

Abstract
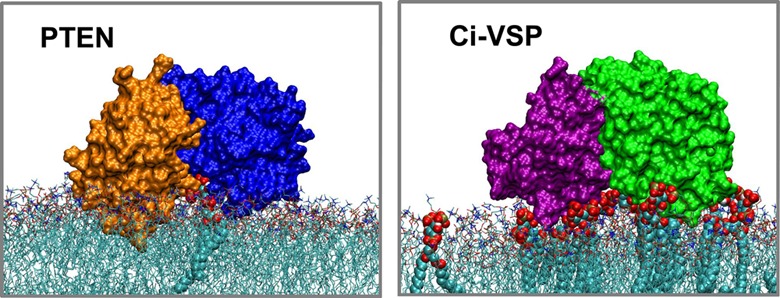
The phosphatase and tensin homologue (PTEN) and the Ciona intestinalis voltage sensitive phosphatase (Ci-VSP) are both phosphatidylinositol phosphate (PIP) phosphatases that contain a C2 domain. PTEN is a tumor suppressor protein that acts as a phosphatase on PIP3 in mammalian cell membranes. It contains two principal domains: a phosphatase domain (PD) and a C2 domain. Despite detailed structural and functional characterization, less is known about its mechanism of interaction with PIP-containing lipid bilayers. Ci-VSP consists of an N-terminal transmembrane voltage sensor domain and a C-terminal PTEN domain, which in turn contains a PD and a C2 domain. The nature of the interaction of the PTEN domain of Ci-VSP with membranes has not been well established. We have used multiscale molecular dynamics simulations to define the interaction mechanisms of PTEN and of the Ci-VSP PTEN domains with PIP-containing lipid bilayers. Our results suggest a novel mechanism of association of the PTEN with such bilayers, in which an initial electrostatics-driven encounter of the protein and bilayer is followed by reorientation of the protein to optimize its interactions with PIP molecules in the membrane. Although a PIP3 molecule binds close to the active site of PTEN, our simulations suggest a further conformational change of the protein may be required for catalytically productive binding to occur. Ci-VSP interacted with membranes in an orientation comparable to that of PTEN but bound directly to PIP-containing membranes without a subsequent reorientation step. Again, PIP3 bound close to the active site of the Ci-VSP PD, but not in a catalytically productive manner. Interactions of Ci-VSP with the bilayer induced clustering of PIP molecules around the protein.
Many cell signaling events are triggered by the association of peripheral membrane proteins with the membrane.1−5 The cell membrane acts both as a scaffold for the localization of peripheral proteins and as a two-dimensional platform that allows diffusion on the membrane surface, resulting in the formation of protein–lipid complexes.6 Association of peripheral proteins with specific lipids in the membrane (e.g., phosphatidylinositol phosphates or PIPs) occurs via lipid-binding modules.7−13 Indeed, it has been shown that the majority of human kinases contain at least one lipid-binding module,4 demonstrating the importance of the peripheral protein–lipid association in many cellular events. The main binding modules that have been identified in mammals are the C1, C2, FERM, PX, and PH domains.14 This work will focus on two related proteins that contain a C2 domain and catalyze the dephosphorylation of PIPs: the intensively studied PTEN (phosphatase and tensin homologue) tumor suppressor and the less well characterized voltage sensitive phosphatase from Ciona intestinalis (Ci-VSP).
C2 domains possess an antiparallel β-sheet architecture with variable loops connecting the eight β-sheets.15,16 They can be grouped into two types: C2 domains that associate with the membrane in a Ca2+-dependent manner and C2 domains that bind to the membrane in a Ca2+-independent manner.17 Both types of C2 domains have been shown to interact with anionic lipids, such as phosphatidylserine (PS) and PIPs, in the plasma membrane.6,7,11,18 PTEN and related proteins (e.g., Ci-VSP and auxilin) contain Ca2+-independent C2 domains,19,20 the loops of which are thought to form direct interactions with anionic lipids. For example, recent simulation studies of the auxilin PTEN-like domain have shown that the loops of its C2 domain determine its orientation relative to the membrane and promote PIP clustering around the bound protein.21
PIPs serve as second messengers in many signaling events and are involved in several pathological defects.22 PIPs have an inositol headgroup that can be phosphorylated at different positions, creating different PIP species. For example, PI(4,5)P2 and PI(3,4,5)P3 are the major PIPs in the plasma membrane.23 The exact percentage of different PIPs in the plasma membrane is difficult to determine because of the reversible turnover of PI(4,5)P2 to PI(3,4,5)P3 and other PIP species. It is generally stated that PI(4,5)P2 comprises ∼5% of all phospholipids in the cytoplasmic leaflet of the plasma membrane.24,25 For comparison, phosphatidylserine is the most abundant anionic phospholipid in eukaryotic cells and comprises approximately 20% of plasma membrane lipids.26
PTEN is a cytosolic enzyme that when bound to the inner leaflet of the plasma membrane catalyzes dephosphorylation of PI(3,4,5)P3 to PI(4,5)P2.27 By reducing the level of PI(3,4,5)P3 in the inner membrane leaflet, PTEN negatively regulates the phosphatidylinositol 3-kinase (PI3K) signaling pathway, leading to a reduced level of cell proliferation.28,29 For this reason, PTEN is a tumor suppressor and is one of the most commonly mutated protein in human sporadic tumors.30 Mutations in PTEN may also lead to Cowden disease, Lhermitte-Duclos disease, and Bannayan-Zonana syndrome.31
PTEN has four domains: an N-terminal PIP2-binding module, a phosphatase domain (PD), a C2 domain, and a C-terminal tail (Figure 1A). The N-terminal binding module specifically binds PIP2 molecules, ensuring selective binding of PTEN to the inner leaflet of the plasma membrane. The PD contains the PI(3,4,5)P3 catalytic binding site, including the P-loop consisting of the signature HCXXGXXR motif, common with many protein tyrosine phosphatases (PTPs).32 The proposed binding site for PI(3,4,5)P3 corresponds to the site of a bound tartrate molecule in the crystal structure and is supported by mutational data.27,33,34 The positively charged face of the C2 domain of PTEN is believed to bind to anionic lipids, e.g., phosphatidylserine and PIPs, in the membrane. PTEN lacks the aspartate residues associated with Ca2+-dependent binding.19 The C-terminal tail of PTEN is not needed for phosphatase activity but has been shown to have regulatory features.29,35−38
Figure 1.
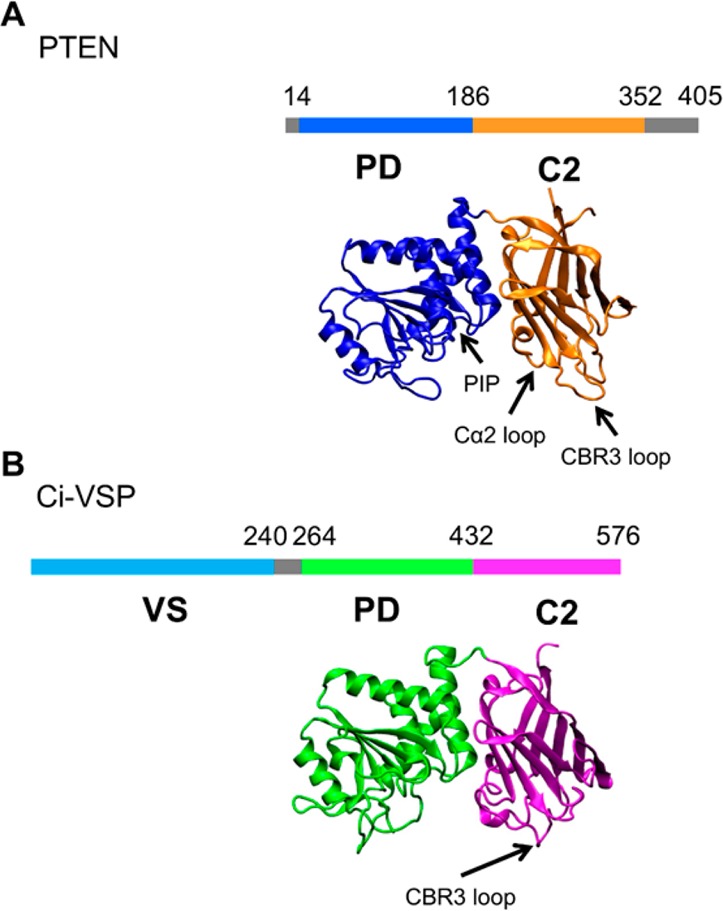
Domain organization of PTEN (A) and Ci-VSP (B) showing the location of the phosphatase domain (PD) and the C2 domains, and of the voltage sensor (VS) domain in Ci-VSP. The crystal structures of the two proteins used in the simulations are also shown: Protein Data Bank (PDB) entry 1D5R for PTEN and PDB entry 3V0H for Ci-VSP.
There are more than 2000 mutations in PTEN associated with cancer and other diseases (http://www.sanger.ac.uk/perl/genetics/CGP/cosmic). These mutations appear in all four domains of the protein. For example, the R130G mutation (identified in endometrial, ovarian, lung, and central nervous system cancer cells) is in the PD, while the R335L mutation (associated with Cowden syndrome) is located in the C2 domain. Mutations can affect PTEN by reducing or abolishing phosphatase activity or by preventing binding to the bilayer.39 The productive orientation of PTEN on the membrane has been suggested to involve association of the C2 domain loops with the lipids.9,19,40 Other proteins (e.g., SUMO1) may form complexes with PTEN and regulate its association with the membrane.41,42
The C. intestinalis voltage sensitive phosphatase (Ci-VSP) catalyzes the dephosphorylation of PI(3,4,5)P3 and PI(4,5)P2 at the 5′-phosphate,43,44 and PI(3,4)P2 at the 3′-phosphate.45 Ci-VSP is expressed in Ciona sperm tail membranes46 and has been suggested to function in digestive system cells and blood cells.47 Mammalian orthologs of Ci-VSP (namely, TPTE,48 TPIP,49 and PTEN250) are expressed in the testis.51 The phosphatase activity of Ci-VSP is regulated by the plasma membrane potential. The membrane potential is sensed by the voltage-sensing (VS) domain, a four-α-helix transmembrane domain at the N-terminus of the protein (Figure 1B). The VS domain is homologous to the VS domain of potassium-selective voltage-gated channels52 and is joined to the cytosolic section by a linker domain. The cytosolic catalytic region is comprised of a PD and a C2 domain and is similar in structure to PTEN itself, with a 44% identical sequence and a high degree of conservation of catalytically significant residues53 (Figure 1B).
It has been suggested that membrane depolarization activates Ci-VSP by moving the PD and C2 domain toward the membrane into the optimal orientation for PIP dephosphorylation.52 However, more recently, it has also been proposed that conformational changes in a “gating loop” (residues 398–413) result in voltage-triggered activity due to the movement of E411, which competes with the substrate for the active site.54 Indeed, a crystal structure of Ci-VSP with E411 in a hydrophobic cavity and not in the active site [Protein Data Bank (PDB) entry 3V0H] contains a bound IP3 molecule at a site on Ci-VSP equivalent to that for binding of tartrate on PTEN.54
The C2 domain of Ci-VSP is similar in structure to that of PTEN and thus is thought to contribute to membrane binding in a Ca2+-independent manner. However, despite similar C2 domains, there are notable differences between the PTEN CBR3 loop and the analogous Ci-VSP loop (P519–P526). While the PTEN loop contains four positively charged residues and points toward the membrane,19 the Ci-VSP CBR3 loop contains only one positive arginine residue and points toward the PD, forming hydrogen bonds with a loop in the PD.55 This indicates a possible regulatory role for the C2 domain. The Ci-VSP PD also shares similarities with the PTEN PD: both contain a P-loop. Ci-VSP, however, has a tyrosine residue (Y522) that fills a shallow pocket present in the PTEN active site and a glycine at residue 365, rather than the equivalent alanine in PTEN.
Molecular dynamics simulations may be used to probe the interactions of both integral and peripheral membrane proteins with lipid bilayers.56 Such simulations have yielded initial models of the likely mode of interaction of PTEN with anionic lipid bilayers that are in good agreement with experimental biophysical data.9,38,40 Furthermore, the resultant models of PTEN–model bilayer interactions are in good agreement with, e.g., recent mutational studies of PTEN that used exogenous expression in Dictyostelium to probe membrane association and PTEN function in living cells.57 There have also been simulations of a homology model of the complete Ci-VSP (including the PTEN domain) bound to the surface of a PIP-containing lipid bilayer. These have focused in particular on the role of the linker region between the voltage sensor and PTEN domains.58 However, a number of aspects of the detailed molecular interactions of PTEN and of the PTEN domain of Ci-VSP with PIP3 molecules in lipid bilayers remain unclear. In this work, we use multiscale molecular dynamics simulations59 to improve our understanding of the similarities and differences in the mechanism(s) of recognition of the PIP-containing lipid bilayer by PTEN and by the PTEN domain from Ci-VSP.
Methods
Structures Used in the Simulations
The PTEN crystal structure (PDB entry 1D5R, resolution of 2.10 Å) consisted of the PD and the C2 domain,19 as did the Ci-VSP crystal structure (PDB entry 3V0H, resolution of 1.85 Å)54 (see Figure 1). The Ci-VSP structure was crystallized with inositol 1,4,5-triphosphate (IP3) bound.
Lipid Bilayers
All lipid bilayers were generated by self-assembly coarse-grained MD simulations. In these simulations, POPC lipids (without any protein) were randomly placed in a simulation box and solvated with coarse-grained water molecules and ions to neutralize the system. Subsequently, a production simulation was performed for 100 ns. After the first ∼15 ns of the simulation, the bilayer was formed with an equal distribution of lipids in the two leaflets. The bilayers for the pten_away, vsp_away, and pten_bound simulations contained 466, 476, and 680 lipids, respectively (see Tables 1 and 2). Prior to the simulations described in this study, using a locally written code the POPC lipids were randomly replaced with POPS or PIP lipids in both leaflets to reach the final concentration of lipids required for each system (see Tables 1 and 2 for the final lipid concentration). In the pten_bound simulations, PIP lipids were placed in the desired position, as shown in Figure 3A, using the GROMACS genbox command.
Table 1. Summary of Simulations with the PTEN.
| simulation | bilayer composition | no. of PIP3s | duration |
|---|---|---|---|
| coarse-grained | |||
| pten_away-1 | POPC/POPS (80:20) | one in each leaflet | 6 × 3 μs |
| pten_away-2 | POPC/POPS (80:20) | two in each leaflet | 6 × 4 μs |
| pten_away-3 | POPC/POPS (80:20) | four in each leaflet | 6 × 3 μs |
| pten_bound-1 | POPC/POPS (80:20) | one in the PTEN-bound leaflet | 6 × 1 μs |
| pten_bound-2 | POPC/POPS (80:20) | four in the PTEN-bound leaflet | 6 × 1.5 μs |
| pten_bound-3 | POPC/POPS (80:20) | one in the PTEN-bound leafleta | 6 × 1 μs |
| atomistic | |||
| pten_AT-1 | POPC/POPS (80:20) | one in the PTEN-bound leaflet | 3 × 50 ns |
| pten_AT-2 | POPC/POPS (80:20) | four in the PTEN-bound leaflet | 3 × 50 ns |
In these simulations, an in silico mutant PTEN (K164E, K269E, K327E, and K330E) was employed.
Table 2. Summary of Simulations with the Ci-VSP.
| simulation | bilayer composition | duration |
|---|---|---|
| coarse-grained | ||
| vsp_away-1 | POPC/POPS (80:20) | 6 × 1 μs |
| vsp_away-2 | POPC/POPS (60:40) | 6 × 1 μs |
| vsp_away-3 | POPC/POPS/PIP2 (75:20:5) | 6 × 1 μs |
| vsp_away-4 | POPC/POPS/PIP3 (75:20:5) | 6 × 1 μs |
| atomistic | ||
| vsp_AT-1 | POPC/POPS/PIP2 (75:20:5) | 3 × 50 ns |
| vsp_AT-2 | POPC/POPS/PIP3 (75:20:5) | 2 × 50 ns |
Figure 3.
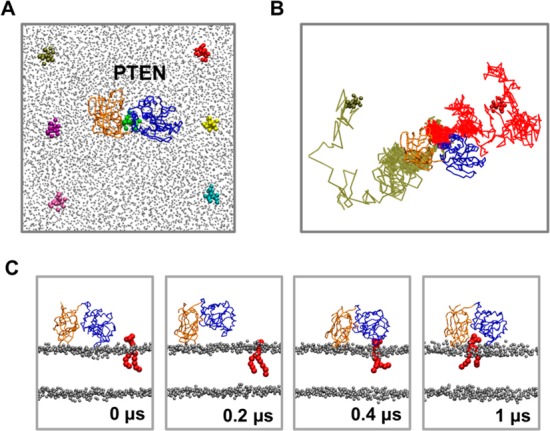
(A) Seven positions in which the PIP3 molecules were placed in the pten_bound simulations (see Table 1). PTEN was placed in the center of the bilayer. POPC and POPS headgroups are shown as gray spheres. (B) Routes from two of the simulations (in one of which the PIP3 lipids reached the PTEN catalytic side via the C2 domain and one via the PD) are colored red and brown. (C) Snapshots from one of the simulations (red in panel B) are shown at 0, 0.2, 0.4, and 1 μs. Note that the systems shown in the figure were fully solvated with CG waters (omitted for the sake of clarity).
Coarse-Grained Molecular Dynamics Simulations (CG-MD)
Coarse-grained molecular dynamics (CG-MD) simulations were performed using GROMACS version 4.5.460 with the Martini 2.1 force field.61,62 In these coarse-grained models, groups of approximately four atoms are represented by a single particle. An elastic network model (ENM)63 was applied to the backbone particles of the protein. Within the network, a harmonic potential was applied to all pairs of backbone particles falling within a cutoff distance of 7 Å to model the secondary and tertiary structure of the protein. All CG-MD simulations were equilibrated for 5 ns and then run using a time step of 20 fs. During the equilibration stage, the protein backbone particles were restrained (force constant of 10 kJ mol–1 Å–2). A Berendsen thermostat64 and a Berendsen barostat were used for temperature and pressure. The LINCS algorithm was used to constrain bond lengths.65 Note that the protein and the bilayer were fully solvated in all simulations using the standard (nonpolarizable) Martini CG water particles.61 For the simulations with the PTEN and the Ci-VSP displaced from the bilayer, the dimensions of the simulation box were 12.5 nm × 12.5 nm × 28 nm, containing ∼30000 CG waters. In the simulations with the PTEN bound to one leaflet of the bilayer (Figure 3), the dimensions of the simulation box were 14.5 nm × 14.5 nm × 13 nm, containing ∼14500 CG waters.
Atomistic Molecular Dynamics (AT-MD) Simulations
AT-MD simulations were conducted as described in Tables 1 and 2. The final snapshots of selected CG-MD simulations were converted to AT representations using a CG-to-AT fragment-based protocol.67 For the AT-MD simulations, the PME (particle mesh Ewald) method68 was used to model long-range electrostatics. A V-rescale thermostat was used for temperature coupling (temperature of 323 K), and the Parrinello–Rahman barostat69 was used for semiisotropic pressure coupling (pressure of 1 bar). The LINCS algorithm was used to constrain bond lengths.65 Prior to the production simulations, the system was equilibrated for 2.5 ns. During the equilibration stage, the protein backbone atoms were restrained (force constant of 10 kJ mol–1 Å–2). Note that no restraints, other than the LINCS algorithm that constrains bond lengths, were imposed during the production AT-MD simulations. Analysis was conducted using GROMACS, PyMol (http://www.pymol.org), VMD,70 and locally written codes. For these simulations, the SPC water model71 was used. Note that in all systems the protein–lipid complex was fully solvated. For all AT-MD simulations (containing either the PTEN or the Ci-VSP), a 15 nm × 15 nm × 14 nm simulation box was used, including ∼63000 water molecules.
Results
Association of PTEN with PIP-Containing Bilayers
Examination of the electrostatic surface of PTEN (Figure S1 of the Supporting Information) reveals an extended positively charged surface formed by the catalytic site of the PD and the Cα2 and CBR3 loops of the C2 domain (Figure S1A of the Supporting Information). As shown by recent heterologous expression studies, mutations removing the positive charges of the CBR3 loop lead to a loss of membrane localization of PTEN in living cells.57 This positively charged surface was also implicated in membrane binding in previous biophysical, modeling, and simulation studies.9,38,40,57 However, a dynamic model of the mechanism of PTEN–membrane association has not been developed.
To study the association of PTEN with PIP3-containing bilayers, a series of three simulations [pten_away-1–3 (see Table 1)] was performed, in which the PTEN molecule was placed ∼12 nm (i.e., 120 Å) from the center of mass of a lipid bilayer containing increasing numbers of PIP3 molecules. In these simulations, PTEN initially diffused in the aqueous environment before interacting with the bilayer (Figure 2A). Overall, increasing the concentration of PIP3 lipids in the bilayer enhanced the formation of the PTEN–bilayer complex. In particular, when only one PIP3 lipid was added to each bilayer leaflet (pten_away-1), just two of six repeat simulations resulted in binding of PTEN to the bilayer (Figure S3A of the Supporting Information), and of these two simulations that yielded a PTEN–lipid complex, only one demonstrated movement of PTEN into a productive orientation (Figure S3D of the Supporting Information) (a productive PTEN–bilayer complex is considered to be one that agrees with the previous experimental and computational data9,38,40).
Figure 2.
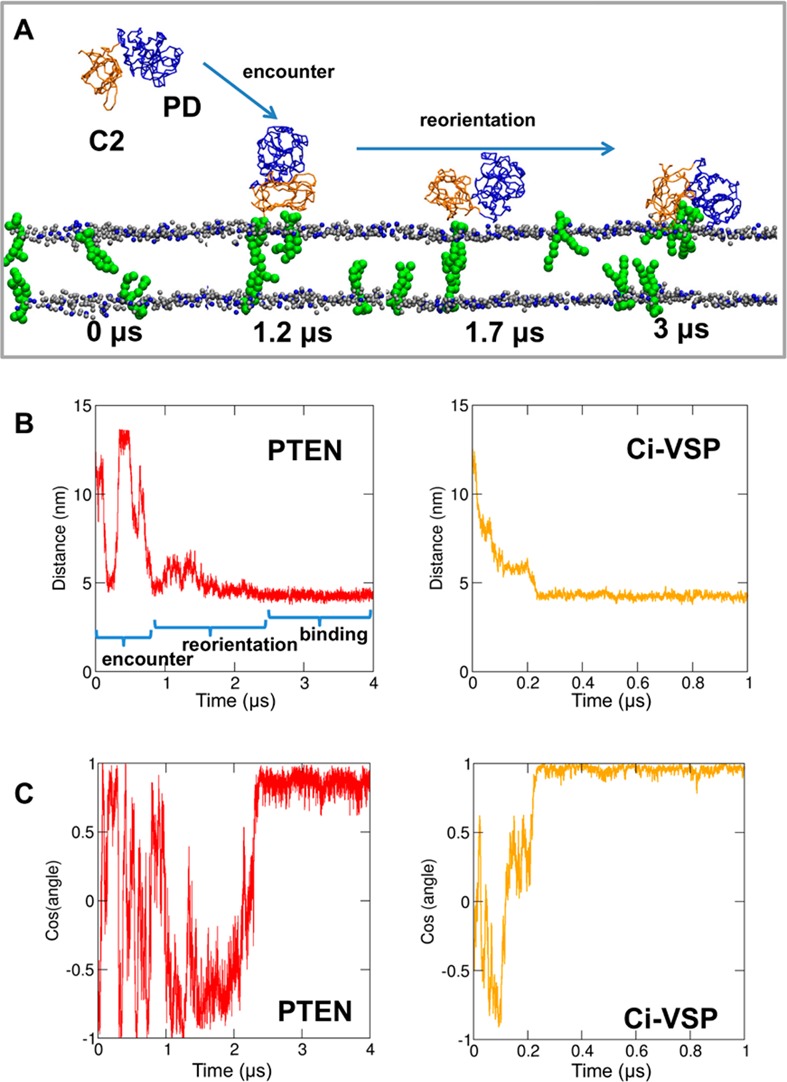
(A) Snapshots of a selected simulation (pten_away-2 in Table 1) with the PTEN molecule displaced from a bilayer that contained two PIP3 molecules (green) in each leaflet. Simulation snapshots are shown at 0, 1.2, 1.7, and 3 μs. The blue arrows indicate the process of initial encounter of the protein and bilayer followed by reorientation of the protein at the bilayer surface. All the systems shown in this figure were fully solvated with CG water particles. However, for the sake of clarity, these are not shown. (B and C) Progress of selected CG-MD simulations of PTEN and Ci-VSP with PIP3-containing bilayers [simulations pten_away-2 and vsp_away-4 (Tables 1 and 2)]. Panel B shows the distance between the center of mass of the protein and the center of mass of the bilayer as a function of time. Panel C shows the cosine of the angle between the protein plane (as defined by the protein’s principal z axis) and the bilayer plane. This angle is equal to 0° (and hence the cosine is equal to 1) if the protein is in the “correct” binding orientation (see the text for more information).
In the systems where the number of PIP3 lipids in each leaflet was increased to two (pten_away-2) and four (pten_away-3), a productive PTEN–bilayer complex was reached in four of six simulations in each case (Figure S3 of the Supporting Information). Note that in all cases PTEN did not initially bind to the bilayer in the productive orientation but the initial encounter was followed by rotational reorientation (see Figure 2) to form more specific interactions via the residues implicated by mutational and computational studies.9,38,40 The resultant productive orientation was retained for the remainder of the simulation. To further determine the stability of the final orientation, all six repeat simulations of the pten_away-2 system were extended from 3 to 4 μs, resulting in PTEN adopting the productive orientation in five of six simulations. Analysis of the radial distribution functions of lipids (Figure S4 of the Supporting Information) indicated preferential interactions of PTEN with PIP3 and, to a lesser extent, POPS.
When similar simulations were run with the PTEN displaced from a POPC/POPS/PIP2 (75:20:5) bilayer, the same mechanism for the formation of the PTEN–bilayer complex was observed. In particular, PTEN associates with the bilayer in a nonproductive orientation, but the initial encounter was followed by rotational reorientation to move PTEN in a productive orientation. Productive association of PTEN with the bilayer resulted in clustering of PIP2 lipids around the protein in the adjacent bilayer leaflet. See Figure S9 and the text of the Supporting Information for more details.
Interactions of a PTEN–Bilayer Complex with PIP3 Molecules
Following definition of the association mechanism to form a “productive” PTEN–bilayer complex, we investigated how PIP3 might interact with a preformed PTEN–bilayer complex. To this end, simulations [pten-bound-1–3 (Table 1)] were initiated with PTEN bound to a POPC/POPS bilayer in a productive orientation (as derived from the simulations described above), but with PIP3 molecules placed in the bilayer away (∼60 Å) from the bound PTEN molecule (Figure 3A). In the first case, a single PIP3 lipid was placed in seven different positions in the bilayer. Four of six simulations in which the PIP3 was displaced from PTEN revealed PIP3 moving into a cleft between the PD and the C2 domain (Figure 3 and Figure S5 of the Supporting Information). The cleft was comprised mainly of PTEN residues K128 (from the active site P-loop), K164, V166, and T167 (from the T1 loop of the PD), and K327 and K330 from the C2 domain Cα2 loop. Interestingly, immediately after the initiation of the simulations, the PD partly dissociated from the bilayer and rebound to the bilayer only after the formation of the PTEN–PIP3 complex (Figure 3C).
Two different routes of the PIP3 to the PD–C2 cleft were observed in the four simulations, two via an initial interaction with the PD (Figure 3B) and two in which the initial interaction occurs via the C2 domain (Figure 3B). Both routes lead to PIP3 binding to the PD–C2 cleft described above and were exploited an equal number of times. Detailed analysis of the routes, however, shows that on its way to the cleft, the PIP3 did not interact with exactly the same residues on the PD or on the C2 domain. Simulations of in silico mutations that reversed the charge of the positive residues in the cleft and surrounding regions (i.e., K164E, K269E, K327E, and K330E; system pten_bound-3 in Table 1) exhibited no binding of PIP3 to the cleft site (see the Supporting Information).
Similar CG-MD simulations were then run with four PIP3 molecules in the PTEN-bound leaflet (pten_bound-2). This resulted in four of six simulations with a PIP3 bound in the aforementioned cleft, three of which went via the C2 domain route and one via the PD route. The interactions with the cleft were again predominantly those with residues K330 and K164. When no PIPs interacted with the cleft or the PD, PTEN again showed transient interaction of the PD with the bilayer. Once interacting with PTEN, the PIP3 molecules tended to remain bound to the same region of the protein for extended periods of time.
To determine whether PTEN can differentiate between PIP2 and PIP3 molecules, similar simulations (compared to those described above) were run with a bilayer containing both four PIP2 molecules and four PIP3 molecules. Two of six simulations showed binding of PIP3 and three of six binding of PIP2 (the remaining simulation showed no binding) to the cleft. Therefore, it seems that both PIP types can bind to the PTEN cleft.
Atomistic Simulations
Atomistic systems were derived from the CG systems described above to study in more detail the interactions of PIP3 with PTEN. AT-MD simulations were set up using the final snapshot from one of the pten_bound-1 simulations and one of the pten_bound-2 simulations. In both simulations, the PIP3 lipid had bound in the aforementioned cleft. Three repeats (with different initial velocities) for each system were run for 50 ns (Table 1). In both systems, PIP3 moved up partially out of the bilayer to interact with PTEN (Figure 4A,B) and then remained there for the remainder of the simulation. The contacts made between PTEN and PIP3 formed in the corresponding CG-MD simulations were maintained during the (short) atomistic simulations (Figure S5 of the Supporting Information). In particular, PIP3 can be seen to form interactions with the P loop (residues 124–129) and T1 loop (residues 164–171) of the PD active site, although the PIP3 headgroup is displaced slightly from the active site formed by the H123CXXGXXR130 motif of the P loop (Figure 5A). Thus, the three major areas of frequent contact between PTEN and PIP3 were residues K164 and T167 of the T1 loop and residue K330 of the C2 domain. PTEN continued to interact with the bilayer in the productive orientation described for the CG-MD simulations with the C2 domain loops penetrating into the bilayer (Figure 6A). Comparison of the PTEN structure from the atomistic simulations with the crystal structure suggests a degree of conformational change corresponding to a rotation of ∼10° of the PD relative to the C2 domain (Figure 5B), although more extended simulations may be needed to capture the full extent of this transition.
Figure 4.
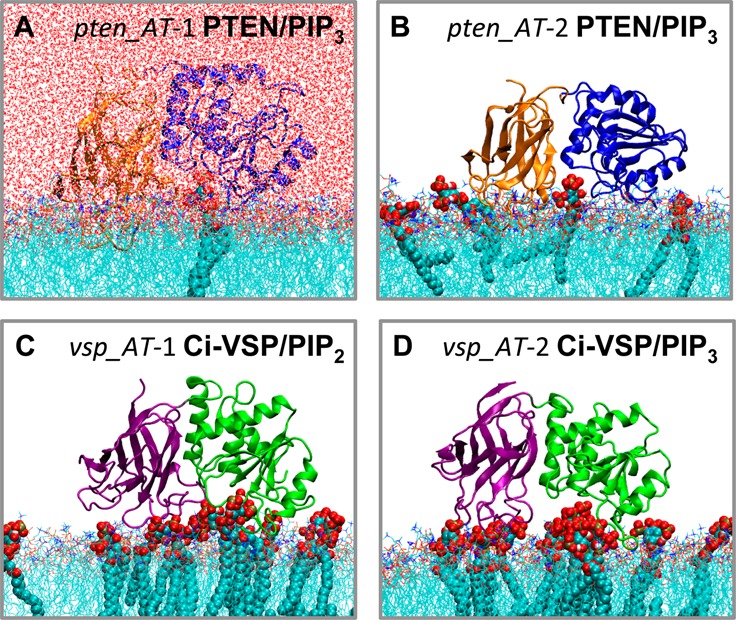
Snapshots of the lipid-bound PTEN and Ci-VSP domains, from the end of all-atom simulations in the presence of PIP2 or PIP3 lipids. (A and B) Snapshots from simulations pten_AT-1 and pten_AT-2, respectively. The PTEN C2 domain is colored orange and the PD blue. (C and D) Snapshots from simulations vsp_AT-1 and vsp_AT-2, respectively. The Ci-VSP C2 domain is colored green and the PD purple. PIP lipids are shown in VDW format. Note that the water molecules included in the simulation box are shown only for the pten_AT-1 simulation system. For the other systems, the waters were included in the simulation (see Methods), but for the sake of clarity, they are not shown in this figure.
Figure 5.
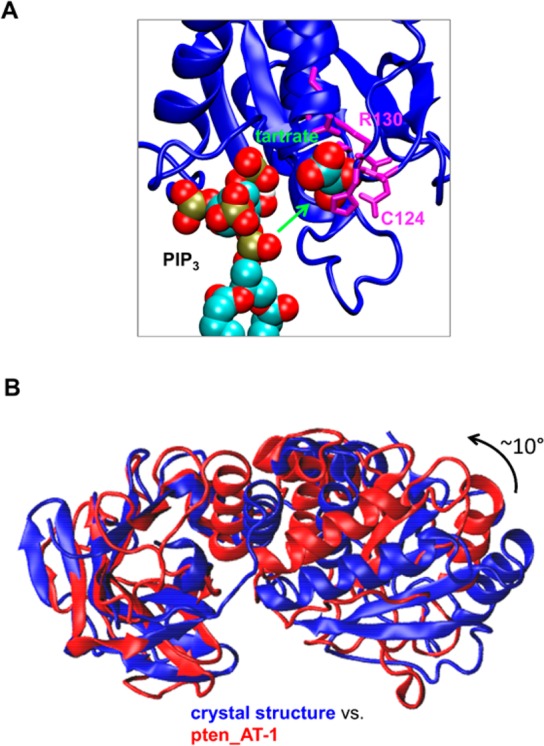
(A) Comparison of the AT-MD simulation (pten_AT-1) and crystal structure in terms of the location of PIP3 and tartrate relative to the PTEN active site defined by catalytic residues C124 and R130. (B) Comparison of the PTEN structure of the AT-MD simulation in panel A (red; pten_AT-1) and the crystal structure (blue) reveals a rotation of ∼10° of the PD relative to the C2 domain (the two structures are superimposed via their C2 domains).
Figure 6.
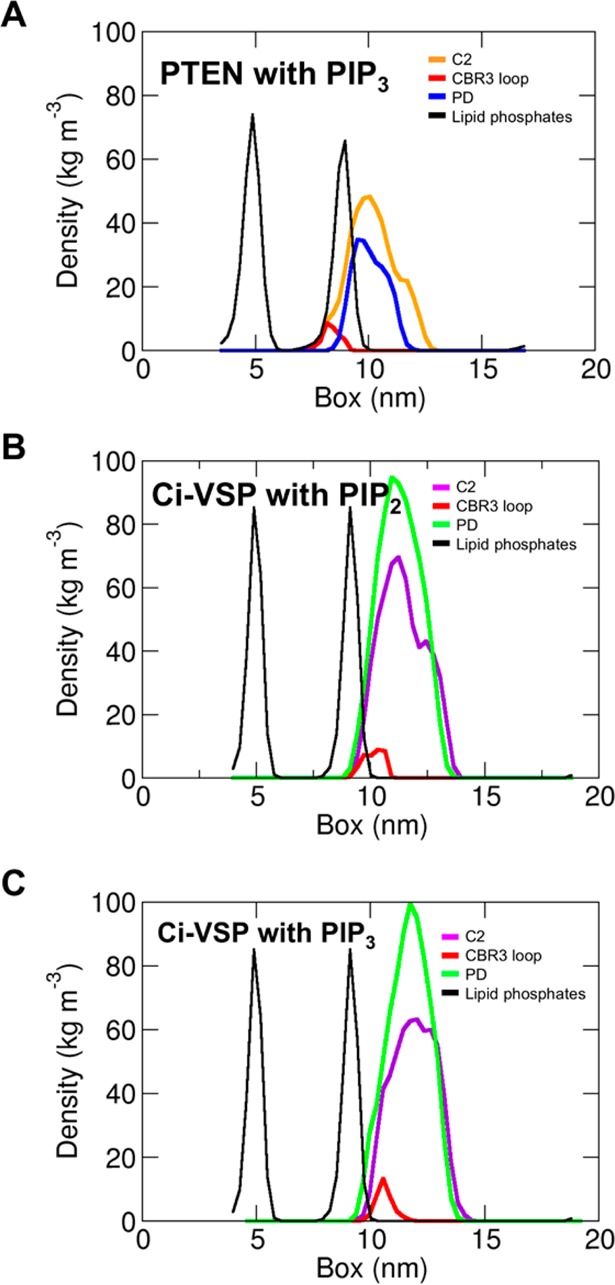
Density profiles along the membrane normal for the C2 domain (orange for PTEN to purple for Ci-VSP), the PD (blue for PTEN to green for Ci-VSP), and the C2 CBR3 loop region (red; residues 261–266 for PTEN to residues 516–524 for Ci-VSP) relative to the positions of the lipid (POPC and POPS) phosphate groups (black), from the (A) pten_AT-1, (B) vsp_AT-1, and (C) vsp_AT-2 simulations.
Interaction of Ci-VSP with Membranes
Ci-VSP is a rather specialized example of a PTEN-like enzyme. Unlike PTEN, Ci-VSP is permanently anchored to the bilayer via an N-terminal voltage-sensor (VS) domain, more generally seen in voltage-gated potassium, sodium, and related ion channels. The PTEN domain from Ci-VSP has an electrostatic surface somewhat different from that of PTEN (Figure S1 of the Supporting Information), with a molecular dipole; its positive pole points directly to the presumed location of the membrane surface (Figure S2 of the Supporting Information). Furthermore, despite the overall conservation of fold, the lipid-binding loops of the C2 domain differ between the two structures. Therefore, we performed a series of simulations of Ci-VSP PTEN (Table 2) comparable to those described above for PTEN itself.
In the absence of PIP molecules, the Ci-VSP PTEN domain did not exhibit any interactions with the bilayer (Figure S6 of the Supporting Information). The presence of PIP2 or PIP3 resulted in interactions with the bilayer such that the Ci-VSP domain adopted an orientation comparable to the productive mode of PTEN discussed above (Figure S6 of the Supporting Information). Interestingly, in the simulations that resulted in a Ci-VSP–bilayer complex, the final orientation of Ci-VSP relative to the bilayer was similar to that of PTEN (Figure 2). However, the Ci-VSP PD made strong interactions with the bilayer unlike the PTEN PD. This is consistent with the difference in the electrostatic potential surface between the two proteins. Calculation of the orientation of Ci-VSP relative to the bilayer (Figure 2B,C and Figure S6 of the Supporting Information) revealed binding without subsequent reorientation of the domain relative to the bilayer surface. Of course, in vivo Ci-VSP PTEN is more likely to be bound to the membrane as it is tethered by the nearby N-terminal VS domain. Overall, these simulations provide support for a model in which PTEN of Ci-VSP interacts all of the time with the bilayer and is voltage-activated via a conformational change rather than a voltage-driven on and off movement of the PTEN domain to and from the membrane.54
Atomistic simulations were performed, using the end points from the vsp_away-3 and vsp_away-4 simulations as starting configurations (Figure 4C,D). Calculation of the contacts between Ci-VSP and the lipids demonstrated that the three regions of Ci-VSP PTEN forming the principal contacts were the same for both PIP2 and PIP3 (Figure S7 of the Supporting Information). These regions included contributions from both the PD and the C2 domain. The most conserved site of protein–PIP interaction between the PIP2 and PIP3 simulations was between residues 362 and 369 (particularly K364 and G365), i.e., corresponding to the H362SxxGxxR369 catalytic loop. As for PTEN (see above) in the atomistic simulations, the PIP2 or PIP3 molecule is close to the active site but is not found directly between the two catalytic residues (S363 and A369), unlike IP3 in the X-ray structure. It is interesting to note that unlike PTEN, the corresponding domain from Ci-VSP does not penetrate beyond the surface of the bilayer. In particular, whereas for PTEN the CBR3 loop penetrates deep into the headgroup phosphates, for Ci-VSP the CBR3 loop sits closer to the phosphate–aqueous interface (Figure 6). This is suggestive of the possibility of a further conformational change linked to tighter binding of the PIP3 substrate, although more extended simulations might be needed to test this.
Discussion
Our simulations have allowed us to model the nature of the PTEN–membrane association process. The predicted final orientation of PTEN relative to the membrane is in good agreement with previous experimental data and computational studies.9,19,38,40,72,73 An increased concentration of PIP3 resulted in more frequent association of PTEN with the model bilayer, and PTEN could achieve its optimal orientation without SUMOylation.41 To reach the preferred orientation, PTEN was observed to be able to reorient at the membrane surface following an initial encounter with the lipid bilayer. It should be noted that in our simulations only a low concentration of PIP3 lipids in the bilayer was required for the formation of a productive PTEN–membrane complex. Recent lipidomics data suggest that the global PIP3 concentration is expected to be lower than, for example, the PIP2 concentration in the plasma membrane, although it should be noted that it is difficult to be certain of the exact concentration of the PIP3 and PIP2 molecules because of the reversible turnover of PIP2 to PIP3 and other PIP species. However, the local concentration of PIP3 is not known, and it remains possible that a high local concentration of PIP3 may drive the formation of the PTEN–membrane complex. Simulation with bilayers containing the more abundant PIP2 at physiological concentrations also resulted in the same mechanism for the formation of the PTEN–membrane complex. A similar encounter-plus-reorientation mechanism has been observed for, for example, the auxilin PTEN-like domain.21 Interestingly, the interaction of the PTEN domain from Ci-VSP did not generally require a reorientation step. In simulations starting with prebound PTEN at the bilayer surface, PIP3 molecules were able to enter the PD–C2 cleft via lateral diffusion along with some degree of “loosening” followed by “tightening” of the PTEN–bilayer interaction.
In CG simulations linked to subsequent atomistic simulations, the PIP3 molecule interacted with residues close the PTEN active site [identified via a tartrate bound in the crystal structure (Figure 5A and Figure S5 of the Supporting Information)]. However, the bound PIP3 in the simulations was somewhat displaced from the optimal binding mode at the active site suggested by the crystal structure (Figure S8 of the Supporting Information). Furthermore, molecular dynamics simulations with a PIP3 molecule initially docked using the tartrate coordinates as a guide did not result in the PIP3 molecule remaining bound to this site.9 Interestingly, docking of PTEN inhibitors to the tartrate binding site has been demonstrated after the removal of the C2 domain.74 Because the C2 domain is more cationic than the PD, it is perhaps not surprising that the negatively charged PIP3 molecule binds initially to a site formed between the PD and the C2 domain and contains multiple lysine residues. It is also possible that a degree of conformational change is needed, and indeed, some limited change in the relative orientation of the PD and the C2 domain is seen during our (short) atomistic simulations. However, it is likely that more extended atomistic simulation studies, using, for example, metadynamics75 for better sampling, will be required to explore this aspect in more detail.
In the case of Ci-VSP, the structural similarity to PTEN76 would suggest a similar orientation of binding to the membrane. The simulations described here are in agreement with this (Figure 4), indicating that the Ci-VSP C2 domain binds to the membrane via a Ca2+-independent mechanism as is the case for PTEN19 and auxilin.20 Ci-VSP PTEN adopted a preferred orientation in simulations directly after initial membrane binding, without a need for subsequent reorientation, reflecting a difference in the molecular dipole orientation between PTEN and Ci-VSP (Figure S2 of the Supporting Information). In simulations with PIPs present, the residues that formed a substantive number of contacts with PIP2 or PIP3 molecules are in the proximity of the IP3 substrate in the crystal structure (see Figure S8 of the Supporting Information). However, as for PTEN, PIP3 did not interact directly with the active site, again suggesting the need for some degree of conformational change and/or more exhaustive sampling of interactions in simulations. In this context, it is of interest that crystal structures of Ci-VSP PTEN have indicated conformational changes may be required for binding of the substrate to the active site of the enzyme.54
It is important to consider possible limitations of the current simulation studies. In particular, one must consider the approximations implicit in the use of a CG-MD-based approach. This has been shown to correctly predict interactions of PIPs with integral membrane proteins77,78 and also the interactions of peripheral proteins with anionic lipid bilayers.79 However, the use of an elastic network model within the coarse-grained simulations means that possible conformational changes of the protein are not accurately represented. The use of a multiscale approach, with subsequent atomistic simulations, in part addresses this limitation. However, as noted above, the AT-MD simulations are relatively short and more extended simulations and/or sampling is likely to be needed to more fully address possible conformational changes linked to, for example, binding of PIP3 to the PTEN active site.
A more “biological” limitation is the focus on the core PTEN domains (i.e., PD and C2 domain), thus excluding the N- and C-terminal tails of PTEN, and the VS and linker region of Ci-VSP. This is necessitated by limited structural data, although there have been simulations of a homology model of the complete Ci-VSP protein.58 There are not any structural data for the PTEN N-terminal domain or for the PTEN C-terminal tail. In particular, the C-terminal tail is thought to be a long, unstructured, and flexible region, which makes modeling or simulation rather difficult. It seems that the C-terminal tail may adopt different dynamic conformations in the membrane-bound and membrane-unbound states of PTEN.38,57 In particular, recent data suggest that the C-terminal tail may interact with those regions of PTEN that otherwise can interact with lipids in the plasma membrane, thus providing an autoinhibitory mechanism.57 Consequently, in future studies, it will require rather detailed and careful simulations, possibly via an enhanced sampling approach, to fully capture the conformational energy landscape of the C-terminal tail. This will be a prerequisite for creating a predictive dynamic model of full length PTEN and of its interaction with the membrane. It will also be important to treat more fully some of the lipid compositional complexities of mammalian cell membranes.81
To summarize, our multiscale simulation approach offers novel insights into the molecular mechanism of association of PTEN proteins with model membranes, providing details of the interactions of these proteins with PIP3. As discussed by, for example, Nguyen et al.,57 the majority of PTEN in a cell is not bound to the membrane, so an improved understanding of the mechanism of PTEN–bilayer interaction offers a longer term prospect of developing activators of PTEN function for therapeutic applications.
Supporting Information Available
Details of additional simulations and analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by Wellcome Trust Grant 092970.
Supplementary Material
References
- Golebiewska U.; Nyako M.; Woturski W.; Zaitseva I.; McLaughlin S. (2008) Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 19, 1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kölsch V.; Charest P. G.; Firtel R. A. (2008) The regulation of cell motility and chemotaxis by phospholipid signaling. J. Cell Sci. 121, 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney K. F.; Huang C.-H.; Devreotes P. N. (2010) Eukaryotic Chemotaxis: A Network of Signaling Pathways Controls Motility, Directional Sensing, and Polarity. Annu. Rev. Biophys. 39, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard T. A.; Hurley J. H. (2011) Regulation of protein kinases by lipids. Curr. Opin. Struct. Biol. 21, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi C.; Falasca M. (2012) Phosphoinositides signalling in cancer: Focus on PI3K and PLC. Adv. Biol. Regul. 52, 166–182. [DOI] [PubMed] [Google Scholar]
- Ziemba B. P.; Falke J. J. (2013) Lateral diffusion of peripheral membrane proteins on supported lipid bilayers is controlled by the additive frictional drags of (1) bound lipids and (2) protein domains penetrating into the bilayer hydrocarbon core. Chem. Phys. Lipids 172–173, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. H.; Falke J. J. (2007) Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 16176–16181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb C. N.; He J.; Xue Y.; Stansfeld P. J.; Stahelin R. V.; Kutateladze T. G.; Sansom M. S. P. (2011) Biophysical and computational studies of membrane penetration by the GRP1 pleckstrin homology domain. Structure 19, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb C. N.; Sansom M. S. P. (2013) Defining the membrane-associated state of the PTEN tumour suppressor protein. Biophys. J. 104, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates L. A.; Lumb C. N.; Brahme N. N.; Bird L. E.; De Colibus L.; Owens R. J.; Calderwood D. A.; Sansom M. S. P.; Gilbert R. J. C. (2012) Structural and functional characterisation of the kindlin-1 pleckstrin homology domain. J. Biol. Chem. 287, 43246–43261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipsborn A. v., and Bastmeyer M. (2007) Mechanisms of gradient detection: A comparison of axon pathfinding with eukaryotic cell migration. In International Review of Cytology (Kwang W. J., Ed.) pp 1–62, Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Lai C.-L.; Landgraf K. E.; Voth G. A.; Falke J. J. (2010) Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain: A combined molecular dynamics and experimental study. J. Mol. Biol. 402, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Suzuki H.; Ozawa K.; Deng J.; Lehel C.; Fukamachi H.; Anderson W. B.; Kawakami Y.; Kawakami T. (1997) Interactions between Protein Kinase C and Pleckstrin Homology Domains: Inhibition by Phosphatidylinositol 4,5-Bisphosphate and Phorbol 12-Myristate 13-Acetate. J. Biol. Chem. 272, 13033–13039. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111. [DOI] [PubMed] [Google Scholar]
- Perisic O.; Fong S.; Lynch D. E.; Bycroft M.; Williams R. L. (1998) Crystal structure of a calcium-phospholipid binding domain from cytosolic phospholipase A2. J. Biol. Chem. 273, 1596–1604. [DOI] [PubMed] [Google Scholar]
- Verdaguer N.; Corbalan-Garcia S.; Ochoa W. F.; Fita I.; Gomez-Fernandez J. C. (1999) Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 18, 6329–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. (2001) Membrane Targeting by C1 and C2 Domains. J. Biol. Chem. 276, 32407–32410. [DOI] [PubMed] [Google Scholar]
- Lomasney J. W.; Cheng H.-F.; Roffler S. R.; King K. (1999) Activation of phospholipase C δ1 through C2 domain by a Ca2+-enzyme-phosphatidylserine ternary complex. J. Biol. Chem. 274, 21995–22001. [DOI] [PubMed] [Google Scholar]
- Lee J. O.; Yang H.; Georgescu M. M.; Di Cristofano A.; Maehama T.; Shi Y.; Dixon J. E.; Pandolfi P.; Pavletich N. P. (1999) Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334. [DOI] [PubMed] [Google Scholar]
- Guan R.; Han D.; Harrison S. C.; Kirchhausen T. (2010) Structure of the PTEN-like region of auxilin, a detector of clathrin-coated vesicle budding. Structure 18, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A.; Morgan G.; Sansom M. S. P. (2013) Interactions of the PTEN-like domain of auxilin-1 with lipid bilayers revealed by multiscale MD simulation. Biophys. J. 105, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann M. P.; Schneiter R. (2008) Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 9, 162–176. [DOI] [PubMed] [Google Scholar]
- Di Paolo G.; De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. (2008) Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio J. L.; Gerl M. J.; Klose C.; Ejsing C. S.; Beug H.; Simons K.; Shevchenko A. (2011) Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. U.S.A. 108, 1903–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventis P. A.; Grinstein S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427. [DOI] [PubMed] [Google Scholar]
- Maehama T.; Dixon J. E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378. [DOI] [PubMed] [Google Scholar]
- Sun H.; Lesche R.; Li D.-M.; Liliental J.; Zhang H.; Gao J.; Gavrilova N.; Mueller B.; Liu X.; Wu H. (1999) PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. S.; Salmena L.; Pandolfi P. P. (2012) The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296. [DOI] [PubMed] [Google Scholar]
- Simpson L.; Parsons R. (2001) PTEN: Life as a tumor suppressor. Exp. Cell Res. 264, 29–41. [DOI] [PubMed] [Google Scholar]
- Liaw D.; Marsh D. J.; Li J.; Dahia P. L. M.; Wang S. I.; Zheng Z.; Bose S.; Call K. M.; Tsou H. C.; Peacoke M.; Eng C.; Parsons R. (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 16, 64–67. [DOI] [PubMed] [Google Scholar]
- Tonks N. K. (2006) Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846. [DOI] [PubMed] [Google Scholar]
- Redfern R. E.; Daou M.-C.; Li L.; Munson M.; Gericke A.; Ross A. H. (2010) A mutant form of PTEN linked to autism. Protein Sci. 19, 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.-Y.; Kato H.; Kato S.; Suzuki T.; Shibata H.; Ishii S.; Shiiba K.-i.; Matsuno S.; Kanamaru R.; Ishioka C. (2000) Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 60, 3147–3151. [PubMed] [Google Scholar]
- Gericke A.; Munson M.; Ross A. H. (2006) Regulation of the PTEN phosphatase. Gene 374, 1–9. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Chaffotte A.; Terrien E.; Préhaud C.; Theillet F.-X.; Delepierre M.; Lafon M.; Buc H.; Wolff N. (2012) Ordered phosphorylation events in two independent cascades of the PTEN C-tail revealed by NMR. J. Am. Chem. Soc. 134, 20533–20543. [DOI] [PubMed] [Google Scholar]
- Vazquez F.; Ramaswamy S.; Nakamura N.; Sellers W. R. (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 20, 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy S. S.; Nanda H.; Lösche M. (2012) Membrane association of the PTEN tumor suppressor: Electrostatic interaction with phosphatidylserine-containing bilayers and regulatory role of the C-terminal tail. J. Struct. Biol. 180, 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A.; Nakano T.; Mak T. W.; Sasaki T. (2008) Portrait of PTEN: Messages from mutant mice. Cancer Sci. 99, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy S.; Shekhar P.; Heinrich F.; Daou M.-C.; Gericke A.; Ross A.; Lösche M. (2012) Membrane association of the PTEN tumor suppressor: Molecular details of the protein-membrane complex from SPR binding studies and neutron reflection. PLoS One 7, e32591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Yan J.; Zhang J.; Zhu S.; Wang Y.; Shi T.; Zhu C.; Chen C.; Liu X.; Cheng J.; Mustelin T.; Feng G.-S.; Chen G.; Yu J. (2012) SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 3, 911. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Santamaria J.; Campagna M.; Ortega-Molina A.; Marcos-Villar L.; de la Cruz-Herrera C. F.; Gonzalez D.; Gallego P.; Lopitz-Otsoa F.; Esteban M.; Rodriguez M. S.; Serrano M.; Rivas C. (2012) Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 3, e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H.; Murata Y.; Kim Y.; Hossain M. I.; Worby C. A.; Dixon J. E.; McCormack T.; Sasaki T.; Okamura Y. (2008) A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. U.S.A. 105, 7970–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaszovich C. R.; Schreiber D. N.; Oliver D. (2009) Ci-VSP is a depolarization-activated phosphatidylinositol-4,5-bisphosphate and phosphatidylinositol-3,4,5-trisphosphate 5′-phosphatase. J. Biol. Chem. 284, 2106–2113. [DOI] [PubMed] [Google Scholar]
- Kurokawa T.; Takasuga S.; Sakata S.; Yamaguchi S.; Horie S.; Homma K. J.; Sasaki T.; Okamura Y. (2012) 3′ Phosphatase activity toward phosphatidylinositol 3,4-bisphosphate [PI(3,4)P2] by voltage-sensing phosphatase (VSP). Proc. Natl. Acad. Sci. U.S.A. 109, 10089–10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y.; Iwasaki H.; Sasaki M.; Inaba K.; Okamura Y. (2005) Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243. [DOI] [PubMed] [Google Scholar]
- Ogasawara M.; Sasaki M.; Nakazawa N.; Nishino A.; Okamura Y. (2011) Gene expression profile of Ci-VSP in juveniles and adult blood cells of ascidian. Gene Expression Patterns 11, 233–238. [DOI] [PubMed] [Google Scholar]
- Chen H.; Rossier C.; Morris M. A.; Scott H. S.; Gos A.; Bairoch A.; Antonarakis S. E. (1999) A testis-specific gene, TPTE, encodes a putative transmembrane tyrosine phosphatase and maps to the pericentromeric region of human chromosomes 21 and 13, and to chromosomes 15, 22, and Y. Hum. Genet. 105, 399–409. [DOI] [PubMed] [Google Scholar]
- Walker S. M.; Downes C. P.; Leslie N. R. (2001) TPIP: A novel phosphoinositide 3-phosphatase. Biochem. J. 360, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Dowbenko D.; Pisabarro M. T.; Dillard-Telm L.; Koeppen H.; Lasky L. A. (2001) PTEN 2, a Golgi-associated testis-specific homologue of the PTEN tumor suppressor lipid phosphatase. J. Biol. Chem. 276, 21745–21753. [DOI] [PubMed] [Google Scholar]
- Tapparel C.; Reymond A.; Girardet C.; Guillou L.; Lyle R.; Lamon C.; Hutter P.; Antonarakis S. E. (2003) The TPTE gene family: Cellular expression, subcellular localization and alternative splicing. Gene 323, 189–199. [DOI] [PubMed] [Google Scholar]
- Villalba-Galea C. A.; Miceli F.; Taglialatela M.; Bezanilla F. (2009) Coupling between the voltage-sensing and phosphatase domains of Ci-VSP. J. Gen. Physiol. 134, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout S. C.; Bell S. C.; Liu L.; Xu Q.; Minor D. L.; IsacoffEhud Y. (2010) Electrochemical coupling in the voltage-dependent phosphatase Ci-VSP. Nat. Chem. Biol. 6, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Kohout S. C.; Xu Q.; Müller S.; Kimberlin C. R.; Isacoff E. Y.; Minor D. L. (2012) A glutamate switch controls voltage-sensitive phosphatase function. Nat. Struct. Mol. Biol. 19, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M.; Takeshita K.; Kurokawa T.; Sakata S.; Suzuki M.; Yamashita E.; Okamura Y.; Nakagawa A. (2011) Crystal structure of the cytoplasmic phosphatase and tensin homolog (PTEN)-like region of Ciona intestinalis voltage-sensing phosphatase provides insight into substrate specificity and redox regulation of the phosphoinositide phosphatase activity. J. Biol. Chem. 286, 23368–23377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. (2011) Molecular simulation approaches to membrane proteins. Structure 19, 1562–1572. [DOI] [PubMed] [Google Scholar]
- Nguyen H. N.; Afkari Y.; Senoo H.; Sesaki H.; Devreotes P. N.; Iijima M. (2013) Mechanism of human PTEN localization revealed by heterologous expression in Dictyostelium. Oncogene DOI: 10.1038/onc.2013.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobiger K.; Utesch T.; Mroginski M. A.; Seebohm G.; Friedrich T. (2013) The linker pivot in Ci-VSP: The key to unlock catalysis. PLoS One 8, e70272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Jefferies E. E.; Sansom M. S. P. (2012) Multiscale simulations reveal conserved patterns of lipid interactions with aquaporins. Structure 21, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S. J. (2008) The MARTINI coarse grained force field: Extension to proteins. J. Chem. Theory Comput. 4, 819–834. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada J.; Yefimov S.; Tieleman D. P.; de Vries A. H. (2007) The MARTINI forcefield: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Atilgan A. R.; Durell S. R.; Jernigan R. L.; Demirel M. C.; Keskin O.; Bahar I. (2001) Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. (2011) From coarse-grained to atomistic: A serial multi-scale approach to membrane protein simulations. J. Chem. Theory Comput. 7, 1157–1166. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Parrinello M.; Rahman A. (1981) Polymorphic transitions in single-crystals: A new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual Molecular Dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., Gunsteren W. F., and Hermans J. (1981) Interaction models for water inrelation toprotein hydration. In Intermolecular Forces (Pullman B., Ed.) pp 331–342, Springer, Dordrecht, The Netherlands. [Google Scholar]
- Georgescu M. M.; Kirsch K. H.; Kaloudis P.; Yang H. J.; Pavletich N. P.; Hanafusa H. (2000) Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 60, 7033–7038. [PubMed] [Google Scholar]
- Das S.; Dixon J. E.; Cho W. W. (2003) Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. U.S.A. 100, 7491–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Wei Y.; Mottamal M.; Roberts M. F.; Krilov G. (2010) Understanding the stereospecific interactions of 3-deoxyphosphatidylinositol derivatives with the PTEN phosphatase domain. J. Mol. Graphics Modell. 29, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limongelli V.; Bonomi M.; Parrinello M. (2013) Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. U.S.A. 110, 6358–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura Y.; Dixon J. E. (2011) Voltage-sensing phosphatase: Its molecular relationship with PTEN. Physiology 26, 6–13. [DOI] [PubMed] [Google Scholar]
- Stansfeld P. J.; Hopkinson R. J.; Ashcroft F. M.; Sansom M. S. P. (2009) The PIP2 binding site in Kir channels: Definition by multi-scale biomolecular simulations. Biochemistry 48, 10926–10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. R.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. (2013) Simulation-based prediction of phosphatidylinositol 4,5-bisphosphate binding to an ion channel. Biochemistry 52, 279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Wegener K. L.; Anthis N. J.; Campbell I. D.; Sansom M. S. P. (2010) The structure of the talin/integrin complex at a lipid bilayer: An NMR and MD simulation study. Structure 18, 1280–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A.; Simons K. (2010) Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 11, 593–598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.