

Abstract
Cholesterol-rich caveolar microdomains and associated caveolins influence sarcolemmal ion channel and receptor function and protective stress signaling. However, the importance of membrane cholesterol content to cardiovascular function and myocardial responses to ischemia-reperfusion (I/R) and cardioprotective stimuli are unclear. We assessed the effects of graded cholesterol depletion with methyl-β-cyclodextrin (MβCD) and lifelong knockout (KO) or overexpression (OE) of caveolin-3 (Cav-3) on cardiac function, I/R tolerance, and opioid receptor (OR)-mediated protection. Langendorff-perfused hearts from young male C57Bl/6 mice were untreated or treated with 0.02–1.0 mM MβCD for 25 min to deplete membrane cholesterol and disrupt caveolae. Hearts were subjected to 25-min ischemia/45-min reperfusion, and the cardioprotective effects of morphine applied either acutely or chronically [sustained ligand-activated preconditioning (SLP)] were assessed. MβCD concentration dependently reduced normoxic contractile function and postischemic outcomes in association with graded (10–30%) reductions in sarcolemmal cholesterol. Cardioprotection with acute morphine was abolished with ≥20 μM MβCD, whereas SLP was more robust and only inhibited with ≥200 μM MβCD. Deletion of Cav-3 also reduced, whereas Cav-3 OE improved, myocardial I/R tolerance. Protection via SLP remained equally effective in Cav-3 KO mice and was additive with innate protection arising with Cav-3 OE. These data reveal the membrane cholesterol dependence of normoxic myocardial and coronary function, I/R tolerance, and OR-mediated cardioprotection in murine hearts (all declining with cholesterol depletion). In contrast, baseline function appears insensitive to Cav-3, whereas cardiac I/R tolerance parallels Cav-3 expression. Novel SLP appears unique, being less sensitive to cholesterol depletion than acute OR protection and arising independently of Cav-3 expression.
Keywords: cardioprotection, caveolae, caveolin-3, cholesterol, contractility, ischemia-reperfusion, membrane microdomains, opioid receptors
sarcolemmal microdomains are critical regulators of cellular function and signaling (34) and may be important in sex-, age-, and disease-dependent differences in cardiac and vascular function (3, 10, 11, 14, 24, 49, 62). Caveolae are lipid rich (i.e., cholesterol and glycosphingolipid) microdomains containing scaffolding proteins, caveolins, that present as distinct molecular platforms for the regulation of cytoprotective and other signaling (52). However, the importance of membrane cholesterol and microdomains to the maintenance of myocardial and coronary function as well as intrinsic responses to insult/stress and protective stimuli is unclear.
Experimental findings from different in vitro cell models have suggested the potential dependence of myocardial and coronary vascular function on membrane cholesterol content (or caveolar integrity). In isolated cardiomyocytes, cholesterol depletion with methyl-β-cyclodextrin (MβCD) induces a negative inotropic effect at rest (6) while enhancing contractile responses to β2-adrenergic receptors (ARs) (6, 7, 26) and β1-ARs (1). In coronary smooth muscle, MβCD impairs endothelin-1 and serotonin responses while enhancing phenylephrine contraction (40). In other vascular smooth muscles, MβCD enhances Ca2+-activated Cl− currents (51), and in uterine myocytes, MβCD amplifies large-conductance Ca2+-activated K+ (BKCa) channel currents (50). In the vascular endothelium, MβCD also enhances BKCa activity to induce hyperpolarization (43) and represses agonist-mediated Ca2+ entry (30).
Despite effects of cholesterol depletion on cardiac and vascular myocyte and endothelial function in vitro, recent studies (8, 53) have reported MβCD insensitivity of coronary and myocardial contractile function in intact hearts. Given in vitro evidence that cholesterol and caveolar depletion significantly influence molecular determinants of cardiovascular function and control, in the present study, we assessed the impacts of acute graded cholesterol depletion with MβCD and lifelong modulation of caveolin-3 (Cav-3) [knockout (KO) and overexpression (OE)] on myocardial function, ischemia-reperfusion (I/R) tolerance, and cardioprotection via conventional, acute opoid receptor (OR) activation (acute ligand-activated preconditioning) versus novel, sustained OR activation [sustained ligand-activated preconditioning (SLP)] (35, 36, 38). The data revealed that cholesterol depletion markedly suppresses myocardial contractile function and coronary perfusion, impairs intrinsic I/R resistance, and negates cardioprotection via acute and sustained morphine treatment. In contrast, Cav-3 KO and OE modified I/R tolerance without altering normoxic function and do not influence protection via novel SLP.
MATERIALS AND METHODS
Animals.
All experiments were approved by and performed in accordance with guidelines of the Animal Ethics Committee of Griffith University (accredited by the Queensland Government, Department of Primary Industries and Fisheries, under the guidelines of “The Animal Care and Protection Act 2001, Section 757”) and the Institutional Animal Care and Use Committee of the Veterans Affairs San Diego Healthcare System. The generation and phenotypic details of Cav-3 KO and OE mice have been previously outlined in detail (12, 54).
Chemicals.
Morphine pellets were obtained via the National Institute of Drug Abuse (Bethesda, MD) or Murty Pharmaceuticals (Lexington, KY). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Induction of SLP.
Mice were briefly anesthetized with isoflurane, a small incision was made at the base of the neck, and placebo or morphine (75 mg) pellets were inserted aseptically in the dorsal subcutaneous space with the site closed with 9-mm wound clips. Pellets were left in place for 5 days, after which mice were euthanized for heart excision/perfusion (35, 36, 38).
Perfused heart preparation.
Mice were anesthetized with 60 mg/kg pentobarbital sodium, and hearts removed and perfused as previously described (16, 37, 58). Briefly, hearts were rapidly excised into ice-cold perfusion fluid, the aorta was cannulated, and the coronary circulation was perfused in Langendorff mode at a pressure of 80 mmHg with modified Krebs-Henseleit solution containing (in mM) 120 NaCl, 25 NaHCO3, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 KH2PO4, 15 d-glucose, and 0.5 EDTA. The perfusion fluid was maintained at 37°C and bubbled with a mix of 95% O2 and 5% CO2 at 37°C to provide a pH of 7.4 and Po2 of ∼600 mmHg at the aortic cannula over a flow range of 1–5 ml/min. The perfusate was continuously passed through a 0.45-μm filter. The left ventricle (LV) was vented with a polyethylene apical drain, and a fluid-filled polyvinyl chloride plastic film balloon inserted into the ventricle via the mitral valve. Balloons were connected by fluid-filled tubing to a P23 XL pressure transducer (Viggo-Spectramed, Oxnard, CA) permitting continuous assessment of contractile function. Balloon volume was increased to give an end-diastolic pressure of 5 mmHg during stabilization and was not further adjusted. Coronary flow was monitored via a flow probe in the aortic perfusion line connected to a T206 flowmeter (Transonic Systems, Ithaca, NY). Functional data were recorded at 1 kHz on an eight-channel MacLab data-acquisition system (AD Instruments) connected to an Apple iMac computer. LV pressure signals were digitally processed to yield diastolic, systolic, and developed pressures and heart rate.
After preparation, hearts were immersed in perfusion fluid at 37°C in a water-jacketed organ bath. Temperatures of the perfusion fluid and organ bath were continuously monitored by two needle thermistor probes connected to a TH-8 digital thermometer (Physitemp Instruments, Clifton, NJ). Hearts were excluded from the study if they met one of the following exclusion criteria after stabilization: 1) coronary flow > 5 ml/min, 2) unstable (fluctuating) contractile function, 3) LV systolic pressure < 100 mmHg, or 4) significant cardiac arrhythmias. This amounted to <4% of all hearts perfused.
Perfused heart experiments.
Hearts were isolated from control and SLP mice. Perfused hearts were then either untreated or subjected to 25 min of treatment with 0.02–1.0 mM MβCD after a stabilization period (20 min). Simultaneously, perfused hearts were switched to ventricular pacing at 420 beats/min (Grass S9 stimulator, Quincy, MA), with the rate normalized to permit comparison of rate-dependent measures of contractile function. Hearts were then either subjected to a 10-min period of morphine treatment (10 μM) or vehicle before 25 min of normothermic global zero-flow ischemia followed by 45 min of aerobic reperfusion. Pacing and drug infusion were terminated on the initiation of ischemia with pacing only resumed at 1.5 min of reperfusion (16, 37). Cell death in response to I/R was assessed via a spectrophotometric enzymatic assay of lactate dehydrogenase (LDH) released over the entire reperfusion period. Samples were stored at −80°C until enzymatic analysis (Cytotox 96, Promega, Madison, WI).
Simulated ischemia in HL-1 cells.
To simulate I/R in cultured HL-1 myocytes, cells were switched to “ischemic” buffer containing 20 mM HEPES (pH 6.6), 125 mM NaCl, 8 mM KCl, 1.2 mM KH2PO4, 1.25 mM MgSO4, 1.2 mM CaCl2, 6.25 mM NaHCO3, and 5 mM Na-lactate in a sealed hypoxic chamber (STEMCELL, Vancouver, BC, Canada) equilibrated with 95% N2-5% CO2 for 3 h. Reperfusion was achieved by the substitution of ischemic buffer with a normoxic solution containing 20 mM HEPES (pH 7.4), 110 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.25 mM MgSO4, 1.2 mM CaCl2, 25 mM NaHCO3, and 15 mM glucose equilibrated with 95% O2-5% CO2 for a further 5 h. Cells were untreated or treated with 1 mM MβCD for 60 min before simulated I/R. LDH activity was assayed using a CytoTox 96 assay kit (Promega) according to the manufacturer's instructions. Normoxic control cells incubated with MβCD showed no detectable cytotoxicity based on LDH changes (data not shown).
Cholesterol assay.
Total cholesterol content was assayed in membrane-enriched myocardial subcellular fractions using an Amplex Red Cholesterol Assay kit (Invitrogen) according to the manufacturer's instructions. After 60 min of incubation (37°C, in the dark), fluorescence was measured using an Infinite M200 Pro microplate reader (Tecan Australia) at 530/25-nm excitation and 590/35-nm emission wavelengths. Samples were assayed in duplicate, and cholesterol levels were determined from cholesterol standard curves run daily (content normalized to micrograms of protein).
Electron microscopy.
Whole hearts were perfused with standard Karnovsky's fixative of 4% paraformaldehyde and 1.5% glutaraldehyde in 0.1 M cacodylate buffer. Samples were further postfixed in 1% osmium tetroxide and en bloc stained with uranyl acetate. After dehydration, hearts were embedded in a longitudinal orientation in LX-112 (Ladd Research, Williston, VT) and polymerized at 60°C for 48 h. Blocks were then trimmed to regions of matching longitudinal orientation and thin sectioned, with sections stained in uranyl acetate and lead citrate and observed with an electron microscope (JOEL 1200 EX-II, JEOL USA, Peabody, MA, or Philips CM-10, Philips Electronic Instruments, Mahwah, NY).
Statistical analysis.
All data are expressed as means ± SE. Differences between groups were tested via one-way ANOVA, with a Newman-Keuls post hoc test used for paired comparisons when significant effects were detected. Significant differences were accepted for P < 0.05.
RESULTS
Cholesterol depletion with MβCD depresses normoxic function in murine hearts.
There were no significant differences in normoxic contractile function or coronary flow in isolated perfused hearts before MβCD treatment (Table 1). A 25-min period of treatment with MβCD concentration dependently depressed ventricular pressure development (Fig. 1, A and B), involving select inhibition of systolic force at 20 μM and combined diastolic and systolic actions at higher MβCD concentrations. Coronary vascular function was much less sensitive to MβCD, with flow only falling significantly (by ∼30%) at the highest concentration (1 mM; Fig. 1C). The insensitivity of coronary perfusion to 20–200 μM MβCD demonstrates that MβCD depresses contraction through a mechanism independent of flow.
Table 1.
Baseline function of ex vivo hearts before MβCD infusion
| Number of Hearts | LVEDP, mmHg | LVDP, mmHg | Heart Rate, beats/min | Coronary Flow, ml/min | |
|---|---|---|---|---|---|
| Control | |||||
| Untreated | 10 | 4 ± 1 | 118 ± 5 | 376 ± 7 | 2.7 ± 0.2 |
| AM | 7 | 5 ± 1 | 126 ± 10 | 377 ± 19 | 3.1 ± 0.4 |
| SLP | 9 | 4 ± 1 | 134 ± 10 | 353 ± 8 | 2.5 ± 0.3 |
| 20 μM MβCD | |||||
| Untreated | 11 | 4 ± 1 | 104 ± 9 | 322 ± 17 | 2.5 ± 0.3 |
| AM | 9 | 4 ± 1 | 109 ± 9 | 316 ± 19 | 2.2 ± 0.3 |
| SLP | 7 | 3 ± 1 | 138 ± 6 | 376 ± 34 | 2.1 ± 0.1 |
| 200 μM MβCD | |||||
| Untreated | 6 | 7 ± 1 | 131 ± 7 | 388 ± 9 | 2.8 ± 0.3 |
| AM | 6 | 5 ± 1 | 136 ± 12 | 378 ± 16 | 2.9 ± 0.4 |
| SLP | 6 | 5 ± 3 | 141 ± 2 | 373 ± 16 | 3.0 ± 0.2 |
| 1 mM MβCD | |||||
| Untreated | 6 | 5 ± 1 | 129 ± 7 | 378 ± 10 | 2.8 ± 0.4 |
Data are means ± SE. Values were recorded before pacing and methyl-β-cyclodextrin (MβCD) infusion. LVEDP, left ventricular (LV) end-diastolic pressure; LVDP, LV developed pressure; AM, acute morphine treatment; SLP, sustained ligand-activated preconditioning.
Fig. 1.

Effects of methyl-β-cyclodextrin (MβCD; 25 min) on normoxic contractile and vascular function in perfused hearts. Data are shown for percent changes from baseline values for end-diastolic pressure (EDP; A), left ventricular developed pressure (LVDP; B), and coronary flow rate (C). Values are means ± SE; for all groups, n ≥ 6. **P < 0.01 vs. untreated [control (CTRL)] hearts.
Cholesterol depletion with MβCD depresses I/R tolerance in murine hearts and HL-1 cells.
A 25-min ischemic insult in untreated control hearts resulted in a profound elevation in postischemic end-diastolic pressure (to 28 mmHg) and blunted recovery of contractile function (LV developed pressure reaching ∼50% of baseline function) after 45 min of reperfusion (Fig. 2, A and B). Pretreatment with MβCD depleted sarcolemmal cholesterol content in postischemic hearts in a concentration-dependent manner (by ∼10–30%; Fig. 2C). Electron microscopy revealed a progressive loss of caveolar structures with increasing concentrations of MβCD, with virtually no morphologically identifiable caveolae at 200 μM MβCD (Fig. 3). Functional recoveries in response to I/R were also impaired by MβCD, although I/R tolerance was less sensitive to MβCD than normoxic function: recoveries for LV diastolic function (Fig. 2A) and pressure development (Fig. 2B) were unaltered by 20 μM MβCD and progressively worsened with 200 μM and 1 mM MβCD. To test whether MβCD reduces ischemic tolerance at the level of the myocyte, HL-1 myocytes (untreated or incubated with 1 mM MβCD) were subjected to simulated I/R (Fig. 4). Pretreatment with MβCD doubled the impact of I/R on cellular damage, as indicated by LDH efflux.
Fig. 2.

Impact of increasing concentrations of MβCD on postischemic recovery and sarcolemmal cholesterol content assayed in membrane fractions after 25 min of ischemia and 45 min of reperfusion from untreated (CTRL) hearts and hearts subjected to 25 min of preischemic treatment with 20 μM, 200 μM, or 1 mM MβCD. Data are shown for recovery of EDP (in mmHg; A), recovery of LVDP (in %baseline; B), and membrane cholesterol content (in ng/μg protein; C). Values are mean ± SE; for all groups, n ≥ 6. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. CTRL hearts.
Fig. 3.

Qualitative electron microscopy showing decreasing caveolar density with 20 or 200 μM MβCD treatment under normoxic conditions.
Fig. 4.

Cellular disruption/death in HL-1 myocytes maintained under normoxic conditions or subjected to simulated ischemia-reperfusion (sI-R) with or without 1 mM MβCD. Values are means ± SE; for all groups, n ≥ 3. **P < 0.01 vs. normoxic conditions; ΦP < 0.01 vs. simulated ischemia-reperfusion.
MβCD treatment attenuates OR-mediated cardioprotection.
Preischemic treatment of perfused hearts with morphine (10 μM for 10 min) significantly reduced postischemic diastolic and contractile dysfunction (Fig. 5, A and B). Morphine-induced SLP provided even greater improvements in diastolic and contractile outcomes (Fig. 5, A and B). Protection with acute morphine was abolished by pretreatment with ≥20 μM MβCD, whereas protection via SLP was less sensitive, only declining with ≥200 μM MβCD.
Fig. 5.
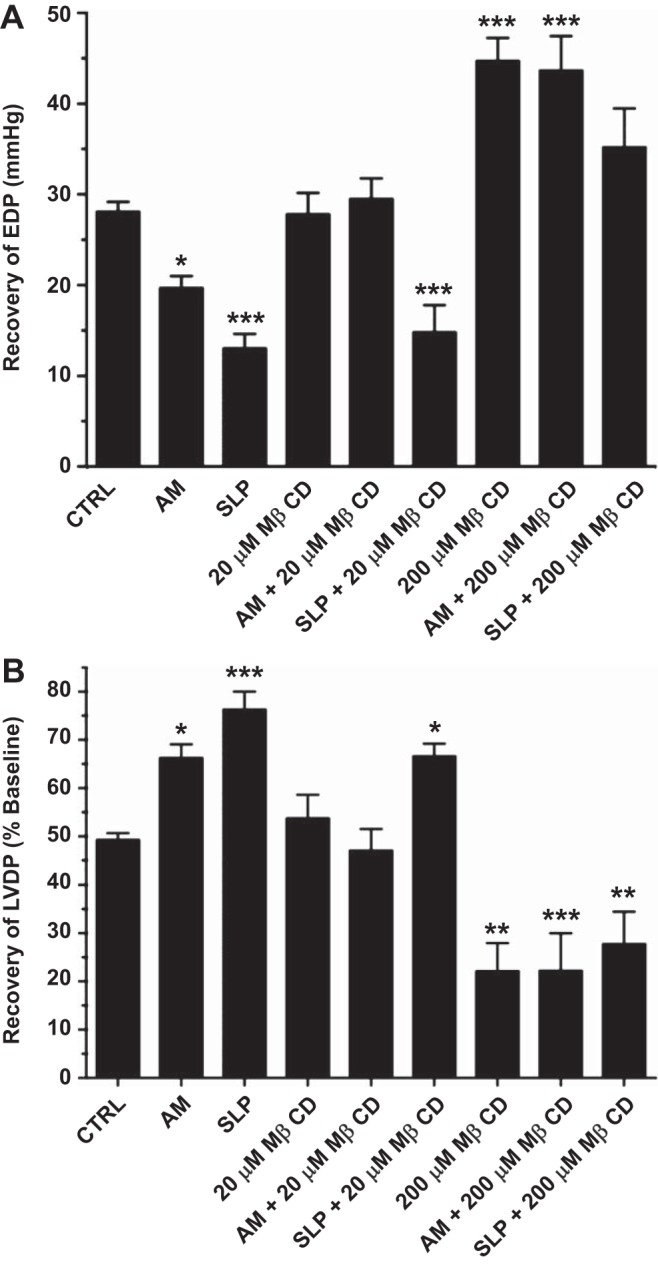
Effects of MβCD on opioid receptor (OR)-mediated cardioprotection. Postischemic recoveries after 45 min of reperfusion are shown for EDP (in mmHg; A) and LVDP (%baseline; B). Hearts were untreated (CTRL), treated acutely for 10 min with 10 μM morphine before ischemia [acute morphine (AM)], or removed from mice implanted with morphine pellets for 5 days [sustained ligand-activated preconditioning (SLP)]. Subsets of hearts were pretreated with 20 or 200 μM MβCD. Values are means ± SE; for all groups, n ≥ 6. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. CTRL hearts.
The SLP phenotype is Cav-3 independent.
Selective effects of 20 versus 200 μM MβCD on I/R tolerance and cardioprotection could reflect more select caveolar disruption with modest cholesterol depletion during low-level MβCD treatment, whereas higher levels may additionally disrupt noncaveolar membrane lipid rafts and essential membrane signaling. To assess the effects of more specific caveolar perturbation, we examined the impacts of Cav-3 KO and transgenic OE; ablation of myocardial Cav-3 eliminates caveolar structures, whereas Cav-3 OE increases caveolar density (18, 19, 54, 55).
Neither KO or OE of Cav-3 altered normoxic contractile function or coronary flow in isolated perfused hearts (Table 2), in contrast to the inhibitory effects of MβCD. However, Cav-3 KO significantly exaggerated postischemic diastolic dysfunction and LDH efflux in perfused hearts (Fig. 6, A–C). Conversely, Cav-3 OE significantly improved postischemic functional outcomes without altering LDH efflux (Fig. 7, A–C). Deletion of Cav-3 failed to attenuate the cardioprotective effects of morphine-dependent SLP (Fig. 6), and SLP significantly improved postischemic recoveries in Cav-3 OE hearts (Fig. 7). Manipulation of Cav-3 expression and treatment with MβCD thus induced both common and distinct cardiac outcomes: MβCD repressed normoxic function, whereas Cav-3 KO did not, and MβCD and Cav-3 KO both impaired protection via acute OR agonism, whereas SLP was only sensitive to MβCD.
Table 2.
Function of ex vivo hearts from wild-type, Cav-3 knockout, and Cav-3-overexpressing mice
| Treatment | Number of hearts | LVEDP, mmHg | LVDP, mmHg | Heart Rate, beats/min | Coronary Flow, ml/min |
|---|---|---|---|---|---|
| Wild-type mice | |||||
| Placebo | 9 | 4 ± 1 | 104 ± 4 | 314 ± 16 | 2.0 ± 0.2 |
| SLP | 7 | 4 ± 1 | 120 ± 8 | 320 ± 19 | 2.0 ± 0.2 |
| Cav-3 knockout mice | |||||
| Placebo | 7 | 5 ± 1 | 122 ± 9 | 305 ± 20 | 2.4 ± 0.3 |
| SLP | 7 | 5 ± 1 | 115 ± 8 | 324 ± 20 | 2.1 ± 0.3 |
| Cav-3-overexpressing mice | |||||
| Placebo | 7 | 3 ± 1 | 104 ± 8 | 372 ± 16 | 3.2 ± 0.9 |
| SLP | 7 | 4 ± 1 | 115 ± 7 | 346 ± 8 | 2.5 ± 0.3 |
Data are means ± SE. Values were recorded before pacing. Cav-3, caveolin-3.
Fig. 6.

Effects of caveolin-3 (Cav-3) knockout (KO) on ischemia-reperfusion (I/R) tolerance and protection via SLP. Data are shown for postischemic EDP (in mmHg; A), LVDP (in %; B), and LDH release (C). Responses were assessed in wild-type (CTRL) and Cav-3 KO hearts from untreated or SLP-treated mice (5-day morphine pellet). Values are means ± SE; for all groups, n ≥ 7. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. CTRL mice; †P < 0.05 vs. Cav-3 KO mice.
Fig. 7.

Effects of Cav-3 overexpression (OE) on I/R tolerance and protection via SLP. Data are shown for postischemic EDP (in mmHg; A), LVDP (in %; B), and LDH release (C). Responses were assessed in wild-type (CTRL) and Cav-3 OE hearts from untreated or SLP-treated mice (5-day morphine pellet). Values are means ± SE; for all groups, n = 7. **P < 0.01, and ***P < 0.001 vs. CTRL mice; †P < 0.05 vs. Cav-3 OE mice; #P < 0.05 vs. SLP.
DISCUSSION
The results of the present study highlight the importance of sarcolemmal cholesterol and membrane microdomains to the maintenance of myocardial contractile function and resistance to I/R. The ability to protect the heart against I/R injury via OR-targeted stimuli is also selectively modified by changes in membrane cholesterol and Cav-3. Distinguishing novel SLP from conventional protection (e.g., via acute OR activation), this conditioning response was shown to be less sensitive to cholesterol depletion and appears entirely Cav-3/caveolae independent.
Role of membrane cholesterol in the maintenance of cardiovascular function.
The present data revealed significant contractile and vascular alterations in hearts perfused with the cholesterol-depleting agent MβCD (Fig. 2). Inhibitory effects of MβCD are concentration and parameter dependent: the myocardial response specifically involves systolic depression at low MβCD (with both diastolic and systolic changes at higher concentrations) and is substantially more MβCD sensitive than coronary function. These findings contrast with recent reports of unaltered normoxic function in perfused hearts treated with 200 μM (8) or 1 mM (53) MβCD. The basis for this difference is unclear, with all studies using isolated perfused rodent hearts and similar ranges of MβCD concentration. However, repression of contractile function is consistent with the significant influences of membrane cholesterol and caveolae on a variety of ion channels, receptor function and signaling, and other physiological processes (3, 4, 13, 22).
The membrane cholesterol dependence of function in intact hearts (Fig. 1) agrees with prior in vitro observations, although mixed effects of cholesterol depletion with MβCD have been reported. For example, L-type Ca2+ channels, β2-AR, adenylyl cyclase, and PKA colocalize in caveolar macrocomplexes governing channel function and cell signaling in ventricular myocytes. Similarly, cholesterol depletion has been reported to reduce myocyte Ca2+ currents and contraction under baseline conditions (6) and inhibit effects of β2-AR activity (2, 6). Nonetheless, other studies have identified amplified β1-AR or β2-AR signaling upon myocyte cholesterol depletion, with enhanced PKA/adenylyl cyclase phosphoregulation of target proteins (e.g., phospholamban and troponin I), and activation of Ca2+ currents and myocyte contractile function (1, 7, 26). While we do not address β-AR responsiveness, our data for intact perfused hearts are consistent with inhibitory rather than stimulatory effects of cholesterol depletion on Ca2+-dependent myocyte function (3, 6). The absence of an effect of Cav-3 KO or OE on normoxic function suggests that cholesterol depletion impacts cardiac function via mechanisms additional to caveolar disruption. However, it is also possible that adaptation within the membrane signaling compartment occurs with lifelong changes in Cav-3, compensating for cardiac functional changes (whereas acute disruption with MβCD cannot be accommodated in such a manner). While MβCD has also been reported to induce cytotoxicity in some cell types (17), this is an unlikely explanation for contractile depression since cardiac effects of MβCD can arise at a low concentration (20 μM), three orders of magnitude lower than levels inducing cytotoxicity in vitro (17). Moreover, functional effects of MβCD are selective, impacting systolic versus diastolic force without influencing coronary function.
Caveolae and cholesterol are implicated in vascular control, and we observed repression of coronary flow with the high concentration (1 mM) of MβCD. In coronary and other vascular smooth muscles, MβCD treatment modifies ion channel function (BKCa and Ca2+-activated Cl− currents) and receptor-dependent control of intracellular Ca2+ concentration and vascular tone (40, 47, 51). MβCD also modifies BKCa currents in nonvascular smooth muscle (50), together with BKCa activity (43) and agonist-mediated Ca2+ entry (30) in the vascular endothelium. Interestingly, MβCD treatment diminishes store-operated Ca2+ channel function (where the reduction in intracellular Ca2+ stores activates Ca2+ influx) and attenuates the hypertensive phenotype (associated with elevated caveolae/caveolin levels) in pulmonary arteries from patients with idiopathic pulmonary arterial hypertension (33). The basis of the reduction in coronary perfusion, observed at the highest level of MβCD treatment, is unclear. Coronary sensitivity to the cholesterol-depleting agent is substantially lower than cardiac sensitivity; however, cardiac changes could contribute to the vascular response. The substantial ∼20-mmHg rise in diastolic pressure with 1 mM MβCD will limit coronary perfusion via diastolic compression, whereas the 40% fall in ventricular pressure development will reduce myocardial work and thereby O2 demand. Thus, MβCD may selectively impair myocardial versus coronary vascular function, with reduced coronary flow reflecting cardiac compression coupled with reduced “metabolic” vasodilatation.
Importance of membrane cholesterol and Cav-3 to I/R tolerance.
The present study revealed common and qualitatively differing effects of MβCD and Cav-3 KO on myocardial I/R tolerance. Since MβCD depletes membrane cholesterol (impacting caveolae together with planar lipid rafts), whereas Cav-3 KO selectively depletes caveolae (without altering other cholesterol-dependent domains, such as lipid rafts, which may still be intact and regulate components of the protective signaling pathway), these data support distinct roles for different membrane compartments and highlight the importance for membrane cholesterol content in the regulation of myocardial I/R tolerance. Acute depletion of membrane cholesterol by ≥20% worsens both postischemic diastolic contracture and contractile depression (Figs. 2 and 5). In contrast, chronic Cav-3 depletion selectively exaggerates diastolic dysfunction and LDH release without impacting force development (Fig. 6). These differences suggest important requirements for Cav-3/caveolae in limiting cell death and diastolic contracture (the latter directly or as a result of altered cell death), whereas non-caveolar domains are additionally important in influencing contractile depression or “stunning.” Curiously, while increased Cav-3 expression did improve I/R tolerance (Fig. 7), this involved enhanced contractile outcomes with no shift in cell death. Thus, although Cav-3/caveolae appear critical to intrinsic resistance to cell death/diastolic dysfunction, an excess of Cav-3 represses mechanical stunning without providing further protection against death.
The declining I/R tolerance is not unexpected given the influences of caveolae and cholesterol on key determinants of stress resistance (8, 18, 31, 32, 44, 52–57). For example, MβCD consistently inhibits BKCa channel function across different cell types, and the channel has been implicated in cytoprotection and I/R injury. Similarly, β2-AR function is consistently perturbed by cholesterol depletion, and these receptors are also cardioprotective (5, 9). Nonetheless, the present study contrasts with those of Das et al. (8) and Sun et al. (53), in which no changes in I/R tolerance (or normoxic function) were observed after MβCD treatment. The reasons for this discrepancy are unclear, although we note consistent exaggeration of I/R injury in both intact hearts and cultured HL-1 cells (Fig. 4), supporting a select cardiac effect.
Importance of membrane cholesterol and Cav-3 in OR-mediated cardioprotection.
Growing evidence indicates that caveolins and caveolae are crucial determinants of the efficacy of cytoprotective stimuli (15, 18, 32, 54–56). We examined the effects of membrane disruption with MβCD on two cardioprotective responses initiated by ORs yet mediated by distinct mechanisms (36): acute (conventional) OR-induced protection and novel OR-induced SLP. Acute OR protection was lost in hearts exposed to ≥20 μM MβCD, whereas protection via SLP was more resistant, only declining at ≥200 μM MβCD (Fig. 5). This supports mechanistically distinct protection via chronic versus acute OR activation. Previous studies have shown that cardioprotective responses induced via ischemic preconditioning, acute OR agonism, and volatile anesthetic treatment are all negated by MβCD treatment (18, 31) or caveolar disruption through caveolin KO (32, 54). This may reflect select disruption of caveolae (and dependent G protein-coupled receptors) at low MβCD levels versus more widespread disruption of membrane lipid rafts at higher concentrations. Non-caveolar planar lipid rafts may be equally important in the membrane signal transduction underlying cytoprotective responses (44, 52). It is also important to note that acute and sustained OR stimuli differ in terms of the timing of OR involvement. For SLP, we here assessed the effects of acute MβCD on the signaling mediating protection downstream of and subsequent to OR agonism (36). In contrast, in the case of acute morphine, we tested the effects of MβCD on both the initiation of OR activity and subsequent signaling events mediating protection. Thus, the greater sensitivity of acute OR protection to MβCD likely reflects the disruption of initial caveolae/Cav-3-dependent OR activity (blocking the initial induction of protection). In contrast, since ORs are not involved in the mediation of protection in SLP hearts (36), there was no impact of low-level MβCD. The effects of higher MβCD on both acute and SLP responses likely reflect “noncaveolar” effects involving other membrane-dependent determinants of cardioprotection.
The observations from the MβCD experiments concur with the effects of Cav-3 KO. SLP dramatically improved postischemic functional recovery in KO hearts, indicating that the efficacy of SLP is neither Cav-3 or caveolae dependent. This is consistent with the additivity of cardioprotection via SLP and Cav-3 OE. A previous report (38) has indicated that SLP is also additive with different adenosinergic stimuli. Moreover, SLP is PKA dependent (36). Wang et al. (57) reported that simultaneous activation of AMP-activated protein kinase and PKA in Cav-3 KO mice significantly reduced infarct size and contractile dysfunction, providing some insight into the potential mechanisms of SLP in Cav-3 KO hearts.
Importantly, the efficacy of SLP in Cav-3 KO mice (and resistance to low-dose MβCD) indicates that the δ-ORs engaged during morphine pretreatment (38) are Cav-3 independent and that the mediation of protection during I/R also involves Cav-3-independent signaling. Data remain mixed regarding the caveolae and Cav-3 dependence of δ-ORs. We have shown that myocardial δ-ORs are localized with Cav-3 in caveolae, with caveolar disruption with MβCD (31) and Cav-3 KO (55) eliminating acute δ-OR-mediated protection. This is consistent with the elimination of acute morphine protection with MβCD in the present study (Fig. 5). Huang et al. (20) also reported that MβCD impairs δ-OR activity in neuronal cells (where the receptor localizes to lipid rafts) yet enhances signaling in transfected Chinese hamster ovary cells. In contrast, δ-ORs expressed in human embryonic kidney-293 cells are reportedly insensitive to MβCD, whereas μ-OR function is cholesterol dependent (23). The caveolar/Cav-3 dependence of δ-OR function is clearly cell type specific. Since myocardial δ-ORs appear to be MβCD/Cav-3 dependent (31, 55), the preservation of SLP in Cav-3 KO mice implicates noncardiac δ-ORs in the induction of cardioprotection. This is consistent with evidence showing that peripheral ORs can mediate cardiac preconditioning (48) and with the potent cardioprotective effects of central/spinal cord ORs (25, 59, 60). The potential extracardiac localization of δ-ORs engaged in protective SLP warrants further investigation.
Our data demonstrate that mechanisms mediating protection with SLP are distinct from those in conventional protection, being less sensitive to cholesterol depletion than the acute OR response and apparently independent of Cav-3 expression and caveolae. Thus, while β2-ARs and PKA have been implicated in SLP (36, 38), the present observations indicate that the effects of such mediators are Cav-3/caveolae independent yet sensitive to high levels of MβCD/cholesterol depletion. This seems at odds with data demonstrating the dependence of β2-AR signal transduction on cholesterol-rich caveolae (29, 45, 61). However, other studies have indicated that only a small fraction of β2-ARs concentrate within caveolae (21, 39), with cholesterol depletion augmenting (rather than limiting) β2-AR signaling (21, 27, 28, 41, 45, 46). Further work is clearly required to resolve the influences of cholesterol levels and caveolae on cardiac β2-AR signaling and the mechanisms underpinning β2-AR/PKA-dependent yet Cav-3/caveolae-independent SLP. Since cholesterol is an important component of the entire membrane, including non-caveolar domains, the present data indicate that other functional compartments play an important role.
Summary.
The present study reveals that myocardial contractile function and I/R tolerance are highly sensitive to membrane cholesterol depletion, whereas lifelong/chronic disruption of caveolae (via Cav-3 manipulation) selectively modifies I/R tolerance without influencing cardiac function. These data indicate that shifts in membrane cholesterol content and/or caveolar function, with age or disease for example, may be important determinants of myocardial dysfunction and injury processes. Finally, the present data confirm a distinct mechanistic basis for protective SLP, which is less sensitive to cholesterol depletion than conventional protection and arises independently of caveolae/Cav-3. Further investigation is required to clarify the mechanistic basis of, and role of membrane cholesterol in, cardioprotective SLP.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-091071 and HL-107200 (to H. H. Patel) and Veterans Affairs Merit Grants BX001963 (to H. H. Patel) and BX000783 (to D. M. Roth). L. E. See Hoe was supported by a scholarship from the National Heart Foundation of Australia. J. N. Peart was supported by a Future Fellowship from the Australian Research Council. J. P. Headrick was supported by a fellowship from the Queensland state government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.E.S.H., C.J.K., K.J.A., J.P.H., H.H.P., and J.N.P. conception and design of research; L.E.S.H., J.M.S., E.T., C.J.K., A.R.B., I.R.N., J.P.H., and J.N.P. performed experiments; L.E.S.H., J.M.S., E.T., C.J.K., A.R.B., I.R.N., K.J.A., J.P.H., H.H.P., and J.N.P. analyzed data; L.E.S.H., J.M.S., E.T., C.J.K., A.R.B., I.R.N., E.F.D.T., K.J.A., D.M.R., J.P.H., H.H.P., and J.N.P. interpreted results of experiments; L.E.S.H., J.P.H., H.H.P., and J.N.P. drafted manuscript; L.E.S.H., J.M.S., C.J.K., E.F.D.T., K.J.A., D.M.R., J.P.H., H.H.P., and J.N.P. edited and revised manuscript; L.E.S.H., J.M.S., E.F.D.T., K.J.A., D.M.R., J.P.H., H.H.P., and J.N.P. approved final version of manuscript; J.N.P. prepared figures.
REFERENCES
- 1.Agarwal SR, MacDougall DA, Tyser R, Pugh SD, Calaghan SC, Harvey RD. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J Mol Cell Cardiol 50: 500–509, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc Natl Acad Sci USA 103: 7500–7505, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balijepalli RC, Kamp TJ. Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol 98: 149–160, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bastiaanse EM, Höld KM, Van der Laarse A. The effect of membrane cholesterol content on ion transport processes in plasma membranes. Cardiovasc Res 33: 272–283, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Bhushan S, Kondo K, Predmore BL, Zlatopolsky M, King AL, Pearce C, Huang H, Tao YX, Condit ME, Lefer DJ. Selective β2-adrenoreceptor stimulation attenuates myocardial cell death and preserves cardiac function after ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol 32: 1865–1867, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calaghan S, White E. Caveolae modulate excitation-contraction coupling and β2-adrenergic signalling in adult rat ventricular myocytes. Cardiovasc Res 69: 816–824, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Calaghan S, Kozera L, White E. Compartmentalisation of cAMP-dependent signalling by caveolae in the adult cardiac myocyte. J Mol Cell Cardiol 45: 88–92, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Das M, Gherghiceanu M, Lekli I, Mukherjee S, Popescu LM, Das DK. Essential role of lipid raft in ischemic preconditioning. Cell Physiol Biochem 21: 325–334, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Fajardo G, Zhao M, Berry G, Wong LJ, Mochly-Rosen D, Bernstein D. β2-Adrenergic receptors mediate cardioprotection through crosstalk with mitochondrial cell death pathways. Mol Cell Cardiol 51: 781–789, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fridolfsson HN, Patel HH. Caveolin and caveolae in age associated cardiovascular disease. J Geriatr Cardiol 10: 66–74, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gratton JP, Bernatchez P, Sessa WC. Caveolae and caveolins in the cardiovascular system. Circ Res 94: 1408–1417, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Hagiwara Y, Sasaoka T, Araishi K, Imamura M, Yorifuji H, Nonaka I, Ozawa E, Kikuchi T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet 9: 3047–3054, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Harvey RD, Calaghan SC. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J Mol Cell Cardiol 52: 366–375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hausman N, Martin J, Taggart MJ, Austin C. Age-related changes in the contractile and passive arterial properties of murine mesenteric small arteries are altered by caveolin-1 knockout. J Cell Mol Med 16: 1720–1730, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Head BP, Peart JN, Panneerselvam M, Yokoyama T, Pearn ML, Niesman IR, Bonds JA, Schilling JM, Miyanohara A, Headrick J, Ali SS, Roth DM, Patel PM, Patel HH. Loss of caveolin-1 accelerates neurodegeneration and aging. PLOS ONE 5: e15697, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Headrick JP, Willems L, Ashton KJ, Holmgren K, Peart J, Matherne GP. Ischaemic tolerance in aged mouse myocardium: the role of adenosine and effects of A1 adenosine receptor overexpression. J Physiol 549: 823–833, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinzey AH, Kline MA, Kotha SR, Sliman SM, Butler ES, Shelton AB, Gurney TR, Parinandi NL. Choice of cyclodextrin for cellular cholesterol depletion for vascular endothelial cell lipid raft studies: cell membrane alterations, cytoskeletal reorganization and cytotoxicity. Indian J Biochem Biophys 49: 329–341, 2012 [PubMed] [Google Scholar]
- 18.Horikawa YT, Patel HH, Tsutsumi YM, Jennings MM, Kidd MW, Hagiwara Y, Ishikawa Y, Insel PA, Roth DM. Caveolin-3 expression and caveolae are required for isoflurane-induced cardiac protection from hypoxia and ischemia/reperfusion injury. J Mol Cell Cardiol 44: 123–130, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horikawa YT, Panneerselvam M, Kawaraguchi Y, Tsutsumi YM, Ali SS, Balijepalli RC, Murray F, Head BP, Niesman IR, Rieg T, Vallon V, Insel PA, Patel HH, Roth DM. Cardiac-specific overexpression of caveolin-3 attenuates cardiac hypertrophy and increases natriuretic peptide expression and signaling. J Am Coll Cardiol 57: 2273–2283, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang P, Xu W, Yoon SI, Chen C, Chong PL, Liu-Chen LY. Cholesterol reduction by methyl-β-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol 73: 534–549, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ianoul A, Grant DD, Rouleau Y, Bani-Yaghoub M, Johnston LJ, Pezacki JP. Imaging nanometer domains of β-adrenergic receptor complexes on the surface of cardiac myocytes. Nat Chem Biol 1: 196–202, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Kozera L, White E, Calaghan S. Caveolae act as membrane reserves which limit mechanosensitive ICl,swell channel activation during swelling in the rat ventricular myocyte. PLOS ONE 4: e8312, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levitt ES, Clark MJ, Jenkins PM, Martens JR, Traynor JR. Differential effect of membrane cholesterol removal on μ- and δ-opioid receptors: a parallel comparison of acute and chronic signaling to adenylyl cyclase. J Biol Chem 284: 22108–22122, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loyer X, Oliviero P, Damy T, Robidel E, Marotte F, Heymes C, Samuel JL. Effects of sex differences on constitutive nitric oxide synthase expression and activity in response to pressure overload in rats. Am J Physiol Heart Circ Physiol 293: H2650–H2658, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Hu J, Zhang Y, Dong C. Spinal neuronal NOS activation mediates intrathecal fentanyl preconditioning induced remote cardioprotection in rats. Int Immunopharmacol 19: 127–131, 2014 [DOI] [PubMed] [Google Scholar]
- 26.Macdougall DA, Agarwal SR, Stopford EA, Chu H, Collins JA, Longster AL, Colyer J, Harvey RD, Calaghan S. Caveolae compartmentalise β2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte. J Mol Cell Cardiol 52: 388–400, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miura Y, Hanada K, Jones TL. Gs signaling is intact after disruption of lipid rafts. Biochemistry 40: 15418–15423, 2001 [DOI] [PubMed] [Google Scholar]
- 28.O'Connor SW, Scarpace PJ, Abrass IB. The effect of age and cholesterol on the rat lung β-adrenergic system. Biochim Biophys Acta 778: 497–502, 1984 [DOI] [PubMed] [Google Scholar]
- 29.Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J Biol Chem 276: 42063–42069, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Paffett ML, Naik JS, Riddle MA, Menicucci SD, Gonzales AJ, Resta TC, Walker BR. Altered membrane lipid domains limit pulmonary endothelial calcium entry following chronic hypoxia. Am J Physiol Heart Circ Physiol 301: H1331–H1340, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel HH, Head BP, Petersen HN, Niesman IR, Huang D, Gross GJ, Insel PA, Roth DM. Protection of adult rat cardiac myocytes from ischemic cell death: role of caveolar microdomains and δ-opioid receptors. Am J Physiol Heart Circ Physiol 291: H344–H350, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Patel HH, Tsutsumi YM, Head BP, Niesman IR, Jennings M, Horikawa Y, Huang D, Moreno AL, Patel PM, Insel PA, Roth DM. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J 21: 1565–1574, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, Thistlethwaite PA, Miyanohara A, Farquhar MG, Yuan JX, Insel PA. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J 21: 2970–2979, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Patel HH, Murray F, Insel PA. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol 48: 359–391, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peart JN, Gross GJ. Morphine-tolerant mice exhibit a profound and persistent cardioprotective phenotype. Circulation 109: 1219–1222, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Peart JN, Gross GJ. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am J Physiol Heart Circ Physiol 291: H1746–H1753, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Peart JN, Gross ER, Headrick JP, Gross GJ. Impaired p38-MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J Mol Cell Cardiol 42: 972–980, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peart JN, Hoe LE, Gross GJ, Headrick JP. Sustained ligand-activated preconditioning via δ-opioid receptors. J Pharmacol Exp Ther 336: 274–281, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pontier SM, Percherancier Y, Galandrin S, Breit A, Galés C, Bouvier M. Cholesterol-dependent separation of the β2-adrenergic receptor from its partners determines signaling efficacy: insight into nanoscale organization of signal transduction. J Biol Chem 283: 24659–24672, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prendergast C, Quayle J, Burdyga T, Wray S. Cholesterol depletion alters coronary artery myocyte Ca2+ signalling in a stimulus-specific manner. Cell Calcium 47: 84–91, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prieto JC, Hueso C, Carmena MJ. Modulation of the β-adrenergic stimulation of cyclic AMP accumulation in rat prostatic epithelial cells by membrane fluidity. Gen Pharmacol 21: 931–933, 1990 [DOI] [PubMed] [Google Scholar]
- 42.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Riddle MA, Hughes JM, Walker BR. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am J Physiol Cell Physiol 301: C1404–C1414, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth DM, Patel HH. Role of caveolae in cardiac protection. Pediatr Cardiol 32: 329–333, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem 275: 41447–41457, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Scarpace PJ, O'Connor SW, Abrass IB. Cholesterol modulation of β-adrenergic receptor characteristics. Biochim Biophys Acta 30: 520–525, 1985 [DOI] [PubMed] [Google Scholar]
- 47.Schach C, Firth AL, Xu M, Remillard CV, Patel HH, Insel PA, Yuan JX. Regulation of pulmonary vasoconstriction by agonists and caveolae. Exp Lung Res 34: 195–208, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schultz JJ, Hsu AK, Gross GJ. Ischemic preconditioning is mediated by a peripheral opioid receptor mechanism in the intact rat heart. J Mol Cell Cardiol 29: 1355–1362, 1997 [DOI] [PubMed] [Google Scholar]
- 49.Sharma A, Yu C, Bernatchez PN. New insights into caveolae, caveolins and endothelial function. Can J Cardiol 26: 5A–8A, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Shmygol A, Noble K, Wray S. Depletion of membrane cholesterol eliminates the Ca2+-activated component of outward potassium current and decreases membrane capacitance in rat uterine myocytes. J Physiol 581: 445–456, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sones WR, Davis AJ, Leblanc N, Greenwood IA. Cholesterol depletion alters amplitude and pharmacology of vascular calcium-activated chloride channels. Cardiovasc Res 87: 476–484, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stary CM, Tsutsumi YM, Patel PM, Head BP, Patel HH, Roth DM. Caveolins: targeting pro-survival signaling in the heart and brain. Front Physiol 3: 393, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun J, Kohr MJ, Nguyen T, Aponte AM, Connelly PS, Esfahani SG, Gucek M, Daniels MP, Steenbergen C, Murphy E. Disruption of caveolae blocks ischemic preconditioning-mediated S-nitrosylation of mitochondrial proteins. Antioxid Redox Signal 16: 45–56, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR, Yokoyama U, Head BP, Hagiwara Y, Ishikawa Y, Miyanohara A, Patel PM, Insel PA, Patel HH, Roth DM. Cardiac-specific overexpression of caveolin-3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation 118: 1979–1988, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsutsumi YM, Kawaraguchi Y, Niesman IR, Patel HH, Roth DM. Opioid-induced preconditioning is dependent on caveolin-3 expression. Anesth Analg 111: 1117–1121, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsutsumi YM, Kawaraguchi Y, Horikawa YT, Niesman IR, Kidd MW, Chin-Lee B, Head BP, Patel PM, Roth DM, Patel HH. Role of caveolin-3 and glucose transporter-4 in isoflurane-induced delayed cardiac protection. Anesthesiology 112: 1136–1145, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, Gao E, Ma XL. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol 32: 934–942, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Willems L, Zatta A, Holmgren K, Ashton KJ, Headrick JP. Age-related changes in ischemic tolerance in male and female mouse hearts. J Mol Cell Cardiol 38: 245–256, 2005 [DOI] [PubMed] [Google Scholar]
- 59.Wong GT, Ling Ling J, Irwin MG. Activation of central opioid receptors induces cardioprotection against ischemia-reperfusion injury. Anesth Analg 111: 24–28, 2010 [DOI] [PubMed] [Google Scholar]
- 60.Wong GT, Lu Y, Mei B, Xia Z, Irwin MG. Cardioprotection from remote preconditioning involves spinal opioid receptor activation. Life Sci 91: 860–865, 2012 [DOI] [PubMed] [Google Scholar]
- 61.Xiang Y, Rybin VO, Steinberg SF, Kobilka B. Caveolar localization dictates physiologic signaling of β2-adrenoceptors in neonatal cardiac myocytes. J Biol Chem 277: 34280–34286, 2002 [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto S, Kita S, Iyoda T, Yamada T, Iwamoto T. New molecular mechanisms for cardiovascular disease: cardiac hypertrophy and cell-volume regulation. J Pharmacol Sci 116: 343–349, 2011 [DOI] [PubMed] [Google Scholar]