

Abstract
Prostate cancers at the late stage of castration resistance are not responding well to most of current therapies available in clinic, reflecting a desperate need of novel treatment for this life-threatening disease. In this study, we evaluated the anti-cancer effect of a recently isolated natural compound Alternol in multiple prostate cancer cell lines with the properties of advanced prostate cancers in comparison to prostate-derived non-malignant cells. As assessed by trypan blue exclusion assay, a significant cell death was observed in all prostate cancer cell lines except DU145 but not in non-malignant (RWPE-1and BPH1) cells. Further analyses revealed that Alternol-induced cell death was an apoptotic response in a dose- and time-dependent manner, as evidenced by the appearance of apoptosis hallmarks such as Caspase-3 processing and PARP cleavage. Interestingly, Alternol-induced cell death was completely abolished by reactive oxygen species (ROS) scavengers, N-acetylcysteine (N-Ac) and dihydrolipoic acid (DHLA). We also demonstrated that the pro-apoptotic Bax protein was activated after Alternol treatment and was critical for Alternol-induced apoptosis. Animal xenograft experiments in nude mice showed that Alternol treatment largely suppressed tumor growth of PC-3 xenografts but not Bax-null DU-145 xenografts in vivo. These data suggest that Alternol might serve as a novel anticancer agent for late stage prostate cancer patient.
Keywords: Alternol, prostate cancer, apoptosis, Bax, Oxidative stress, tumor suppression
Introduction
Although a steady decline of prostate cancer-related death in the past decade and numerous advances in early diagnosis and monitoring, advanced disease at the castration-resistant stage is still a big challenge for the community (1). While a few chemical compounds were approved for clinical use, currently castration-resistant prostate cancers (CRPC) are virtually not curable due to lack of substantial response to most of therapeutic agents that are clinically available (2). Therefore, development of effective therapies is an urgent need for this patient population.
The novel compound Alternol was isolated from fermenting products by microorganism named as Alternaria alternata var. monosporus, which was obtained from the bark of yew tree in Kunming, China (3-5). This source is similar as the current anti-cancer drug Paclitaxel (6), which has been effectively used for lung, breast and gastric cancers, and its derivative Docetaxel has been approved for use in prostate cancers (7). It has been shown that Alternol induced cell cycle arrest, interrupted epithelial-to-mesenchymal transition and apoptotic cell death in human cancer cell lines (3-5, 8, 9). A recent publication showed that Alternol preferentially kills prostate cancer cells over non-malignant prostatic epithelial cells, indicating a potential benefit for prostate cancer patients (10).
Reactive oxygen species (ROS) is a collective term embracing a variety of oxygen-containing, reactive and short-lived molecules. Essentially there are two types of ROS: free radical ROS, such as superoxide (O2-), hydroxyl radical (OH•) and nitric oxide (NO•), which contain one or more unpaired electron(s); and non-radical ROS, such as hydrogen peroxide (H2O2) or singlet oxygen, which do not contain unpaired electrons but are highly reactive and can give rise to radical forms of ROS (11). It has become increasingly evident that certain anticancer agents induce intracellular oxidative stress that is either the primary mechanism of cell death or is a secondary indirect effect that may lead to cell death (11, 12). Furthermore, cancer cells are more vulnerable to oxidative stress caused by ROS-inducing agents. Due to differential redox states between normal and cancer cells, the therapeutic strategies would be reconsidered to selectively utilize these ROS-inducing agents for cancer therapy (11).
Bax, the Bcl-2-associated X protein, is a cardinal pro-apoptotic member of BCL-2 family proteins, which regulates the critical balance between cell survival and death (13). It has been demonstrated that Bax transforms into a lethal mitochondrial oligomer in response to cellular stress and becomes activated to cause mitochondrial damage, a key step for the intrinsic pathway to apoptosis (14-16). The mechanisms that push Bax to mitochondria are not entirely clear but previous reports have shown that oxidative stress-induced dimerization promoted Bax translocation to mitochondria and that ROS acts upstream of calpain-mediated mitochondrial Bax cleavage (17-20).
In attempt to develop novel chemotherapy for CRPCs, we evaluated the anti-cancer effect of Alternol on multiple prostate cancer cell lines in comparison to non-malignant prostate-derived epithelial cells. In this report we demonstrated that Alternol induced massive cell killing in prostate cancer cells but less toxic to non-malignant prostate epithelial cells through an oxidative stress-dependent mechanism. We also confirmed that the pro-apoptotic protein Bax was activated in response to oxidative stress, which is critical in Alternol-induced apoptotic cell death. Animal experiments in nude mice indicated that Alternol treatment largely suppressed xenografts tumor growth in vivo. These data suggest that Alternol might be developed as a novel therapeutic agent for CRPC patients.
Materials and Methods
Cell culture, antibodies, and reagents
RWPE-1,LNCaP, DU145,PC-3and 22RV1 cells were purchased from ATCC (Manassas, VA)in February 2002 and maintained in a humidified atmosphere of 5% CO2, RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and antibiotics except that RWPE-1 cells were maintained in Keratinocyte-SFM media (Invitrogen, Carlsbad, CA). LAPC-4 was obtained from UCLA and C4-2 was purchased from UroCor (Oklahoma City, OK) in May 2002 as described in our previous publications (21, 22). BPH1 cell line was kindly provided by Dr Long-cheng Li (Department of Urololgy, University of California at San Francisco) in August 2011. These cell lines were authenticated using PromegaPowerPlex® technology and carried out at Genetica® DNA Laboratories (Burlington, NC) in January 2014. Alternol (99.9% purity) was a kind gift from Strand Biotech Co (Shantou, China) and its structural scheme is shown in Fig 1A. It was dissolved in dimethyl sulfoxide (DMSO) as a 10 mM stock solution. Antibodies for caspase-3, PARP, Bax, Bif-1, Bcl-2, Bcl-xL and BAD were obtained from Cell Signal (Danvers, MA). Pre-validated small interfering RNAs, Actin antibody, CPT-11, n-acetylcysteine (N-Ac), dihydrolipoic acid (DHLA), MG132, E-64, PD150606 and Bax channel blocker were purchased from Santa Cruz Biotech (Santa Cruz, CA). All the reagents were prepared according to the manufacturer's instruction and used as described in the figure legends. Final concentrations of the solvent did not exceed 0.1% of the culture media. Bax expression construct pCEP4-HA-hBax (23) was obtained from Add gene (Cambridge, MA) and the control empty vector was purchased from Invitrogen (Carlsbad, CA). DNA Ladder Detection Kit and Caspase-9 colorimetric activity assay kit were purchased from Millipore (Billerica, MA). Preassembled assay kit for total ROS detection was purchased from Enzo Life Sciences (Farmingdale, NY). Annexin V-FITC Apoptosis Detection kit was purchased from BD PharMingen (San Diego, CA). Mitochondrial membrane potential assay kit with the fluorescent dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl carbocyanine iodide (JC-1), Lipofectamine 2000 and RNAiMAX were purchased from Invitrogen (Carlsbad, CA). DAKO LSAB+ System kit was purchased from DAKO USA (Carpinteria, CA).
Fig 1. Alternol induces cell death preferentially in prostate cancer cells.
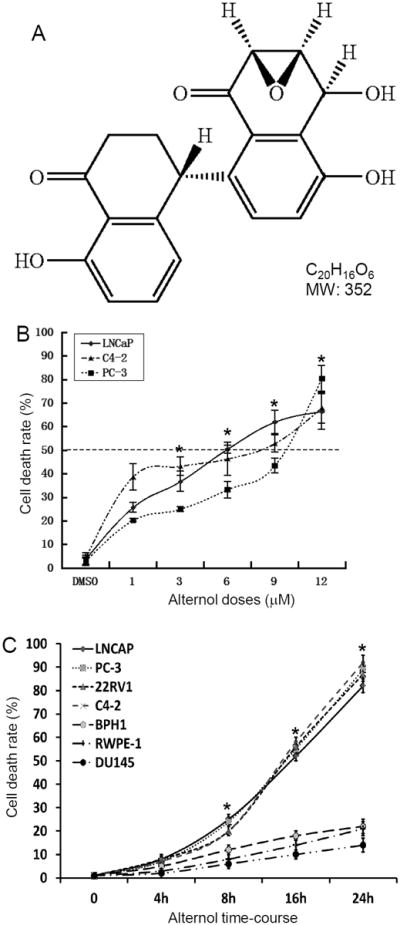
A The structure of Alternol (C20H16O6, MW352). B&C Cells were seeded in 12-well plates overnight and then treated with the solvent or Alternol in different concentration (panel B for 24 h) or for different time period as indicated (panel C, Alternol concentration 10 μM). Cells were harvested and stained in 0.4% trypan blue solution. Dead cells were counted using a hemocytometer under an inverted microscope. Data presented are the mean ± SEM from three independent experiments. The asterisk indicates a significant difference at each dosing level (ANOVA analysis, p < 0.05) compared to the DMSO control (DMSO or 0 h).
Cytotoxicity, flow cytometry and mitochondrial membrane potential assays
Cells were seeded at 3 × 104 cells/well in 12-well plates (trypan-blue assay) or in 6-well plate (flow cytometry assay). The next day, cells were treated with the solvent or Alternol as described in the figure legend. Cell viability was assessed with a trypan blue exclusion assay (22). Apoptotic cell death was evaluated with a flow cytometry-based Annexin V binding and PI staining assay, as described in our previous publication (22).
Mitochondrial Membrane Potential assay was done as previously described (22). Briefly, PC-3 cells were treated with the solvent (DMSO) or Alternol in the presence or absence of the anti-oxidants as indicated in the figures. Then PC-3 cells were incubated with JC-1 (0.3 μg/ml) for 15 min at 37°C. Thereafter, cells were analyzed and microscopic images were taken under a fluorescent microscope (Olympus, Japan), as described in our previous publications (22, 24).
DNA fragmentation and Caspase-9 activity assays
Cells were treated as indicated in the figures. Total genomic DNA was extracted using the DNA ladder detection kit by following the manufacturer's instructions. DNA ladders were analyzed on 1% agarose gel electrophoresis.
For caspases-9 assay, PC-3 cells were treated with the solvent or Alternol as indicated in the figures. Cells were rinsed with ice-cold PBS and lysed on ice in cell lysis buffer from the Caspase-9 colorimetric activity assay kit. Caspase-9 activity was measured by following the manufacturer's manual and presented as a relative value compared to the solvent control that was set as a value of 1.0.
Western blot assay
After treatment, cells were rinsed with ice-cold PBS and lysed on ice in RIPA buffer (Cell Signal, MA). Equal amount of proteins from each lysates was loaded onto SDS-PAGE gels, electrophoresed, and transferred onto PVDF membrane. Following electrotransfer, the membrane was blocked for 2 h in 5% nonfat dried milk; and then incubated with primary antibody overnight at 4°C. Visualization of the protein signal was achieved with horseradish peroxidase conjugated secondary antibody and enhanced chemiluminescence procedures according to the manufacturer's recommendation (Santa Cruz Biotech, Santa Cruz, CA).
Measurement of intracellular reactive oxygen species
The level of intracellular ROS generation was assessed with the total ROS detection kit (Enzo Life) by following the manufacturer's instructions. Cells were seeded in a 24-well culture plate. After 24 h, cells were loaded with the ROS detection solution and incubate under normal culture conditions for 1 h. After carefully removing the ROS detection solution and cells were treated with the solvent or Alternol in the presence or absence of the anti-oxidants as indicated in the figures. There are three replicated wells for each group. After careful wash with the washing buffer cells were immediately observed and microscopic images were taken under a fluorescence microscope (Olympus, Japan).
Mouse xenografts model and Alternol treatment
Athymic NCr-nu/nu male mice (NCI-Frederick, Fort Detrick, VA, USA) were maintained in accordance with the Institutional Animal Care and Use Committee (IACUC) procedures and guidelines. Xenograft tumors were generated as described in our recent publications (24, 25). Briefly, exponentially grown prostate cancer cells (PC-3 and DU145) were trypsinized and resuspended in PBS. A total of 2.0 × 106 cells was resuspended in RPMI-1640 and was injected subcutaneously (s.c.) into the flanks of 6-week-old mice using a 27-gauge needle and 1-ml disposable syringe. For animal treatment, Alternol was dissolved in a solvent that contains 20% DMSO in PBS solution and the dose was set for 20 mg/Kg body weight based on a previous patent publication (US20090203775A1). When tumors were palpable (about 30 mm3), animals were treated twice a week with the solvent or Alternol (about 100 μl in volume) via intraperitoneal injection. Tumor growth was monitored by measuring the length (L) and the width (W), and tumor volumes were calculated (V = [L × W2]/2), as described previously (24). Animal body weight and the wet weights of dissected xenograft tumors were recorded at the end of drug treatment.
Immunohistochemical staining and in situ TUNEL assay
Immunohistochemical staining assay was done as previously described (24-26). Xenograft specimens were fixed in 4% paraformaldehyde and paraffin-embedded. Sections were de-paraffinized and rehydrated, followed by antigen retrieval and endogenous peroxidase blocking. Multiple dilutions of the primary IHC-specific antibody for cleaved caspases-3 (Catalog #9661, Cell Signal, Danvers, MA) were utilized to achieve optimal immunosignals. A negative control was set up for each case by omitting the primary antibody. Immunosignals were detected with DAKO LSAB+ System by following the manufacturer's manual. Apoptosis index in tissue sections was determined by in situ TUNEL analysis with the ApoAlert™ DNA fragmentation assay kit (Clontech, Mountain View, CA), as described previously (24).
Statistical analysis
Images of western blots, Total ROS Detection, JC-1 staining, flow cytometry analysis, DNA fragmentation assay, in situ TUNEL assay and IHC microscopic images were from representative experiments. The mean and standard error of the mean (SEM) are shown for all of the quantitative data. The significance of the differences between treatment and control was analyzed by ANOVA or Student t-test as indicated in the figure legends using SPSS software (SPSS, Chicago, IL). Alternol's IC50 value was calculated with the Four-Parameter Logistic Function using the SPSS software.
Results
Alternol treatment induces cell death in prostate cancer cells but not in normal prostate cells
It was recently reported that Alternol induced growth inhibition and apoptosis in human cancer cells, including prostate cancer cells (4, 10). To further illustrate the anti-cancer property of Alternol on prostate cancer, we tested Alternol on multiple prostate-derived epithelial cells, including malignant and non-malignant cell lines. After Alternol treatment, cell death rate was quantified using trypan-blue exclusion assay. We first conducted a dose-response test in three representative prostate cancer cell lines, androgen responsive LNCaP, castration-resistant C4-2 and androgen receptor (AR)-negative PC-3. As shown in Fig 1B, a clear dose-dependent cell death was observed with an IC50 of 5-10 μM at 24 h. Then, we extended the test to include more malignant (castration-resistant 22RV1 and AR-negative DU145) cell line and non-malignant prostatic epithelial cell lines (RWPE-1 and BPH1). Cells were treated with Alternol for up to 24 h at a dose level of 10 μM. Similar to the first three cell lines tested, 22RV1 showed a similar death rate but not DU145 and two benign cell lines (RWPE-1 and BPH1) only weakly responded to Alternol treatment (Fig 1C). These data indicate that Alternol has a preference in killing prostate cancer cells over non-malignant prostate epithelial cells, which is supported by a recent report (10). The case for cell death resistance in prostate cancer DU145 cells might be due to defect of cell death pathway that will be explained later.
Alternol-induced cell death is mainly an apoptotic response
To determine if Alternol-induced cell death is an apoptotic response as reported in gastric cancer cells (4), we first examined two apoptosis hallmarks, PARP cleavage and caspase-3 processing (27). Exponentially grown cells were treated with either the solvent or Alternol for up to 16 h. As shown in Fig 2A, Alternol treatment induced a typical processing of caspase-3 and a classical pattern of PARP cleavage in malignant LNCaP, C4-2, 22RV1 and PC-3 cells. Consistent with the data shown in Fig 1, caspase-3 processing and PARP cleavage were weakly induced in RWPE-1 and BPH1 cells after Alternol treatment. No sign of caspases-3 processing and PARP cleavage were observed in DU145 cells.
Fig 2. Alternol induces apoptotic cell death in prostate cancer cells.

(A) Cells were treated with the solvent or Alternol (10 μM). Cells were harvested at the indicated time points. Equal amount of whole cell lysates were subjected to Western blot with the primary antibodies as listed on the left side of the panels. Actin blot was served as protein loading control.
(B) PC-3 cells were treated with the solvent or Alternol (10 μM) for the indicated time period. Cells were harvested for Western blot as described above or (bottom panel) genomic DNA was extracted for the DNA ladder detection assay by following the manufacturer's instructions. Total DNAs were analyzed by 1% agarose gel electrophoresis. Lane M, DNA size markers.
(C & D) Cells were either left untreated or treated with Alternol (10 μM) for the indicated time period before harvested for Annexin V-FITC binding/PI staining assay. The percentage numbers showed the positive cells in each boxes. Quantitative data (mean ± SEM) from three independent experiments were summarized in panel D. The asterisk indicates a significant difference (ANOVA analysis, p < 0.05) compared to the control.
(E) PC-3 cells were treated with the solvent or Alternol (10 μM) for the indicated time period. Cells were harvested for caspases-9 activity assay with a caspases-9 colorimetric activity kit. Data (mean ± SEM) are shown as the relative value of assay reading compared to the solvent control (set as value of 1.0) from three independent experiments. The asterisk indicates a significant difference (ANOVA analysis, p < 0.05)
(F & G) PC-3 cells were treated with the solvent or Alternol (10 μM) in the presence or absence of N-Ac (5 mM) or DHLA (0.25 mM) for 16 h. Then PC-3 cells were stained with JC-1 (0.3 μg/ml) for 15 min at 37C. Microscopic images were taken under a fluorescence microscope at a magnification of × 200. Quantitative Data (mean ± SEM) were summarized in panel G. The asterisk indicates a significant difference (ANOVA analysis, p < 0.05) compared to the solvent control.
Then, we further confirmed Alternol-induced apoptosis by assessing DNA fragmentation. As shown in Fig 2B lower panel, in parallel to Alternol-induced caspase-3 processing and PARP cleavage, a typical DNA fragmentation was observed in a time-dependent manner after Alternol treatment. Next, we assessed the integrity of cellular plasma membrane and the exposure of inner phosphatigylserine by a combinational assay of propidium iodide (PI) staining and Annexin V binding. As shown in Fig 2C and 2D, Alternol treatment induced a significant increase of PI/Annexin V-dual positive population in PC-3 cells in a time-dependent manner but not in DU145 and BPH1 cells. In addition, a less than 10% of PC-3 or BPH1 cells were seen with PI staining, indicating a small portion of cells underwent necrotic cell death. We also investigated if caspases-9, the major component of apoptosome for apoptosis execution (28), was activated after Alternol treatment. As shown in Fig 2E, after Alternol treatment caspases-9 activity was significantly increased at 8-16 h post-treatment.
Mitochondria damage is often reported in chemodrug-induced apoptosis (29). Therefore, we investigated whether Alternol treatment could cause any mitochondrial damage. We assessed the integrity of mitochondrial membrane permeability with a fluorescent dye JC-1, which exerts an orange color in healthy mitochondria but a green color when mitochondria are damaged (30). The results shown in Fig 2F and 2G revealed that in the majority of PC-3 cells compared to the solvent control, Alternol-treated cells appeared as green color, which was a clear sign of mitochondrial membrane potential transition (MPT)(31). Taken together, these data demonstrated that Alternol mainly induced apoptotic cell death in prostate cancer cells.
Oxidative stressis essential in Alternol-induced apoptosis
Although a previous report showed Alternol-induced ROS response in mouse leukemia cells, the ROS involvement in cell death was not defined (3). We then went on to investigate if Alternol-induced oxidative stress and what was the significance in prostate cancer cell death. As shown in Fig 3A, when assessed with a total ROS detection assay Alternol treatment induced a dramatic oxidative response in a time-dependent manner. Quantitative analysis revealed that the oxidative stress was gradually increased after Alternol addition, which reached a significant level at 2-4 h post-treatment (Fig 3B). Interestingly, Alternol treatment also induced a significant increase of ROS accumulation in DU145 cells but not in non-malignant BPH1 cells (Fig 3C), indicating that cancer cells are sensitive to ROS insults compared to non-malignant cells.
Fig 3.

Alternol treatment induces a profound oxidative stress.
(A & B) PC-3 cells seeded in 24-well plates were pre-loaded with the total ROS detection solution for 1 h. Then cells were left un-treated or treated with Alternol (10 μM) for the indicated time period. After careful wash, cells were immediately observed under a fluorescence microscope. ROS, green; cellular nuclear were stained with Hoechst 33342 as blue. Quantitative data (mean ± SEM) from three independent experiments were summarized in panel B. The asterisk indicates a significant difference (ANOVA analysis, p < 0.05) compared to the untreated control.
(C) Cells as indicated were seeded in 6-well plates and pre-loaded with the total ROS detection reagent for 1 h. After treatment with the solvent or Alternol (10 mM) for 4 h, ROS positive cells were assessed and analyzed as described in panel B.
(D) PC-3 cells were pre-loaded with the total ROS detection reagent for 1 h and then treated with the solvent or Alternol (10 μM) in the presence or absence of N-Ac (5 mM) or DHLA (0.25 mM) for 4 h. Magnification of microscopic images, × 200.
Next, we determined if oxidative stress was involved in Alternol-induced apoptotic cell death. Two structurally distinct anti-oxidant compounds, N-acetylcysteine (N-Ac) and dihydrolipoic acid (DHLA) that can broadly scavenge ROS species were used to reduce ROS-induced cellular stress (24). As shown in Fig 3D, pre-treatment of the cells with these two compounds abolished Alternol-induced ROS accumulation. Most importantly, when cell death rate was assessed, pre-treatment with N-Ac and DHLA significantly reduced Alternol-induced cell death (Fig 4A). Further analysis revealed that these two ROS scavengers abolished mitochondrial damage as assessed by JC-1 staining (Fig 2F & 2G), PARP cleavage andcaspase-3 processing (Fig 4B & 4C). Alternol-induced casapse-9 activation (Fig 2E) and DNA fragmentation (Fig 4D) were attenuated by N-Ac pre-treatment. These data suggest that oxidative response plays an essential role in Alternol-induced apoptosis.
Fig 4. Alternol-induced apoptosis is abolished by anti-oxidants.
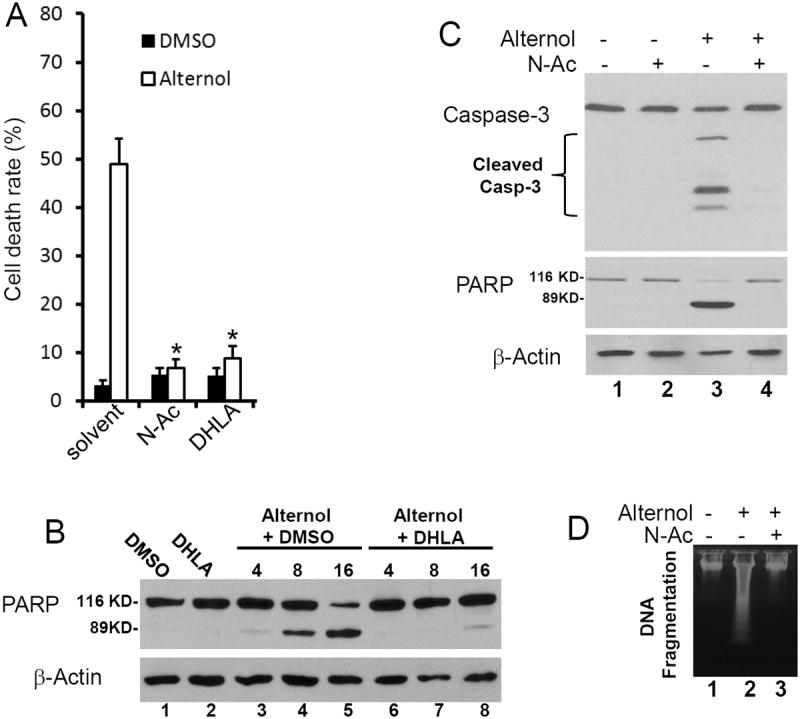
(A) PC-3 cells were treated with the solvent or Alternol (10 μM) in the presence or absence of N-Ac (5 mM) or DHLA (0.25 mM). Cell death rate was determined by trypan blue exclusion assay as described in Fig 1A.
(B & C) PC-3 cells were treated as indicated and harvested at the indicated time points or at 8 h (panel C). Drug concentration was described as above.
(D) PC-3 cells were treated with the solvent or Alternol (10 μM) in the presence of N-Ac. Cells were harvested at 16 h after treatment for DNA fragmentation assay as described in Fig 2B.
Oxidative stress leads to Bax activation after Alternol treatment
Bcl-2 family proteins including anti-apoptotic and pro-apoptotic members are major modulators of apoptosis (16). Alternol was shown to down-regulate anti-apoptotic protein Bcl-2 and Bcl-xL expression in mouse leukemia cells (5). In this study, we assessed the major members of Bcl-2 family proteins during Alternol treatment. Our results (Fig 5A) revealed that in addition to a dramatic decline of Bcl-2 protein level, a moderate increase of Bif-1 protein and a slight reduction of Bcl-xL protein expression, Alternol treatment induced a clear cleavage of Bax protein, a sign of Bax activation (32). Most significantly, DU145 cell line that is lack of Bax expression (33) was sensitive to Alternol-induced ROS accumulation (Fig 3C) but resistant to Alternol-induced cell death (Fig 1C & 2A), indicating a potential role of Bax activation in ROS-dependent apoptosis after Alternol treatment.
Fig 5. Bax plays an essential role in Alternol-induced apoptosis.
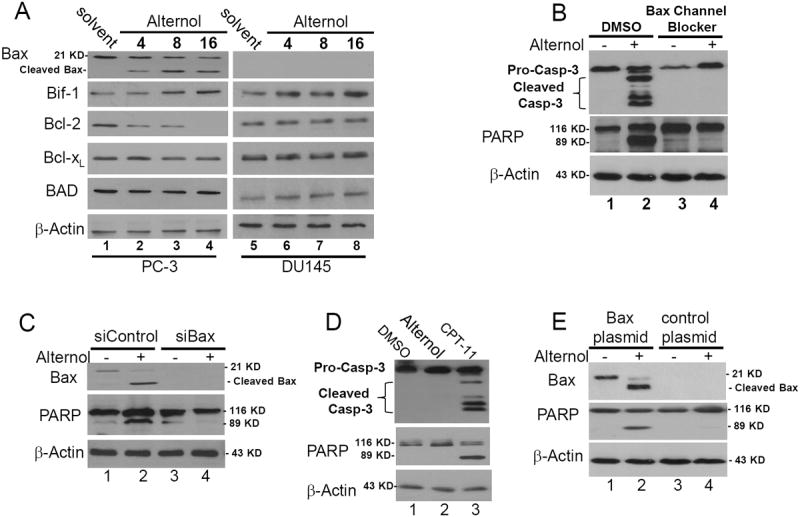
(A) PC-3and DU145 cells were treated with the solvent or Alternol (10 μM) and were harvested at the indicated time point. Equal amount of protein lysates were used for Western blot analysis with the antibodies as indicated. Anti-β-Actin blot served as protein loading control.
(B) PC-3 cells were pre-treated with the solvent or Bax channel blocker (20 μM) for 30 min before Alternol addition (10 μM) Cells were harvested at 16 h after treatment.
(C) PC-3 cells were transfected with Bax siRNAs (siBax, 100 nM) or the control siRNA (100 nM) for 72 hours. Then cells were treated with the solvent or Alternol (10 μM) for 16 h.
(D) DU145 cells were treated with the solvent (DMSO), Alternol (10 μM) or CPT-11 (100 ng/ml) for 16 h before harvested for Western blot assays as described above.
(E) DU145 cells plated in 6-well plates were transfected with control plasmid or Bax expressing plasmid (2 μg DNA/well) for 24 h, followed by treatment with the solvent or Alternol (10 μM) for 16 h. Western blot was conducted as described above. Data represent three independent experiments.
Next, we used two approaches to verify the functional role of Bax cleavage/activation in Alternol-induced ROS-dependent apoptosis in PC-3 cells. First, a Bax channel blocker (34) was used to pre-treat PC-3 cells before Alternol addition. As shown in Fig 5B, pretreatment with the Bax channel blocker abolished Alternol-induced caspase-3 processing and PARP cleavage. Second, Bax siRNAs were used to knock down Bax expression followed by Alternol addition. As shown in Fig 5C, compared to the control siRNA, Bax siRNA largely reduced Alternol-induced PARP cleavage in PC-3 cells.
Then, we determined if the lack of apoptotic response in DU145 cells after Alternol treatment is due to Bax-null status. In contrast to Alternol, topoisomerase I inhibitor CPT-11 (35) that induces intrinsic apoptosis by causing DNA damage, induced a drastic response of caspase-3 processing and PARP cleavage in DU145 cells (Fig 5D), indicating the apoptotic machinery is functional. Then, we restored Bax expression in DU145 cells, followed by Alternol treatment. As shown in Fig 5E, compared to empty control construct, re-installation of Bax expression in DU145 cells sensitized them to Alternol-induced PARP cleavage, indicating the critical role of Bax in Alternol-induced apoptosis.
Since ROS scavengers N-Ac and DHLA abolished Alternol-induced apoptosis, and it has been reported that oxidative stress caused Bax activation (18, 20), (15) we examined if Alternol-induced Bax cleavage was due to oxidative stress. PC-3 cells were pretreated with N-Ac or DHLA before Alternol addition and Bax cleavage was evaluated by western blot assays. As shown in Fig 6A & 6B, either N-Ac or DHLA pre-treatment dramatically reduced Alternol-induced Bax cleavage in PC-3 cells, indicating the causative role of oxidative stress in Bax activation.
Fig 6. Alternol-induced Bax cleavage is abolished by antioxidants but not by calpain inhibitors.
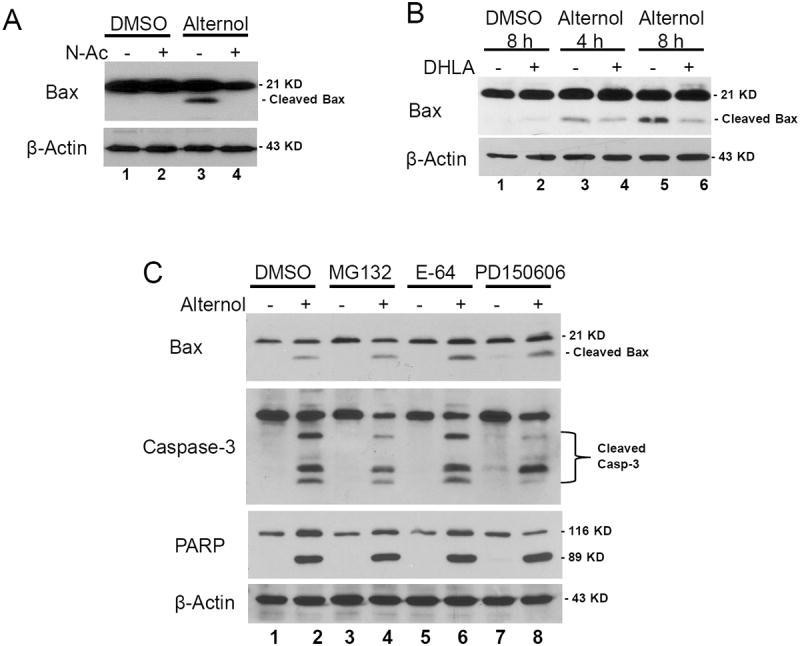
PC-3 cells were treated with the solvent or Alternol (10 μM) in the presence or absence of anti-oxidant N-Ac (5 mM, panel A) and DHLA (0.25 mM, panel B), or protease inhibitors (C). MG132, 10 μM; E-64, 10 μM; PD150606, 20 μM. Cells were harvested at 8 h (A & C) or at the indicated time points (B). Western blots were conducted as described above.
Lastly, we determined if Alternol-induced Bax cleavage is dependent on calpain, which was shown to be involved in Bax cleavage/activation in chemotherapeutic drug-induced apoptosis (32, 36). A broad spectrum inhibitor of cysteine proteases including calpain E-64 (37), a calpain-specific inhibitor PD150606 (38) and a dual calpain/proteasome inhibitor MG-132 (39) were used in PC-3 cells as a pre-treatment before Alternol addition. Compared to the solvent control, these inhibitors had no obvious inhibitory effect on Alternol-induced Bax cleavage, caspase-3 processing and PARP cleavage (Fig 6C). These data demonstrated that Alternol-induced ROS-dependent Bax activation and apoptotic cell death might be a calpain-independent mechanism.
Alternol suppressed xenograft tumor growth in vivo
Finally, we tested if Alternol treatment suppresses tumor growth in vivo. Xenograft tumors were established in nude mice with PC-3 and DU145 cells. Once xenografts were palpable (30 mm3), animals were randomly divided into two groups to receive intraperitoneal injection of the solvent or Alternol twice a week. Xenograft tumors were monitored for 3 weeks. No obvious side effect and body weight loss were observed in both types of xenograft models after Alternol treatment.
As shown in Fig 7A & 7B, in PC-3 xenograft-bearing animals, tumor growth was significantly suppressed in Alternol treatment group compared to the solvent control group. Tumor wet weights were also significantly lower in Alternol group than that in control group. However, there was no significant difference in tumor growth rate in DU145 xenografts between Alternol group and the control group. Further analysis on paraffin-embedded xenograft tissue sections (Fig 7C & 7D) revealed that there was a significant increase of TUNEL-positive cells and the expression of cleaved caspase-3 in PC-3-derived xenografts after Alternol treatment compared to the control solvent. However, DU145-derived xenografts showed no obvious difference in either TUNEL index or cleaved caspase-3 expression between Alternol-treated and the solvent control. These data suggest that Alternol treatment suppressed tumor growth through Bax-dependent apoptosis in vivo.
Fig 7.

Alternol treatment significantly suppressed tumor growth in vivo.
(A & B) PC-3 and DU145 xenograft tumors were established in nude mice as described.(24, 25) Once xenografts were palpable (about 30 mm3 in volume), animals were treated with intraperitoneal injection of the solvent (20% DMSO in PBS) or Alternol (20 mg/kg bodyweight) in a volume of 100 μl twice a week for three weeks (the first and third day of each week). Tumor growth was monitored as a percentage increase of tumor volume (L × W2 × 1/2)(24, 25) in comparison to the initial volume ([tumor volume – initial volume]/initial volume × 100%)(24, 25, 51). Tumor wet weights and animal body weights were recorded at the end of experiment. Data were shown as mean ± SEM. The asterisk indicates a significant difference compared to the control (Student's t-test, p < 0.05, n = 8).
(C & D) Paraffin-embedded tumor tissue sections were prepared from PC-3 and DU145 xenograft tumors. Cleaved caspases-3 expression was evaluated by immunostaining with an IHC-specific anti-cleaved caspases-3 and immunol signals were visualized with DAKO LSAB+ kit. Apoptosis was assessed using the in situ TUNEL assay kit as described (24, 25). Quantitative data (mean ± SEM) were summarized in panel D. The asterisk indicates a significant difference (Student t-test, p< 0.05) between the solvent control and drug treatment.
Discussion
Here we reported the natural compound Alternol for its apoptosis-inducing effect on human prostate cancer cells, which is the first step towards new drug development for advanced prostate cancers. This apoptotic effect was achieved through a dramatic accumulation intracellular ROS species, resulting in a strong death response in either AR-positive or AR-negative prostate cancer cells regardless of their hormone responsiveness. In addition, we confirmed the Alternol preference in killing malignant over non-malignant cells through an oxidative stress-mediated Bax activation-dependent apoptotic pathway in vitro and in vivo. Consistently, Bax-null prostate cancer DU145 cells were resistant to Alternol-induced apoptotic cell death in vitro and in vivo. These data provide a strong proof-of-principle that Alternol is feasible to be further developed as a novel anti-cancer therapy for prostate cancer, especially for the late-stage castration-resistant prostate cancers that are currently without means to cure (1).
Oxidative stress is often induced by chemotherapeutic drugs in cancer cells (12). Meanwhile, compared to non-malignant cells, malignant cells are much more vulnerable to oxidative stress-induced cell death due to elevated production of endogenous ROS (11, 40, 41). Thus far, several cancer-specific ROS inducers were reported with promising results (42-44). Although Alternol was previously reported to induce ROS accumulation in gastric cancer cells, its functional significance on cell death was not determined (3). In this study, we demonstrated that oxidative stress was gradually accumulated after Alternol treatment. Furthermore, two structurally distinct anti-oxidants N-Ac and DHLA abolished Alternol-induced oxidative stress and apoptotic cell death, as evidenced by the completed blockage of caspases-3 processing, PARP cleavage, mitochondrial damage and DNA fragmentation. These data confirm the functional significance of ROS accumulation in Alternol-induced cytotoxicity.
Bcl-2 family proteins are the major regulators of apoptotic cell death, of which the pro-apoptotic Bax protein is the dominant one in triggering apoptosis (13). Bax activation is a highly regulated multistep process, involving its conformational change and then translocation from the cytosol to the mitochondrial outer membrane where it is cleaved and oligomerizes (16, 45). In this study, we presented another important finding that Bax protein was cleaved after Alternol treatment in parallel to intracellular ROS accumulation and subsequent apoptotic cell death. We demonstrated that Bax cleavage is functionally significant because the anti-oxidant agents completely blocked Bax cleavage and cell death. In addition, suppression of Bax function by either Bax channel blocker or Bax-specific siRNA attenuated Alternol-induced apoptosis. Furthermore, Bax-null DU145 cells are resistant to Alternol-induced apoptosis in vitro and in vivo. Nonetheless, Alternol-induced Bax cleavage was not suppressed by calpain inhibitors, although calpain was shown to mediate chemodrug-induced Bax cleavage (32, 36, 46, 47), suggesting a calpain-independent mechanism. Although oxidative stress has been linked to Bax dimerization and activation (18, 20), as we showed in this study, the exact mechanism for Alternol-induced Bax cleavage is still under further investigation by our group.
In this study, we found that the protein levels of anti-apoptotic Bcl-2 but not Bcl-XL significantly decreased while the Bax interacting protein Bif-1 moderately increased after Alternol treatment, which is supported by a previous report (5)(4). As a major Bax inhibitory protein, Bcl-2 down-regulation is postulated to facilitate Bax-mediated apoptotic cell death after Alternol treatment. Similar is true for Bif-1 that has been shown to enhance Bax function (48-50). However, the mechanisms underlying the expression changes of these Bcl-2 family proteins after Alternol treatment needs further investigation.
Finally, we conducted a pilot “proof-of-principle” experiment to assess the in vivo efficiency of Alternol on tumor inhibition. The growth rate of xenografts in nude mice derived from PC-3 but not DU145 showed a significant reduction in Alternol-treated animals compared to the solvent control. Analysis of the tissue section revealed that Alternol treatment induced a dramatic apoptotic response in PC-3 but not DU145-derived xenografts. These results were in line with a previous patent publication (US20090203775A1), showing in vivo anti-cancer effect of Alternol on human gastric cancer cell-derived xenografts. Our xenograft data also were consistent with cell culture data that DU145 cells were resistant to Alternol treatment due to Bax-null status. In spite that only one dose was implicated in our animal experiment, it is suggestive that Alternol possess a great potential as a novel small molecule anti-cancer agent for further pre-clinical and clinical development.
In conclusion, we provided convincing evidences that natural compound Alternol is capable of inducing apoptotic cell death in prostate cancer cells through an oxidative stress-dependent Bax activation mechanism, which provides a proof-of-principle for further development as a novel anti-cancer drug for advanced prostate cancers. Future studies will elucidate the detailed mechanisms responsible for Alternol-induced intracellular ROS accumulation and changes in Bcl-2 family proteins, especially Bax cleavage and activation.
Acknowledgments
The authors are very grateful for the kind gift of Alternol compound from Strand Biotech Co. Ltd. (Shantou, China) and the initial discussion about the Alternol compound with Dr Ji Li (Department of Pharmacology and Toxicology, School of Medicine and Biomedical Sciences, State University of New York at Buffalo). We also thank the KUMC Flow Cytometry Core facility supported by NIH-KU COBRE Grant (P20 RR016443) for technical assistance. This study is partially supported by grants to Dr Benyi Li from KU William L. Valk Endowment Foundation, China Natural Science Foundation (NSFC #81172427) and the “Chutian Scholar” program funded by Hubei Province of China dedicated to Three Gorges University (Yichang, China).
Financial Support: KU William L. Valk Endowment Foundation to B. Li
China Natural Science Foundation (#81172427) to B. Li
Three Gorges University “Chutian Scholar” program from Hubei Province of China to B. Li.
Abbreviations
- AR
androgen receptor
- ATCC
American Type Culture Collection
- CRPC
castration-resistant prostate cancers
- DHLA
dihydrolipoic acid
- DMSO
Dimethyl Sulfoxide
- FBS
fetal bovine serum
- IACUC
Institutional Animal Care and Use Committee
- JC-1
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl carbocyanine iodide
- MPT
membrane potential transition
- N-Ac
N-acetylcysteine
- PAGE
Polyacrylamide gel electrophoresis
- PARP
Poly (ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PI
propidium iodide
- PVDF
Polyvinylidene fluoride
- RIPA
radio-immunoprecipitation assay
- ROS
reactive oxygen species
- s.c.
subcutaneous
- SDS
Sodium dodecyl sulfate
- SEM
standard error of mean
- siRNA
small interfering RNA
- TBS-T
Tris-buffered solution plus Tween 20
- TUNEL
terminal deocynucleotide transferased UTP nick end labeling
Footnotes
Conflict of Interest statement: The authors have no conflicts to disclose.
References
- 1.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 2.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501–11. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu ZZ, Chen JP, Zhao SL, Li CL. Apoptosis-inducing effect of alternol on mouse lymphocyte leukemia cells and its mechanism. Yao Xue Xue Bao. 2007;42:1259–65. [PubMed] [Google Scholar]
- 4.Liu X, Wang J, Sun B, Zhang Y, Zhu J, Li C. Cell growth inhibition, G2M cell cycle arrest, and apoptosis induced by the novel compound Alternol in human gastric carcinoma cell line MGC803. Invest New Drugs. 2007;25:505–17. doi: 10.1007/s10637-007-9057-4. [DOI] [PubMed] [Google Scholar]
- 5.Liu ZZ, Zhu J, Sun B, Liu S, Geng S, Liu X, et al. Alternol inhibits proliferation and induces apoptosis in mouse lymphocyte leukemia (L1210) cells. Mol Cell Biochem. 2007;306:115–22. doi: 10.1007/s11010-007-9560-0. [DOI] [PubMed] [Google Scholar]
- 6.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–7. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 7.Vogelzang NJ. Docetaxel (Taxotere) in hormone-refractory prostate cancer: a new addition to the physicians' toolbag. Semin Oncol. 1999;26:1–2. [PubMed] [Google Scholar]
- 8.Liu L, Zhang B, Yuan X, Wang P, Sun X, Zheng Q. Alternol induces an S-phase arrest of melanoma B16F0 cells. Cell Biol Int. 2014;38:374–80. doi: 10.1002/cbin.10226. [DOI] [PubMed] [Google Scholar]
- 9.Zhu XL, Wang YL, Chen JP, Duan LL, Cong PF, Qu YC, et al. Alternol inhibits migration and invasion of human hepatocellular carcinoma cells by targeting epithelial-to-mesenchymal transition. Tumour Biol. 2013 doi: 10.1007/s13277-013-1224-y. [DOI] [PubMed] [Google Scholar]
- 10.Yeung ED, Morrison A, Plumeri D, Wang J, Tong C, Yan X, et al. Alternol exerts prostate-selective antitumor effects through modulations of the AMPK signaling pathway. Prostate. 2012;72:165–72. doi: 10.1002/pros.21417. [DOI] [PubMed] [Google Scholar]
- 11.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 12.Deavall DG, Martin EA, Horner JM, Roberts R. Drug-induced oxidative stress and toxicity. J Toxicol. 2012;2012:645460. doi: 10.1155/2012/645460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walensky LD, Gavathiotis E. BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci. 2011;36:642–52. doi: 10.1016/j.tibs.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czabotar PE, Colman PM, Huang DC. Bax activation by Bim? Cell Death Differ. 2009;16:1187–91. doi: 10.1038/cdd.2009.83. [DOI] [PubMed] [Google Scholar]
- 15.Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, et al. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell. 2005;16:2424–32. doi: 10.1091/mbc.E04-12-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, et al. Arsenic induces pancreatic beta-cell apoptosis via the oxidative stress-regulated mitochondria-dependent and endoplasmic reticulum stress-triggered signaling pathways. Toxicol Lett. 2011;201:15–26. doi: 10.1016/j.toxlet.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 18.D'Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C, et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J. 2005;19:1504–6. doi: 10.1096/fj.04-3329fje. [DOI] [PubMed] [Google Scholar]
- 19.Oh SH, Lee BH, Lim SC. Cadmium induces apoptotic cell death in WI 38 cells via caspase-dependent Bid cleavage and calpain-mediated mitochondrial Bax cleavage by Bcl-2-independent pathway. Biochem Pharmacol. 2004;68:1845–55. doi: 10.1016/j.bcp.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 20.Jungas T, Motta I, Duffieux F, Fanen P, Stoven V, Ojcius DM. Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2002;277:27912–8. doi: 10.1074/jbc.M110288200. [DOI] [PubMed] [Google Scholar]
- 21.Liao X, Zhang L, Thrasher JB, Du J, Li B. Glycogen synthase kinase-3beta suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand resistance in prostate cancer. Mol Cancer Ther. 2003;2:1215–22. [PubMed] [Google Scholar]
- 22.Liao X, Tang S, Thrasher JB, Griebling TL, Li B. Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol Cancer Ther. 2005;4:505–15. doi: 10.1158/1535-7163.MCT-04-0313. [DOI] [PubMed] [Google Scholar]
- 23.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–82. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 24.Li B, Sun A, Youn H, Hong Y, Terranova PF, Thrasher JB, et al. Conditional Akt activation promotes androgen-independent progression of prostate cancer. Carcinogenesis. 2007;28:572–83. doi: 10.1093/carcin/bgl193. [DOI] [PubMed] [Google Scholar]
- 25.Sun A, Tang J, Terranova PF, Zhang X, Thrasher JB, Li B. Adeno-associated virus-delivered short hairpin-structured RNA for androgen receptor gene silencing induces tumor eradication of prostate cancer xenografts in nude mice: a preclinical study. Int J Cancer. 2010;126:764–74. doi: 10.1002/ijc.24778. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Q, Youn H, Tang J, Tawfik O, Dennis K, Terranova PF, et al. Phosphoinositide 3-OH kinase p85alpha and p110beta are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene. 2008;27:4569–79. doi: 10.1038/onc.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 29.Decaudin D, Marzo I, Brenner C, Kroemer G. Mitochondria in chemotherapy-induced apoptosis: a prospective novel target of cancer therapy (review) Int J Oncol. 1998;12:141–52. [PubMed] [Google Scholar]
- 30.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671–5. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–17. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- 32.Wood DE, Thomas A, Devi LA, Berman Y, Beavis RC, Reed JC, et al. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–78. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- 33.Hemmati PG, Gillissen B, von Haefen C, Wendt J, Starck L, Guner D, et al. Adenovirus-mediated overexpression of p14(ARF) induces p53 and Bax-independent apoptosis. Oncogene. 2002;21:3149–61. doi: 10.1038/sj.onc.1205458. [DOI] [PubMed] [Google Scholar]
- 34.Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, Halazy S. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J Med Chem. 2003;46:4365–8. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- 35.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–8. [PubMed] [Google Scholar]
- 36.Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 37.Sugita H, Ishiura S, Suzuki K, Imahori K. Inhibition of epoxide derivatives on chicken calcium-activated neutral protease (CANP) in vitro and in vivo. J Biochem. 1980;87:339–41. doi: 10.1093/oxfordjournals.jbchem.a132742. [DOI] [PubMed] [Google Scholar]
- 38.Wang KK, Nath R, Posner A, Raser KJ, Buroker-Kilgore M, Hajimohammadreza I, et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci U S A. 1996;93:6687–92. doi: 10.1073/pnas.93.13.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem. 1996;119:572–6. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- 40.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-b. [DOI] [PubMed] [Google Scholar]
- 41.Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, Me LL. Oxidative stress and cancer: an overview. Ageing Res Rev. 2013;12:376–90. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Basu S, Ganguly A, Chakraborty P, Sen R, Banerjee K, Chatterjee M, et al. Targeting the mitochondrial pathway to induce apoptosis/necrosis through ROS by a newly developed Schiff's base to overcome MDR in cancer. Biochimie. 2012;94:166–83. doi: 10.1016/j.biochi.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–4. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Malhi SS, Budhiraja A, Arora S, Chaudhari KR, Nepali K, Kumar R, et al. Intracellular delivery of redox cycler-doxorubicin to the mitochondria of cancer cell by folate receptor targeted mitocancerotropic liposomes. Int J Pharm. 2012;432:63–74. doi: 10.1016/j.ijpharm.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 45.Ghibelli L, Diederich M. Multistep and multitask Bax activation. Mitochondrion. 2010;10:604–13. doi: 10.1016/j.mito.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Yoo JO, Lim YC, Kim YM, Ha KS. Transglutaminase 2 promotes both caspase-dependent and caspase-independent apoptotic cell death via the calpain/Bax protein signaling pathway. J Biol Chem. 2012;287:14377–88. doi: 10.1074/jbc.M111.326074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyota H, Yanase N, Yoshimoto T, Moriyama M, Sudo T, Mizuguchi J. Calpain-induced Bax-cleavage product is a more potent inducer of apoptotic cell death than wild-type Bax. Cancer Lett. 2003;189:221–30. doi: 10.1016/s0304-3835(02)00552-9. [DOI] [PubMed] [Google Scholar]
- 48.Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, et al. Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem. 2001;276:20559–65. doi: 10.1074/jbc.M101527200. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, et al. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol Cell Biol. 2005;25:9369–82. doi: 10.1128/MCB.25.21.9369-9382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Etxebarria A, Terrones O, Yamaguchi H, Landajuela A, Landeta O, Antonsson B, et al. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J Biol Chem. 2009;284:4200–12. doi: 10.1074/jbc.M808050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Q, Yang J, Han S, Liu J, Holzbeierlein J, Thrasher JB, et al. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate. 2011;71:835–45. doi: 10.1002/pros.21300. [DOI] [PubMed] [Google Scholar]