

Abstract
Age at pubertal onset varies substantially in healthy girls. Although genetic factors are responsible for more than half of the phenotypic variation, only a small part has been attributed to specific genetic polymorphisms identified so far. Follicle-stimulating hormone (FSH) stimulates ovarian follicle maturation and estradiol synthesis which is responsible for breast development. We assessed the effect of three polymorphisms influencing FSH action on age at breast deveopment in a population-based cohort of 964 healthy girls. Girls homozygous for FSHR -29AA (reduced FSH receptor expression) entered puberty 7.4 (2.5–12.4) months later than carriers of the common variants FSHR -29GG+GA, p = 0.003. To our knowledge, this is the strongest genetic effect on age at pubertal onset in girls published to date.
Pubertal onset varies substantially among healthy girls and only part of the variation can be explained by racial differences1. Timing of puberty has received considerable attention due to the associations with numerous adverse conditions later in life, e.g. behavioural disorders, breast cancer, obesity, metabolic syndrome, and cardiovascular disease2,3. Although genetic factors seem responsible for more than half of the phenotypic variation, knowledge of specific regulators of pubertal onset is limited4. Abnormal pubertal timing can be caused by rare mutations: e.g. MKRN3, ESR1, KISS1, KISS1R5,6,7,8 whereas common genetic variations in candidate loci (KISS1, KISS1R, LIN28B, LIN28A) do not seem to be involved in idiopathic central precocious puberty9. Additionally, large genome-wide association (GWA) studies have drawn attention to single nucleotide polymorphisms (SNP) affecting pubertal timing10,11,12,13. Nevertheless, only approximately 15% of the variance in age at menarche can be explained by genetic variations identified in GWA studies14.
The majority of clinical studies of pubertal timing are based on recall of age at menarche which is a late pubertal event occuring in average three years after onset of breast development15. Growth of breast tissue is the first clinical sign of the pubertal reactivation of the hypothalamic-pituitary-gonadal axis resulting in ovarian estradiol production. Assessment of breast development is considered the golden standard when evaluating pubertal onset and progression in girls16.
Follicle-stimulating hormone (FSH) is essential for female reproductive maturation. FSH induces ovarian follicle growth and aromatase activity, hence, estradiol synthesis responsible for breast development17, and genetic variants in FSHB and FSHR have been reported as modulators of FSH action18.
FSHB -211G>T is located in the promoter of the gene encoding the FSH beta subunit. Reduced FSH production has been suggested in carriers of FSHB -211TT vs. GG19,20 due to altered affinity of the LHX3 transcription factor binding element21.
FSHR 2039A>G is located in the coding region of the intracellular domain of the FSH receptor. Carriers of FSHR 2039GG have slower intracellular cAMP production (assessed in human granulosa cells)22 and they need more exogenous FSH for sufficient ovarian stimulation than FSHR 2039AA homozygotes23,24.
FSHR -29G>A is located in the promoter region of the gene encoding the FSH receptor. In in-vitro fertilisation patients, the FSH dose had to be increased 83% in order to induce follicular growth in carriers of FSHR -29AA vs. GG25. Despite increased exogenous FSH, the numbers of mature- and retrieved oocytes were reduced 40% and 34%, respectively. The impaired ovarian FSH response in women with FSHR -29AA was supported by DNA topography and expression analyses, which indicated that the A allele was less accessible for binding of transcription factors compared with the G allele. In support, FSH receptor expression at both mRNA and protein level was reduced in granulosa cells from carriers of FSHR -29AA vs. GG25,26.
Recently, we observed that FSHB -211G>T and FSHR 2039A>G may influence age at pubertal onset in girls, but due to the relatively small sample size of 78 girls, no firm conclusions could be drawn from the study27. We have since genotyped a large cohort of 964 healthy Danish girls as well as female patients with idiopathic delayed puberty, including evaluation of the FSHR -29G>A SNP.
We report how the three polymorphisms affect age at pubertal onset evaluated by breast development, revealing a remarkably strong effect of FSHR -29G>A.
Results
Healthy girls
At evaluation, the mean age of the entire study population was 10.8 (9.6–12.1) years (+/− 1SD), and the mean age at pubertal onset was 10.0 (9.9–10.2) years (95% CI).
The girls had the following allele distributions: FSHB -211G>T (GG 650, GT 290, TT 22, minor allele frequency (MAF) 17%), FSHR -29G>A (GG 512, GA 368, AA 74, MAF 27%), and FSHR 2039A>G (AA 242, AG 504, GG 218, MAF 49%). Distributions were consistent with Hardy-Weinberg equilibrium (Pearson's χ2 = 2.47, p = 0.116; χ2 = 0.48, p = 0.488; and χ2 = 2.07, p = 0.151, respectively).
Age at pubertal onset according to the presence of genetic variants of FSHR -29G>A, FSHB -211G>T, and FSHR 2039A>G is listed in Table 1. We observed a strong effect of the SNP in the FSH receptor promoter (FSHR -29G>A). Girls homozygous for the minor allele (FSHR -29AA, n = 74) entered puberty 7.4 (2.5–12.4) months later than carriers of the more common variants FSHR -29GG+GA, 10.6 (10.2–11.0) vs. 10.0 (9.8–10.1), p = 0.003 (Figure 1A). BMI was not affected by genotypes (Table 1), and adjusting for BMI did not alter the effect of FSHR -29AA vs. GG+GA, p = 0.004 (data not shown). The additive model indicated a co-dominant effect of the alleles in which the heterozygotes exhibited intermediate levels; the age at pubertal onset increased 2.4 (0.2–4.5) months per A allele, p = 0.031 (Table 1).
Table 1. Clinical parameters of the study population stratified by genotypes.
| Genotype | Recessive model * | Additive model ¤ | |||
|---|---|---|---|---|---|
| FSHB -211G>T | GG | GT | TT | GG+GT vs. TT | GG vs. GT vs. TT |
| n | 650 | 290 | 22 | ||
| Age (years) | 10.8 (9.6–12.1) | 10.8 (9.5–12.0) | 11.1 (10.1–12.1) | p = 0.346 | p = 0.416 |
| BMI (kg/m2) | 17.3 (15.0–19.8) | 17.3 (15.0–19.9) | 17.6 (15.8–19.6) | p = 0.560 | p = 0.816 |
| Age at pubertal onset | 10.0 (9.8–10.1) | 10.1 (9.8–10.3) | 10.7 (10.5–10.9) | p = 0.246 | p = 0.437 |
| FSHR -29G>A | GG | GA | AA | GG+GA vs. AA | GG vs. GA vs. AA |
| n | 512 | 368 | 74 | ||
| Age (years) | 10.8 (9.6–12.1) | 10.8 (9.6–12.1) | 10.8 (9.6–12.1) | p = 0.736 | p = 0.898 |
| BMI (kg/m2) | 17.2 (15.1–19.5) | 17.5 (15.1–20.2) | 17.2 (14.7–20.0) | p = 0.660 | p = 0.133 |
| Age at pubertal onset (years) | 10.0 (9.8–10.1) | 10.0 (9.8–10.2) | 10.6 (10.2–11.0) | p = 0.003 | p = 0.031 |
| FSHR 2039A>G | AA | AG | GG | AA+AG vs. GG | AA vs. AG vs. GG |
| n | 242 | 504 | 218 | ||
| Age (years) | 10.8 (9.6–12.1) | 10.8 (9.6–12.0) | 10.9 (9.6–12.1) | p = 0.715 | p = 0.443 |
| BMI (kg/m2) | 17.4 (15.0–20.3) | 17.2 (15.1–19.7) | 17.3 (15.2–19.7) | p = 0.960 | p = 0.816 |
| Age at pubertal onset | 10.0 (9.7–10.2) | 10.0 (9.9–10.2) | 10.1 (9.8–10.3) | p = 0.527 | p = 0.579 |
Age: mean (+/− SD), BMI: geometric mean (+/− SD), Age at pubertal onset: mean (95% CI).
In the recessive model, strong effects are expected only in minor allele homozygotes. These are compared with pooled wild-type and heterozygotes (e.g. FSHR -29GG+GA vs. AA).
Differences in ages and BMI levels between genotypes were assessed with independent samples t-test.
Differences in ages at pubertal onset between genotypes were assessed with probit analysis (categorical variable).
The additive model assumes a codominant effect of alleles in which the heterozygotes should exhibit intermediate levels; regressions are calculated over, for example, FSHR -29GG vs. GA vs. AA.
Differences in ages and BMI levels between genotypes were evaluated with One-way ANOVA.
Differences in ages at pubertal onset between genotypes were assessed with probit analysis (continuous variable).
Figure 1. Influence of FSHR -29G>A and FSHB -211G>T on age at pubertal onset in 964 healthy Danish girls.

Left panel: The number of girls and their mean age at pubertal onset according to FSHR -29G>A genotypes (A) and the combined genotypes of FSHR -29G>A and FSHB -211G>T (B). In the combined model, we tested if there was an additive effect of the minor alleles across the genotypes (FSHR -29A and FSHB -211T). The fading green/yellow/red bars indicate intermediate FSH production and FSH receptor expression, respectively, in the heterozygotes compared with the wild-types and the minor allele homozygotes. The colours correspond to the subgroups of combined genotypes: 0 minor alleles: dark green; 1 minor allele: light green; 2 minor alleles: orange; 3 minor alleles: red; No girls had 4 minor alleles (C). Right panel: Mean age at pubertal onset (95% CI) according to FSHR -29G>A genotypes (A) and the combined genotypes of FSHR -29G>A and FSHB -211G>T (B). The combined model revealed additive effect of the minor alleles across the genotypes (B). The outline of the figure is inspired by Tuttelmann et al.48.
Girls with the rare FSHB -211TT genotype (n = 22) seemed to enter puberty later than carriers of FSHB -211GG and GT, but the difference did not reach significant levels in any of the models. Even after adjusting for FSHR -29G>A, there was no clear effect of FSHB -211GG+GT vs TT, p = 0.129 (data not shown). FSHR 2039A>G did not seem to influence age at pubertal onset (Table 1).
In a combined model of FSHR -29G>A and FSHB -211G>T, age at pubertal onset increased with increasing number of minor alleles across the genotypes; i.e. 1.9 (0.2–3.6) months per minor allele, p = 0.025 (Figure 1B).
We estimated that 2.2% and 2.9% of the variance in age at pubertal onset could be explained by FSHR -29G>A alone and in combination with FSHB -211G>T, respectively.
Patients with delayed puberty
Age at breast development for the patients with delayed puberty was 13.8 years (13.0–17.0). Puberty was induced by exogenous estradiol in four patients at age 13.0, 13.1, 14.5, and 17.0 years. The remaining patients entered puberty spontaneously. The SNPs causing later pubertal onset in healthy girls (FSHR -29AA and FSHB -211TT) were significantly more prevalent in patients with idiopathic delayed puberty compared with healthy girls (5/18 patients = 28% vs. 96/964 healthy girls = 10%, p = 0.014). Distribution of genotypes are shown in Figure 2.
Figure 2.
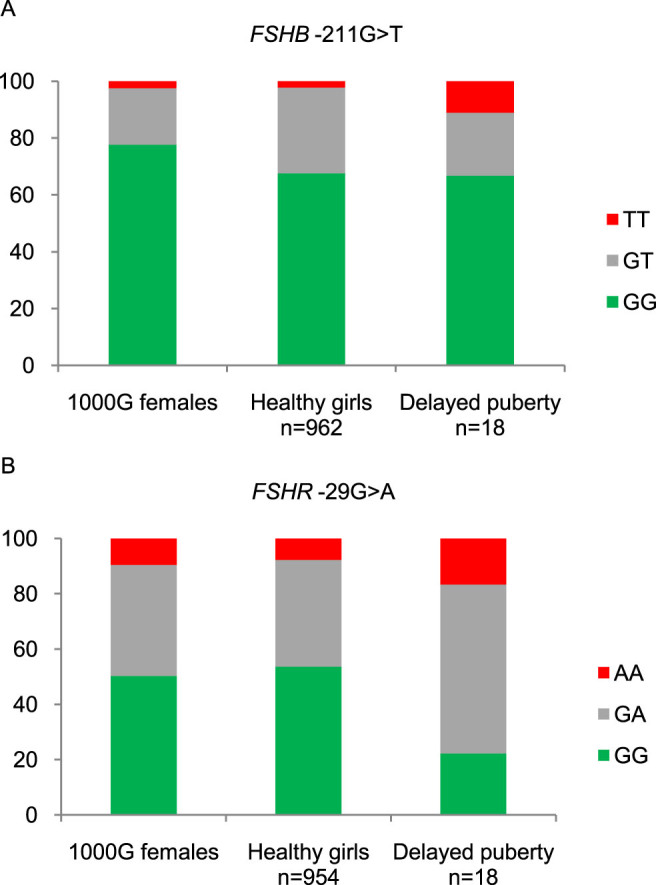
Distribution of genotypes FSHB -211G>T (A) and FSHR -29G>A (B) in Caucasian females (data from 1000 Genomes: www.1000genomes.org), as well as healthy Danish girls and 18 patients with delayed puberty from the present study population.
Discussion
This large population-based study of 964 healthy Danish girls revealed that breast development (marking the onset of puberty) occurred on average 7.4 months later in carriers of FSHR -29AA genotype compared with the common genotype variants FSHR -29GG+GA. Based on a combined model of FSHR -29G>A and FSHB -211G>T, the age at pubertal onset increased with the number of minor alleles across the genotypes. To our knowledge, a stronger genetic influence on pubertal onset in girls has not previously been reported.
Considerable effort has been invested in mapping genetic variations responsible for pubertal timing. From a recent large-scale meta-analysis of 57 GWA studies including data from 182.416 women, 106 genomic loci associated with cellular development, body weight, and hormone pathways were proposed to affect age at menarche14. However, compared with our selective approach of assessing the effect of biological relevant SNPs, the effect estimates in GWA studies were limited and the strongest association was responsible for only a 1.3 months change in age at menarche per allele (LIN28B). All of the suggested loci only explained 15% of the variance in age at menarche. In this perspective, our finding of two SNPs explaining 2.9% of the variance in age at breast development is remarkable. Although we compare how SNPs affect two different milestones of pubertal development (breast development and menarche), estradiol is essential for both events. Furthermore, genes associated with age at menarche were also associated with age at breast development, e.g. LIN28B10. Noteworthy, FSHR -29G>A and FSHB -211G>T were not included in commercial genotyping platforms (e.g. Affymetrix and Illumina) commonly used in GWA studies (http://ncbi.nlm.nih.gov/SNP), which is probably why they have not been identified previously. An indirect way to confirm the observed effect would be to review GWA studies concerning the effect of other SNPs inherited with FSHR -29G>A and FSHB -211G>T (tagSNPs). However, no such tagSNPs exist (http://www.ensembl.org/Homo_sapiens/Variation).
FSH is essential for female reproductive maturation28, and our findings are therefore supported by a plausible biological mechanism. Female FSHB and FSHR knock-out mice and women with loss-of-function mutations in these specific genes present with primary amenorrhea and sterility due to disrupted follicle maturation29,30,31,32. In our study, the main effect on pubertal onset seemed to be driven by genetic variation in the promoter of the gene encoding the FSH receptor. FSHR -29G>A was first described by Wunsch and colleages who did not observe effect on promotor activity in in vitro studies of nonhuman granulosa- and sertoli-cell lines33. However, in vitro studies of human granulosa cell lines as well as cohort studies of IVF patients suggested reduced FSH receptor expression in carriers of FSHR -29AA vs. GG25,26. As in our study, intermediate effect was found in heterozygotes (FSHR -29AG)25.
The effect of FSHB -211TT (reduced FSH production) seemed to be even stronger than FSHR -29AA. Despite our fairly large sample size, we acknowledge that the FSHB -211TT subgroup is small due to the very low minor allele frequency, and the lack of statistical significant effect of the FSHB -211TT (when evaluated separately) may therefore be due to a type II error. The combined model suggests that both the FSHR -29A allele and the FSHB -211T allele contribute to postpone pubertal onset. Our finding of higher prevalence of FSHR -29AA and FSHB -211TT in patients suffering from delayed puberty compared with healthy girls supports that these genotypes delay pubertal onset in girls.
FSHR -29AA was found to be more prevalent among young women suffering from idiopathic premature ovarian insufficiency compared with healthy controls34. None of the girls in the present study carried the combination of FSHR -29AA and FSHB -211TT. Thus, larger studies of adolescents and adult women are needed to clarify if this combination of genotypes causes markedly delayed puberty or even premature ovarian insufficiency.
The SNP in exon 10 of the gene encoding the FSH receptor (FSHR 2039A>G) did not affect pubertal onset either when evaluated separately or in combination with other polymorphisms. We did not include other polymorphisms in the FSH receptor or its promoter due to very low minor allele frequencies33 or high degree of linkage disequilibrium with FSHR 2039A>G24.
Upstream of the pituitary gland, reactivation of the hypothalamus and its connecting neurones drives the onset of puberty through the hypothalamic-pituitary-gonadal (HPG) axis. Hypothalamic GnRH is controlled by multiple pathways; e.g. kisspeptin, neurokinin B, and metabolites from adipose tissue8,35. Much focus has therefore been placed upon childhood growth and adiposity in relation to pubertal timing. BMI is a crude estimate of fat mass, and the effect of FSHR -29G>A and FSHB -211G>T was not driven by differences in BMI in the present study. Thus, our findings indicate that even after central reactivation of the hypothalamus, down-stream processes in the HPG axis influence the peripheral attainment of secondary sexual characteristics.
Due to racial differences concerning age at pubertal onset and distribution of genotypes33,36, we have only included Caucasian girls in the final data analyses. The distribution of genotypes and the age at pubertal onset indicate that the present study population is representative for healthy caucasian girls36,37,38. The HapMap project indicates that FSHR -29A is more prevalent among Chinese (CHB, minor allele frequency 51%) and Japanese (JPT, 47%), and less prevalent among Sub-Saharan Africans (YRI, 24%) compared with Western European (CEU, 30%) (http://hapmap.ncbi.nlm.nih.gov). The distribution fits well with the most recent longitudinal puberty study from the USA. Asian girls entered puberty later and black girls entered puberty at a younger age than white girls (remained after adjusting for BMI)36. Part of the racial difference in age at pubertal onset may be explained by different distributions of genotypes affecting FSH action.
In caucasian girls, pubertal onset has been reduced approximately one year during the past two decades15,36. This strong secular trend cannot be driven by genetic changes alone but is more likely caused by epigenetic changes, lifestyle factors, and/or environmental factors. Our finding of a strong regulatory effect of FSHR -29G>A on age at pubertal onset suggests that the pituitary-gonadal axis is a target for these factors responsible for the observed secular trend.
In this thoroughly characterized population of healthy Danish girls, pubertal onset was evaluated by the presence of palpable breast tissue, which is a current golden standard. This is in contrast to the majority of previous studies based on recollection of age at menarche. Our selective approach of assessing the effect of variation in specific genes affecting FSH-signaling (a biological pathway essential for pubertal onset) minimises the risk of false positive associations. We have not assessed the effect of other polymorphisms.
In conclusion, we demonstrate here for the first time that age at pubertal onset in healthy girls is highly affected by genetic variation in promoters affecting FSH action. Breast development occurred 7.4 months later in healthy girls with the FSHR -29AA genotype compared with carriers of FSHR -29GG+GA. This is the strongest genetic effect on age at pubertal onset in girls published to date.
Methods
Participants
Healthy girls
Participants were recruited as part of two population-based cohort studies of healthy Danish children and adolescents; 1) The COPENHAGEN Puberty Study, detailed information about the study has been described previosuly15,39: a cross-sectional study conducted at ten schools in the Copenhagen area, 2006–2008. All pupils were invited (3102 girls) of whom 35% chose to participate, i.e. 1097 girls aged 6–20 years. 2) The Copenhagen Mother-Child Cohort, detailed information about the study has been described previosuly40,41,42: a longitudinal birth cohort following healthy Danish children. The children were examined during infancy, at mid-childhood and again yearly (one to three times) in 2010–2012 (mean age 11.0 years, range 6.0–15.0).
In the present study-population, all girls (age 8–13 years) from the Copenhagen Mother-Child Cohort (n = 548) and the cross sectional part of the COPENHAGEN Puberty Study (n = 564) were included. No girls from our previously published study of FSH polymorphisms in peripubertal girls (longitudinal part of the COPENHAGEN Puberty Study) were included27. Of the 1112 girls, a total of 148 were excluded from the present dataset because no DNA was available (n = 94), one or both parents originated from a non-European country (n = 46), intracerebral or endocrine disease (n = 3), or due to lack of pubertal staging (n = 5). The remaining 964 girls had no history of gynecological disorders.
Patients with delayed puberty
All girls registered with a diagnosis of delayed puberty (ICD-10: DE 30.0) at the Department of Growth and Reproduction, Rigshospitalet, Denmark were evaluated (n = 168). DNA was available in 41 girls. Girls were excluded due to pubertal onset before age 13 years (n = 14) or due to known cause of delayed puberty (e.g. Turner Syndrome and anorexia nervosa) (n = 9). The remaining 18 girls with idiopathic delayed puberty were included in the study.
Clinical examination
Clinical evaluations were done by trained physicians and included pubertal staging of breast development according to Tanner's classification evaluated by palpation43. Breast stage 2 or more was considered to be a marker of pubertal onset. Height was measured to the nearest 0.1 cm by a wall-mounted stadiometer (Holtain Ltd., Crymych, United Kingdom) and weight was measured to the nearest 0.1 kg using calibrated digital electronic scales (Seca Delta, Germany and Bisco model PERS 200, Denmark). Anthropometrics were available from 963 girls.
Genotyping
Peripheral blood (0.2 ml EDTA-preserved) was used for isolation of genomic DNA using the QuickGene-810 Nucleic Acid Isolation System (Fujifilm, Life Science Products, Tokyo, Japan) and quantified on a NanoDrop ND-1000 spectrophotometer (Saveen Werner, Limhamn, Sweden).
All SNPs were analysed at LGC Genomics (LGC Genomics, Hoddesdon, UK) using their KASP™ SNP genotyping assays, which facilitates bi-allelic discrimination through a competitive PCR and incorporation of a fluorescent resonance energy transfer quencher cassette. KASP™ genotyping assays were designed by LGC Genomics towards the following sequences: FSHB -211G>T (rs10835638), TATCAAATTTAATTT[G/T]TACAAAATCATCAT; FSHR -29G>A (rs1394205), TCTCTGCAAATGCAG[A/G]AAGAAATCAGGTGG; FSHR 2039A>G (rs6166), ATGTAAGTGGAACCA[C/T]TGGTGACTCTGGGA. FSHR 2039A>G was genotyped in all 964 girls. In a few cases genotyping was impossible due to poor DNA quality, i.e. FSHR -29G>A and FSHB -211G>T were genotyped in 954/964 and 962/964 girls, respectively.
A subset of samples (COPENHAGEN Puberty Study, n = 498) were re-genotyped for FSHB -211G>T and FSHR 2039A>G using restriction fragment length polymorphism (RFLP) analysis, according to previously described methods44,45,46. In brief, DNA served as PCR template. Primers: FSHB -211G>T (GGAGCCAGATCATGAAATGTT and CGAAGTCCGATCGTAACCAG) and FSHR 2039A>G (TTTGTGGTCATCTGTGGCTGC and CAAAGGCAAGGACTGAATTATCATT). Fragments digested with TatI (Thermo Scientific, Erembodegem, Belgium) or BsrI (New England Biolabs, Herts, UK). Genotypes were called by banding patterns. Samples with known genotypes were used as positive controls.
Discordance between the two methods were present in 6% (FSHB -211G>T) and 12% (FSHR 2039A>G), respectively. In most cases re-inspection of gels showed ambiguous banding pattern in the RFLP analysis. A few remaining discordant cases were re-analysed, confirming all results in accordance with the KASP™ genotyping.
Statistical analyses
To estimate the mean age (95% CI) at pubertal onset, we used probit analyses taking left, right – and interval censored data into account (SAS: proc lifereg). Probit analysis is a type of regression used with binomial response variables (in this study: breast development, yes/no)47. Based on cross sectional data, the probit analysis provides estimates of proportions of girls having entered puberty at different ages. In this study, longitudinal data of girls entering puberty during follow up is included in the probit analyses (interval censored data). In this way, all available information concerning age at pubertal onset is included. If a given girl entered puberty between two examinations during follow-up, we registered the last pre-pubertal examination (Tanner Stage B1) and the first post-pubertal examination (TS ≥ B2). The time between these examinations contains the true age at onset (interval-censored data, n = 95). If a girl had not entered puberty at her last examination (or only examination if she was only seen once), the current age at examination was known to be a lower bound for the true age at pubertal onset (right censored data, n = 245). If a girl had entered puberty at her first examination, the age at examination was known to be an upper bound for the true age at onset (left censored data, n = 624). We then included the genotypes in the model to estimate the related differences in mean age at pubertal onset (95% CI).
Two genetic models were used to assess the effect of genotypes on age at pubertal onset. In the recessive model, strong effects were expected only in the minor allele homozygotes. These were compared with pooled wild-type and heterozygotes (e.g. FSHR -29GG+GT vs. TT). The additive model assumed a co-dominant effect of alleles in which the heterozygotes should exhibit intermediate effect; regressions were calculated over, for example, FSHR -29GG vs. GA vs. AA (included as a continuous variable).
Based on initial analyses, we evaluated the combined effect of FSHR -29G>A and FSHB -211G>T. We tested if there was an additive effect of the minor alleles by including the subgroups as a continuous variable.
To estimate how much of the overall variance in age at pubertal onset was explained by the SNPs, we compared the results from two models. In the first model, we estimated the variance in the age at pubertal onset in a probit analysis in which genotypes were included as an explanatory variable. Secondly, we estimated the variance in a probit analysis in which genotypes was not included as an explanatory variable. The difference between the two variances is due to the SNPs included in the model.
To assess if the effect of the genotypes was driven by differences in BMI, we compared BMI levels between genotype sub-groups by independent samples t-test (2 groups) and One-way ANOVA (3 groups), and we adjusted the probit analysis for BMI by including this as a continuous variable.
To assess if the prevalence of susceptible genotypes was higher in patients with delayed puberty vs. healthy girls the χ2 test was used.
P ≤ 0.05 was considered statistically significant. Data were analysed separately by two investigators (CPH and JHP).
Ethical considerations
The COPENHAGEN Puberty Study (ClinicalTrials.gov ID: NCT01411527), The Copenhagen Mother-Child Cohort, and the genetic study of girls with delayed puberty were carried out in accordance with the protocols approved by the scientific ethical committee at The Capital Region of Denmark (KF 01 282214; V200.1996/90, KF 01 030/97/KF 01276357/H-1-2009-074, and KF1328087, respectively) as well as the Danish Data Protection Agency (2010-41-5042, 1997-1200-074/2005-41-5545/2010-41-4757, 2006-41-7251, respectively). The study was conducted in accordance with the second Helsinki Declaration. All children and parents received written information, and informed consent was obtained from all participants.
Author Contributions
A.J., K.M.M., L.A., K.S., E.R.M., K.A., C.P.H. were responsible for study design. C.P.H., K.S., L.A., A.M., M.G.M., J.T., C.W.V., K.A. collected data. C.P.H., C.W.V., J.H.P., E.R.M., K.A., A.J. contributed to data analysis. All authors contributed to data interpretation and preparation of the manuscript.
Acknowledgments
The authors are grateful to the participating children and all colleagues who were involved in the COPENHAGEN Puberty Study and The Copenhagen Mother-Child Cohort. Special thanks to H. Kelkeland, M. Dalgaard, B. Hansen, H. Hansen, I. Garn, and the staff of the hormone laboratory for their expert technical assistance. The study received financial support from the Danish Council for Independent Reseach (DFF-1331-00113), Danish Agency for Science, Technology and Innovation (09-067180), The Capital Region of Denmark's Research Fund for Health Research (R129-A3966), and The Kirsten and Freddy Johansen Foundation.
Disclosure statement: The authors have nothing to disclose.
References
- Parent A. S. et al. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr. Rev. 24, 668–693 (2003). [DOI] [PubMed] [Google Scholar]
- Carel J. C. & Leger J. Clinical practice. Precocious puberty. N. Engl. J. Med. 358, 2366–2377 (2008). [DOI] [PubMed] [Google Scholar]
- Golub M. S. et al. Public health implications of altered puberty timing. Pediatrics 121 Suppl 3, S218–S230 (2008). [DOI] [PubMed] [Google Scholar]
- Sorensen K. et al. Birth size and age at menarche: a twin perspective. Hum. Reprod. 28, 2865–2871 (2013). [DOI] [PubMed] [Google Scholar]
- Abreu A. P. et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 368, 2467–2475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaynor S. D. et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor alpha variant. N. Engl. J. Med. 369, 164–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teles M. G. et al. A GPR54-activating mutation in a patient with central precocious puberty. N. Engl. J. Med. 358, 709–715 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu A. K. et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N. Engl. J. Med. 366, 629–635 (2012). [DOI] [PubMed] [Google Scholar]
- Tommiska J. et al. LIN28B, LIN28A, KISS1, and KISS1R in idiopathic central precocious puberty. BMC. Res. Notes 4, 363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K. K. et al. Genetic variation in LIN28B is associated with the timing of puberty. Nat. Genet. 41, 729–733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry J. R. et al. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat. Genet. 41, 648–650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulem P. et al. Genome-wide association study identifies sequence variants on 6q21 associated with age at menarche. Nat. Genet. 41, 734–738 (2009). [DOI] [PubMed] [Google Scholar]
- Elks C. E. et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat. Genet. 42, 1077–1085 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry J. R. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 10.1038/nature13545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksglaede L., Sorensen K., Petersen J. H., Skakkebaek N. E. & Juul A. Recent decline in age at breast development: the Copenhagen Puberty Study. Pediatrics 123, e932–e939 (2009). [DOI] [PubMed] [Google Scholar]
- Tanner J. M. Growth at adolescence(Blackwell & Mott Ltd, Oxford, 1962). [Google Scholar]
- Jenner M. R., Kelch R. P., Kaplan S. L. & Grumbach M. M. Hormonal changes in puberty. IV. Plasma estradiol, LH, and FSH in prepubertal children, pubertal females, and in precocious puberty, premature thelarche, hypogonadism, and in a child with a feminizing ovarian tumor. J. Clin. Endocrinol. Metab 34, 521–530 (1972). [DOI] [PubMed] [Google Scholar]
- Simoni M. & Casarini L. Mechanisms in endocrinology: Genetics of FSH action: a 2014-and-beyond view. Eur. J. Endocrinol. 170, R91–107 (2014). [DOI] [PubMed] [Google Scholar]
- La M. A. et al. The combination of genetic variants of the FSHB and FSHR genes affects serum FSH in women of reproductive age. Hum. Reprod. 28, 1369–1374 (2013). [DOI] [PubMed] [Google Scholar]
- Hoogendoorn B. et al. Functional analysis of human promoter polymorphisms. Hum. Mol. Genet. 12, 2249–2254 (2003). [DOI] [PubMed] [Google Scholar]
- Benson C. A., Kurz T. L. & Thackray V. G. A Human FSHB Promoter SNP Associated with Low FSH Levels in Men Impairs LHX3 Binding and Basal FSHB Transcription. Endocrinology (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarini L. et al. FSHR polymorphism p.N680S mediates different responses to FSH in vitro. Mol. Cell Endocrinol. 393, 83–91 (2014). [DOI] [PubMed] [Google Scholar]
- Nordhoff V. et al. Effects of the FSH receptor gene polymorphism p.N680S on cAMP and steroid production in cultured primary human granulosa cells. Reprod. Biomed. Online. 23, 196–203 (2011). [DOI] [PubMed] [Google Scholar]
- Perez M. M. et al. Ovarian response to follicle-stimulating hormone (FSH) stimulation depends on the FSH receptor genotype. J. Clin. Endocrinol. Metab 85, 3365–3369 (2000). [DOI] [PubMed] [Google Scholar]
- Desai S. S. et al. Follicle-stimulating hormone receptor polymorphism (G-29A) is associated with altered level of receptor expression in Granulosa cells. J. Clin. Endocrinol. Metab 96, 2805–2812 (2011). [DOI] [PubMed] [Google Scholar]
- Nakayama T. et al. Mutation of the follicle-stimulating hormone receptor gene 5'-untranslated region associated with female hypertension. Hypertension 48, 512–518 (2006). [DOI] [PubMed] [Google Scholar]
- Hagen C. P. et al. FSHB-211 and FSHR 2039 are associated with serum levels of follicle-stimulating hormone and antimullerian hormone in healthy girls: a longitudinal cohort study. Fertil. Steril. 100, 1089–1095 (2013). [DOI] [PubMed] [Google Scholar]
- Rosenfield R. L., Bordini B. & Yu C. Comparison of detection of normal puberty in girls by a hormonal sleep test and a gonadotropin-releasing hormone agonist test. J. Clin. Endocrinol. Metab 98, 1591–1601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar T. R., Wang Y., Lu N. & Matzuk M. M. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 15, 201–204 (1997). [DOI] [PubMed] [Google Scholar]
- Dierich A. et al. Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc. Natl. Acad. Sci. U. S. A 95, 13612–13617 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews C. H. et al. Primary amenorrhoea and infertility due to a mutation in the beta-subunit of follicle-stimulating hormone. Nat. Genet. 5, 83–86 (1993). [DOI] [PubMed] [Google Scholar]
- Aittomaki K. et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 82, 959–968 (1995). [DOI] [PubMed] [Google Scholar]
- Wunsch A. et al. Single-nucleotide polymorphisms in the promoter region influence the expression of the human follicle-stimulating hormone receptor. Fertil. Steril. 84, 446–453 (2005). [DOI] [PubMed] [Google Scholar]
- Achrekar S. K., Modi D. N., Meherji P. K., Patel Z. M. & Mahale S. D. Follicle stimulating hormone receptor gene variants in women with primary and secondary amenorrhea. J. Assist. Reprod. Genet. 27, 317–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welt C. K. et al. Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 351, 987–997 (2004). [DOI] [PubMed] [Google Scholar]
- Biro F. M. et al. Onset of Breast Development in a Longitudinal Cohort. Pediatrics (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman-Giddens M. E. et al. Secondary sexual characteristics and menses in young girls seen in office practice: a study from the Pediatric Research in Office Settings network. Pediatrics 99, 505–512 (1997). [DOI] [PubMed] [Google Scholar]
- Sun S. S. et al. National estimates of the timing of sexual maturation and racial differences among US children. Pediatrics 110, 911–919 (2002). [DOI] [PubMed] [Google Scholar]
- Sorensen K., Aksglaede L., Petersen J. H. & Juul A. Recent changes in pubertal timing in healthy Danish boys: associations with body mass index. J Clin Endocrinol Metab 95, 263–270 (2010). [DOI] [PubMed] [Google Scholar]
- Chellakooty M. et al. Inhibin A, Inhibin B, Follicle-Stimulating Hormone, Luteinizing Hormone, Estradiol, and Sex Hormone-Binding Globulin Levels in 473 Healthy Infant Girls. Journal of Clinical Endocrinology Metabolism 88, 3515–3520 (2003). [DOI] [PubMed] [Google Scholar]
- Boisen K. A. et al. Difference in prevalence of congenital cryptorchidism in infants between two Nordic countries. Lancet 363, 1264–1269 (2004). [DOI] [PubMed] [Google Scholar]
- Wohlfahrt-Veje C. et al. Body fat throughout childhood in 2647 healthy Danish children: agreement of BMI, waist circumference, skinfolds with dual X-ray absorptiometry. Eur. J. Clin. Nutr. 68, 664–670 (2014). [DOI] [PubMed] [Google Scholar]
- Marshall W. A. & Tanner J. M. Variations in pattern of pubertal changes in girls. Arch. Dis. Child 44, 291–303 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorova M., Punab M., Ausmees K. & Laan M. FSHB promoter polymorphism within evolutionary conserved element is associated with serum FSH level in men. Hum. Reprod. 23, 2160–2166 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo S. et al. Genetic and functional analyses of polymorphisms in the human FSH receptor gene. Mol. Hum. Reprod. 8, 893–899 (2002). [DOI] [PubMed] [Google Scholar]
- Pengo M. et al. FSH receptor gene polymorphisms in fertile and infertile Italian men. Reprod. Biomed. Online. 13, 795–800 (2006). [DOI] [PubMed] [Google Scholar]
- FINNEY D. J. & STEVENS W. L. A table for the calculation of working probits and weights in probit analysis. Biometrika. 35, 191–201 (1948). [PubMed] [Google Scholar]
- Tuttelmann F. et al. Combined effects of the variants FSHB -211G>T and FSHR 2039A>G on male reproductive parameters. J. Clin. Endocrinol. Metab 97, 3639–3647 (2012). [DOI] [PubMed] [Google Scholar]