isotopically enriched using Chinese hamster ovary cells. Biochemistry 32, 1152–1163. [DOI] [PubMed] [Google Scholar]
- Eghbalnia H. R.; Wang L.; Bahrami A.; Assadi A.; Markley J. L. (2005) Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J. Biomol. NMR 32, 71–81. [DOI] [PubMed] [Google Scholar]
- Pellaud J.; Schote U.; Arvinte T.; Seelig J. (1999) Conformation and self-association of human recombinant transforming growth factor-beta3 in aqueous solutions. J. Biol. Chem. 274, 7699–7704. [DOI] [PubMed] [Google Scholar]
- Radaev S.; Zou Z.; Huang T.; Lafer E. M.; Hinck A. P.; Sun P. D. (2010) Ternary complex of transforming growth factor-beta1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 285, 14806–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S. M.; Sugars R. V.; Wendel M.; Smith A. J.; Waddington R. J.; Cooper P. R.; Sloan A. J. (2009) TGF-beta/extracellular matrix interactions in dentin matrix: a role in regulating sequestration and protection of bioactivity. Calcif Tissue Int. 85, 66–74. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Ling T. Y.; Shieh H. S.; Johnson F. E.; Huang J. S.; Huang S. S. (2001) Identification of the high affinity binding site in transforming growth factor-beta involved in complex formation with alpha 2-macroglobulin. Implications regarding the molecular mechanisms of complex formation between alpha 2-macroglobulin and growth factors, cytokines, and hormones. J. Biol. Chem. 276, 46212–46218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.