

Summary
piRNAs guide an adaptive genome defense system that silences transposons during germline development. The Drosophila HP1 homolog Rhino is required for germline piRNA production. We show that Rhino binds specifically to the heterochromatic clusters that produce piRNA precursors, and that binding directly correlates with piRNA production. Rhino co-localizes to germline nuclear foci with Rai1/DXO related protein Cuff and the DEAD box protein UAP56, which are also required for germline piRNA production. RNA sequencing indicates that most cluster transcripts are not spliced, and that rhino, cuff and uap56 mutations increase expression of spliced cluster transcripts over 100 fold. LacI∷Rhino fusion protein binding suppresses splicing of a reporter transgene, and is sufficient to trigger piRNA production from a trans combination of sense and antisense reporters. We therefore propose that Rhino anchors a nuclear complex that suppresses cluster transcript splicing, and speculate that stalled splicing differentiates piRNA precursors from mRNAs.
INTRODUCTION
Transposons and other repetitive elements are major genome constituents that can mobilize and induce DNA breaks and insertional mutations (McClintock, 1950; Bennetzen, 2000; Beck et al., 2010). The germline transmits the inherited genetic complement, and PIWI-interacting RNAs (piRNAs) have a conserved role in suppressing transposon expression and maintaining genome integrity during germline development (Khurana and Theurkauf, 2010; Siomi et al., 2011; Guzzardo et al., 2013). The 23-30 nt long piRNAs bear a 5′ monophosphate, a 3’ terminal 2′-O-methyl group, and bind to PIWI clade Argonautes (Aubergine, Piwi and Ago-3 in Drosophila) (Grivna et al., 2006; Girard et al., 2006; Aravin et al., 2006; Vagin et al., 2006; Lau et al., 2006; Horwich et al., 2007; Saito et al., 2007). piRNAs bound to PIWI proteins can guide cleavage of complementary targets, which contributes to transposon silencing and generates the precursors of sense strand piRNAs that direct cleavage of antisense precursors and drive a “ping-pong amplification” cycle (Brennecke et al., 2007; Gunawardane et al., 2007).
The primary piRNAs that initiate the ping-pong cycle are derived from clusters of nested transposon fragments, which generally reside in subtelomeric or pericentromeric heterochromatin (Brennecke et al., 2007). In Drosophila, the majority of these heterochromatic domains produce piRNAs from both genomic strands, and piRNAs mapping uniquely to these “dual-strand clusters” are the dominant species in germline cells. A distinct set of clusters produce unique piRNAs from only one genomic strand, and piRNAs from these “uni-strand clusters” dominate in the somatic follicle cells that surround the germline (Brennecke et al., 2007; Malone et al., 2009). It is unclear how the piRNAs precursors produced by dual-strand and uni-strand clusters are distinguished from gene transcripts.
The rapidly evolving Heterochromatin Protein 1 (HP1) homolog Rhino (Rhi) is required for production of primary piRNAs from the germline specific dual-strand clusters (Klattenhoff et al., 2009). Here we show that Rhi binds specifically to dual strand clusters and that binding correlates with germline piRNA production. Significantly, we also present evidence that Rhi functions with the Rai1/DXO related protein Cutoff (Cuff) and the DEAD box protein UAP56 to suppress cluster transcript splicing, and that Rhi binding suppresses splicing of a reporter transgene, and is sufficient to drive piRNA biogenesis from transgenic reporters that express complementary transcripts. Stalled splicing intermediates are precursors for transposon-silencing siRNAs in the pathogenic yeast Cryptococcus (Dumesic et al., 2013). Stalled splicing may therefore have a conserved function in differentiating potentially deleterious RNAs from gene transcripts, and produce the precursors of trans-silencing small RNAs that guide host defense systems.
RESULTS
Rhino marks dual-strand piRNA clusters
The HP1 family protein Rhino (Rhi), also referred to as HP1D, is required for transposon silencing and piRNA production from heterochromatic dual-strand clusters (Klattenhoff et al., 2009). In fission yeast, HP1 appears to bind centromeric transcripts and direct these RNAs to the degradation machinery (Keller et al., 2012). We therefore assayed for cluster transcript binding by Rhi, using co-immunoprecipitation and qPCR conditions that yield significant cluster transcript enrichment by UAP56 (Zhang et al., 2012a). We did not detect significant cluster transcript binding in these studies (F. Zhang and W. Theurkauf, unpublished). By contrast, our earlier Rhi chromatin immunoprecipitation-qPCR (ChIP-qPCR) experiments showed significant enrichment for two regions in the major 42AB piRNA cluster (Klattenhoff et al., 2009). While we cannot rule out RNA binding by Rhi, these observations suggest that this HP1 family protein interacts directly with chromatin. We therefore determined the genome-wide distribution of Rhi by ChIP-Sequencing (ChIP-Seq). These studies indicate that Rhi is enriched at essentially all of the germline-specific dual-strand clusters, but shows only background binding to uni-strand clusters and protein coding genes (Figure 1 and S1A).
Figure 1. Rhi binding correlates with piRNA production.
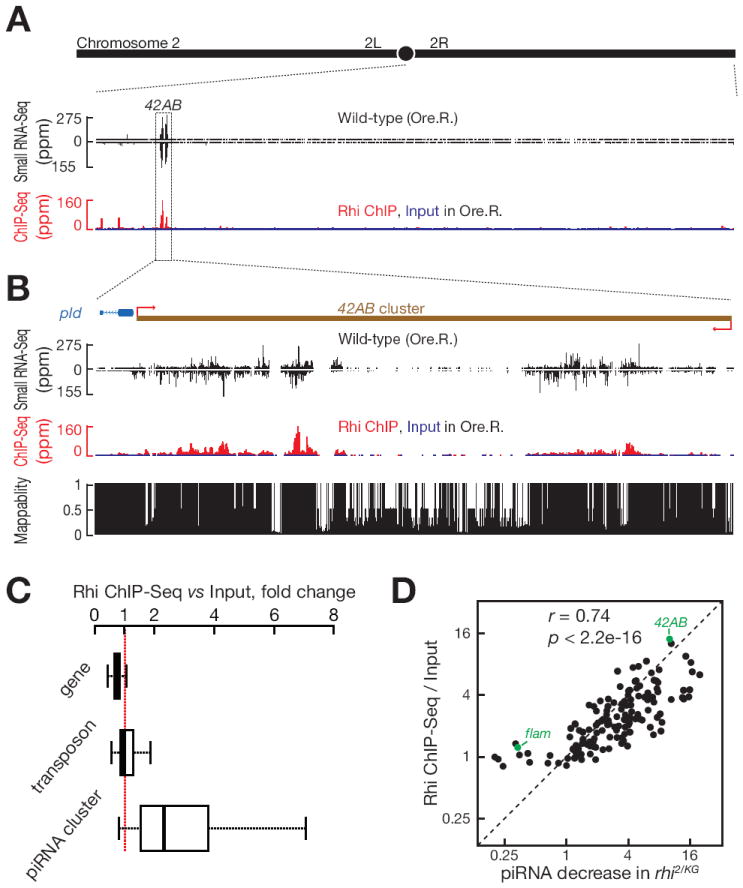
(A) piRNA and Rhi ChIP-Seq signal across the right arm of chromosome 2.
(B) piRNA and Rhi ChIP-Seq signal across the 42AB piRNA cluster, which produces ~ 30% of fly ovary piRNAs.
(C) Boxplots showing Rhi enrichment at protein coding genes, transposons and piRNA clusters. Outliers are not shown.
(D) Rhi binding correlates with Rhi-dependent piRNA production. Scatter plots showing Rhi binding enrichment (y-axis) relative to reduction in piRNA production in rhi2/KG mutants (x-axis). Each point represents a distinct piRNA cluster. Pre-I.S. is Pre-Immune Serum. The major germline piRNA cluster at 42AB and the major somatic cluster (flam) are indicated in green.
See also Figure S1 and S2.
Visual inspection of genome browser profiles revealed a striking correlation between unique Rhi ChIP-Seq and unique piRNA profiles across several of the major germline clusters (Figure 1A, 1B and S1A). We therefore compared Rhi binding to cluster chromatin, assayed by ChIP-Seq, to fold reduction in piRNA production in rhi mutants, assayed by small RNA-Seq. Eleven piRNA clusters, including the two major uni-strand clusters (Cluster 2 and flamenco), showed no decrease in piRNA expression in rhi2/KG mutants and showed only background Rhi binding by ChIP-Seq (Figure 1D and S1A). For the remaining 131 piRNA clusters, by contrast, Rhi binding was significantly correlated with fold reduction in piRNA expression in rhi mutants (Figure 1D, Pearson correlation coefficient r = 0.74; P < 2.2×10-16). ChIP-Seq signal of a matched pre-immune serum control did not correlate with piRNA expression (r = 0.19, P = 0.03; Figure S1B).
The correlation between Rhi binding and piRNA expression raised the possibility that piRNAs direct Rhi to chromatin, perhaps through a process analogous to siRNA guided centromeric heterochromatin assembly in S. pombe (Verdel et al., 2004; Iida et al., 2008; Grewal, 2010). We therefore performed Rhi ChIP-Seq in ovaries mutant for armi (armi1/armi72.1), which is required for production of piRNAs from most dual-strand piRNA clusters (Malone et al., 2009). Rhi binding in the armi mutants and matched controls were essentially identical (r = 0.63~0.84, p < 2.2 ×10-16, Figure S2, shown for the 42AB cluster in Figure S2A), suggests that Rhi localization to clusters is independent of piRNA production. However, maternal piRNAs could localize Rhi to clusters during early development, and this distribution could be epigenetically propagated to the adult stage by a piRNA independent mechanism. It is also possible that the low level of piRNAs expression in armi mutants is sufficient to localize Rhi. The mechanisms that localize Rhi and define cluster location thus remain to be determined.
Cluster transcript splicing
Rhi co-localizes to nuclear foci with the piRNA pathway proteins Cuff and UAP 56, mutations in rhi disrupt localization of both proteins, and mutations in cuff and uap56 disrupt Rhi localization (Pane et al., 2011; Zhang et al., 2012a). The Rhi protein appears to directly associate with chromatin (Klattenhoff et al., 2009), while Cuff and UAP56 are related to well characterized RNA binding proteins, and cluster transcripts co-immunoprecipitate with UAP56 (Pane et al., 2011; Zhang et al., 2012a). These three proteins may therefore have distinct molecular functions at a related step in piRNA precursor biogenesis.
qPCR assays for two regions the 240 kb 42AB piRNA cluster showed reduce RNA expression in rhi mutants (Klattenhoff et al., 2009), suggesting that transcription or transcript stability may be decreased. To extend these findings, we used strand-specific paired-end RNA sequencing (RNA-Seq) to profile the transcriptome in rhi mutants, and in ovaries mutant for cuff and uap56. These studies did not show a consistent reduction in reads mapping to clusters in rhi mutants, or in ovaries mutant for cuff or uap56 (Figure S3). However, visual inspection of several prominent clusters showed that all three mutations produce similar shifts in RNA-Seq signal to a few well-defined peaks (Figure 2A). The regions previously assayed by qPCR in rhi mutants fall between these peaks, explaining the apparent discrepancy with our early findings (Klattenhoff et al., 2009).
Figure 2. Rhi and Cuff suppress splicing at the 42AB and sox102F clusters.
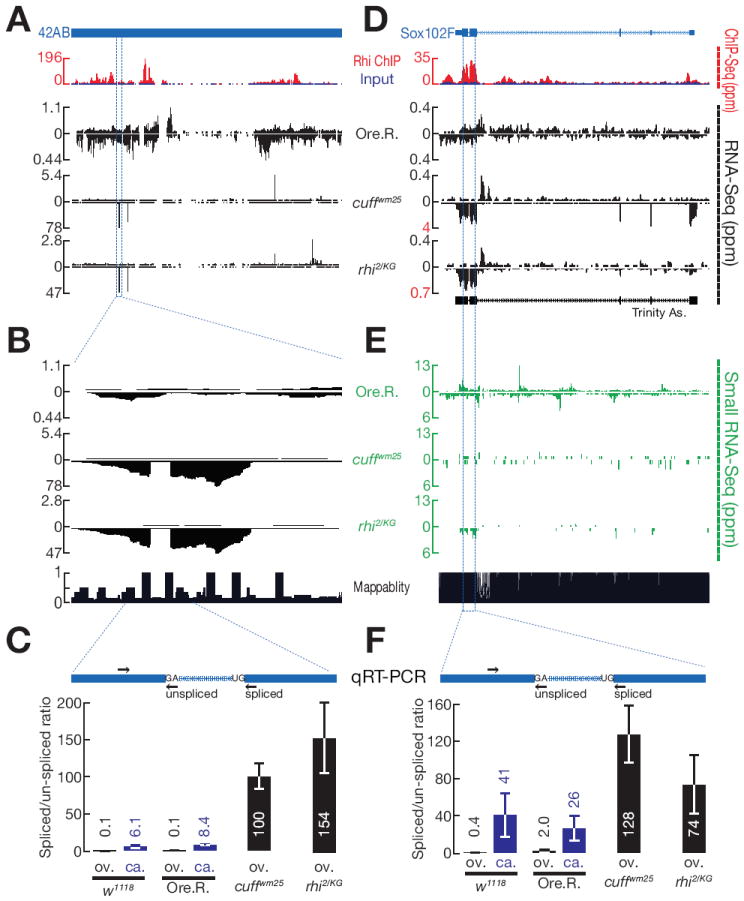
A, D. Rhi ChIP-Seq and RNA-Seq profiles for the 42AB cluster (A) and sox102F locus (D) are shown. For ChIP-Seq, Rhi signal (red) is superimposed on input (blue). RNA-Seq is shown for Oregon R (Ore. R.) control, cuff and rhi mutants. In Ore. R. controls, signal is spread over both 42AB and sox102F. In rhi and cuff mutants, by contrast, signal shifts to distinct peaks (Note that data are scaled to avoid peak clipping). At the sox102F locus, the boundaries correspond to annotated splice sites in the mature somatic transcript, and de novo transcript assembly from these data yields the annotated gene structure (Trinity As.)
(B) A high resolution expansion of the indicated region of 42AB shows that the peak is interrupted by a region with little signal, defined by very sharp boundaries characteristic of intron removal.
(C, F) qRT-PCR quantify the splicing efficiency at 42AB (C) and sox102F (F) loci. The diagrams show the putative introns (blue) and the position of primers (arrow) used to assay unspliced and spliced transcripts at 42AB (C) and sox102F (F). Both spliced and unspliced transcripts are amplified using the same forward primers. Reverse Primers for unspliced transcripts span the splice site and for spliced transcripts span the mature junction. Bar graphs show the ratio of spliced to unspliced RNAs in ovary (ov.) and carcass (ca.) in two different control strains (w1118 and Ore. R.) and in cuff and rhi mutants. In ovaries, splicing at 42AB and sox102F increases over 80 fold in both cuff and rhi mutants. The sox102F locus is expressed in somatic tissue present in the carcass, and the transcripts are spliced. Data are mean ± standard deviation for 3 independent biological samples.
(E) piRNA production from sox102F locus in Ore.R., cuff and rhi mutants.
See also Figure S3, S4 and S5.
RNA-Seq reads that cross splice junctions map to two genomic locations separated by the intron length. In genome browser views, these split reads produce signal profiles that are interrupted by sharply defined gaps. Surprisingly, the cluster peaks in rhi, cuff and uap56 mutants are often interrupted by sharply defined gaps that precisely map to consensus splice donor and acceptor sites. Figure 2B shows an example in the 42AB cluster. qPCR studies confirmed that RNAs crossing this unique donor-acceptor site junction increase over 100 fold in both cuff and rhi mutants (Figure 2C). The spliced peak in 42AB is in the sense orientation of a gypsy12 mobile element that could be activated in the mutant strains, and increased expression of the spliced RNA could therefore reflect activation of an active element. We therefore assayed a second putative intron in a chromosome 4 cluster that is antisense to the telomeric transposon TART. Utilization of this intron cannot be explained by expression of the active element. qPCR confirmed that spliced transcripts mapping to this intron increase in rhi, cuff and uap mutants (F. Zhang and W. Theurkauf, unpublished observations). Mutations in rhi, cuff and uap56 thus lead to accumulation of spliced transcripts from two major germline piRNA clusters.
To analyze splicing across the transcriptome, we computationally identified all split reads mapping to consensus splice donor and acceptor sites in three control stains (Oregon R, w1 and cn,bw) and in rhi, cuff and uap56 mutants. To filter potential sequencing artifacts, we restricted our analysis to introns defined by a minimum of 10 reads mapping across the splice junctions, and with a minimum splicing entropy of 2 (the entropy cutoff controls for potential PCR amplification artifacts; (Graveley et al., 2011). To calculate splicing efficiency, we divided the number of split reads (defining spliced RNAs) by the number of reads crossing the corresponding splice sites (defining unspliced RNAs). As shown in the scatter plots in Figure 3A-D, rhi, cuff and uap56 did not alter global splicing efficiency for protein coding genes (black points) or for the rare cluster mapping introns (red points). Consistent with these observations, the mutations did not alter global gene expression (Figure S3).
Figure 3. rhi, cuff and uap56 mutations do not alter global splicing efficiency, but lead to splicing of novel cluster introns.
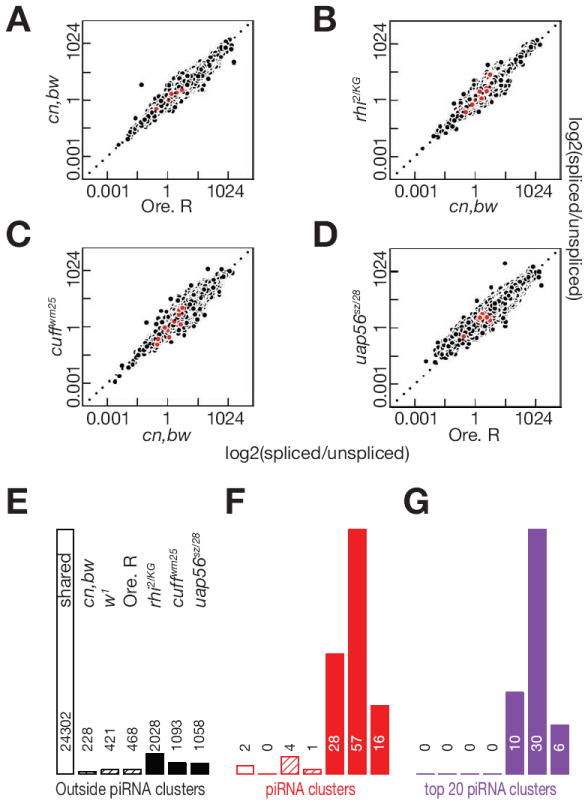
A-D. Scatter plots showing splicing efficiency at introns shared between the two indicated genotypes. Each point is one intron. Cluster mapping introns are in red and introns mapping outside clusters in black. E-G. Bar graphs quantifying shared and genotype specific introns. Introns outside of piRNA clusters are in black, introns mapping to all clusters are in red, and introns mapping to the top 20 clusters are in purple. Introns shared by the cn,bw, w1 and Oregon R control strains are indicated by the open bars. Mutant specific introns are in solid bars. For each set of bar graphs, the genotypes are ordered as in E, and the number of introns detected is above or within the bar.
Splicing efficiency was determined for introns that were utilized in both mutant and control strains, but the cluster introns that we detected by visual inspection were often spliced in one or more mutant strain, but not in any of the control strains. We therefore quantified introns that were used in only the control or mutants strains. We detected over 24,000 introns that were used in all of the strains, but only 230 to 469 introns were used in one or more of the control strains, but in none of the mutant strains (Figure 3E and F, black cross hatched bars). Five of these introns mapped to piRNA clusters, with the balance mapping to protein coding genes. However, none of cluster introns, and only 20 of the genic introns, were used in all of the control strains. Rhi, Cuff and UAP56 thus do not promote splicing of clusters transcripts, or of the vast majority of genic pre-mRNAs.
We detected between 1058 and 2028 genic introns that were utilized in one or more mutant strains but not in any of the control strains (Figure 3E, black bars), but only 117 of these introns were used in all three mutants. Rhi, Cuff and UAP56 could directly regulate splicing at these introns, but these events are very rare relative to splicing of the 24,000 genic introns that are used in all of the control and mutant strains. In striking contrast, we detected only 2 cluster mapping introns that were used in all of the strains, and only 5 additional cluster introns that were used in a one or more control strain but not in the mutants (Figure 3F). In the control strains, most cluster transcripts thus do not appear to be spliced. In rhi, cuff and uap56 mutants, by contrast, we detected 16 to 57 cluster mapping introns (Figure 3F), and these introns were enriched in the top 20 clusters, which make the majority of piRNAs in the ovary (Figure 3G). The most striking increase in cluster transcript splicing was in cuff mutants, where we detected 57 introns were not in any of the control strains, and 30 of these introns mapped to the top 20 piRNA clusters. In addition, 5 of these cluster-mapping introns were used in uap56, cuff and rhi mutants. This represents a minimum estimate of cluster introns that are expressed in the mutants, because the repeated sequences that make up most of these loci cannot be uniquely mapped. These findings suggest that Rhi, Cuff and UAP56 suppress splicing of cluster transcripts, or destabilize spliced transcripts from these loci. However, the repeated nature of the clusters, noted above, made independent verification of these splicing events difficult. In addition, some of the changes in intron utilization could be secondary to transposon mobilization and genome instability. To overcome these limitations and more rigorously explore rhi, cuff and uap56 function in cluster transcript processing, we exploited a unique germline piRNA cluster that is also a somatic protein coding gene, and developed a transgenic reporter system that allowed us to directly assay Rhi function in wild type ovaries, in the absence of genome instability.
Rhino, Cuff and UAP56 convert a somatic protein-coding gene to a germline piRNA cluster
The sox102F locus is largely composed of unique sequences and cDNA and RNA-Seq data indicate that this locus produces four distinct spliced primary transcripts in somatic cells (FlyBase Genome Annotators, FlyBase.org; Figure 2D). In the germline, by contrast, we find that this locus produces piRNAs from both genomic strands (Figure 2E and S4). Mutations in rhi, cuff and uap56 disrupt production of these piRNAs (Figure 2E and S4). Consistent with this pattern of piRNA production, RNA sequencing on control ovaries revealed transcripts mapping across both strands of the locus, and to both the introns and exons of the somatic gene (Figure 2D). ChIP-Seq shows that Rhi binds to this locus (Figure 2D and S4). Intriguingly, the Rhi ChIP signal correlates with somatic exons, despite the absence of splicing, suggesting that RNA processing signals may be recognized in the germline, and recruit Rhi to chromatin (Figure 2D and S4). These findings indicate that the sox102F locus is a protein coding gene in the soma, and a Rhi-dependent piRNA cluster in the ovary.
In ovaries mutant for rhi, cuff and uap56, spliced reads mapping to annotated sox102F donor and acceptor sites increase over 100 fold, and strand specific qPCR confirmed this increase in spliced pre-mRNA expression (Figure 2D, 2F and Figure S4). The increase in splicing-specific reads was particularly pronounced in cuff mutants, where all of the annotated introns were efficiently excised and de novo transcript assembly generated an mRNA that precisely matches the somatic sox102F transcript (Figure 2D). To determine if background mutations contribute to the increase in splicing, we expressed a wild type rhi transgene in rhi mutants and used qPCR to assay spliced and unspliced transcript levels at both sox102F and 42AB (Figure S5A and data not shown). The transgene fully suppressed expression of the spliced transcripts from this locus (Figure S5A). Transposon mobilization and DNA damage are common to all piRNA pathway mutants, and could contribute to the splicing defects. We therefore analyzed cluster splicing in ovaries mutant for qin, which disrupt a component of the cytoplasmic nuage required for piRNA amplification and transposon silencing (Zhang et al., 2011). In contrast mutations in rhi, cuff and uap56, the qin mutant combination did not increase spliced transcripts from the sox102F locus (Figure S4). The increase in spliced cluster transcripts in rhi, cuff and uap56 mutant ovaries thus do not appear to be caused by background mutations or DNA damage.
Increased expression of spliced cluster transcripts in mutant ovaries could be due to enhanced splicing, or preferential stabilization of spliced transcripts. If the mutations increase splicing efficiency, accumulation of spliced transcripts should be coupled to reduced expression of unspliced transcripts. By contrast, preferential stabilization of spliced transcripts would increase expression of processed RNAs without altering unspliced transcript levels. We therefore quantified RNA-Seq reads mapping across spliced and unspliced RNAs from the sox102F locus (Table S1). In cn;bw, Ore. R and w1 control strains, 23, 12 and 15 reads mapped across the splice sites of unprocessed transcripts, and 6, 0 and 6 reads mapped to splice junctions. In cuff mutants, only 2 reads mapped across splice sites, and no reads mapping across splice sites were recovered in rhi and uap56 mutants. By contrast, 110 reads mapped across mature splice junctions in cuff, and 18 and 5 reads mapped to these junctions in rhi and uap56. Consistent with these data, qPCR indicated unspliced reads decrease and spliced reads increase in rhi mutants (Figure S5B and S5C). Furthermore, we found that expression of a wild type rhi transgene in the mutant background restored expression of unspliced transcripts and suppresses accumulation of spliced transcripts (Figure S5B and 5C). These finding are indirect, but support the hypothesis that rhi, cuff and uap56 function to suppress cluster transcript splicing.
To determine if unspiliced transcripts are preferentially processed into piRNAs, we compared expression of spliced and unspliced long RNAs to expression of piRNA reads mapping to the splice junction and splice sites in the sox102F locus (Figure S5D). In the control w1118 strain, RNA sequencing and qPCR assays indicate that the ratio of unspliced to spliced transcripts is approximately 3:1 (Table S1). By contrast, we identified forty-nine reads, representing 11 species, mapping to sox102F splice sites, but no reads mapping to mature junctions. These data, while limited, suggest that unspliced cluster transcripts are preferentially processed into piRNAs.
Rhi tethering suppresses splicing and promotes piRNA production from complementary transcripts
To determine if Rhi binding is sufficient to suppress splicing and induce piRNA production, we used a transgenic LacO-LacI system to “tether” Rhino to an ectopic locus. For these experiments, we generated transgenic flies harboring a reporter transgene containing 36 LacO DNA-binding sites upstream a truncated vasa promoter, which drives EGFP expression in the germline and somatic follicle cells of the ovary. The transcription unit contains the 84B α-Tubulin 5′ UTR and first intron, followed by an exon encoding EGFP (Figure 4A). We then introduced transgenes carrying LacI or a LacI∷Rhi fusion proteins, under the control of inducible UASp promoters. Germline expression was induced using that nanos-Gal4-VP16 driver and EGFP expression was assayed by laser scanning confocal microscopy and western blotting (Figure 5A and S6). Germline expression of the LacI control did not block EGFP accumulation in the nurse cells, or in somatic follicle cells (Figure 5A). Immunolabeling confirmed that LacI was expressed in the nurse cells (Figure 5A). Germline expression of the LacI∷Rhi fusion, by contrast, silenced EGFP in the nurse cells, but not in the somatic follicle cells (Figure 5A). Rhi mediated EGFP silencing was confirmed by western blotting (Figure S6). Rhi binding upstream of the reporter promoter thus silences expression of the downstream transcription unit.
Figure 4. Rhi “tethering” leads to spreading through the target transcription unit, but does not reduce Pol-II occupancy.
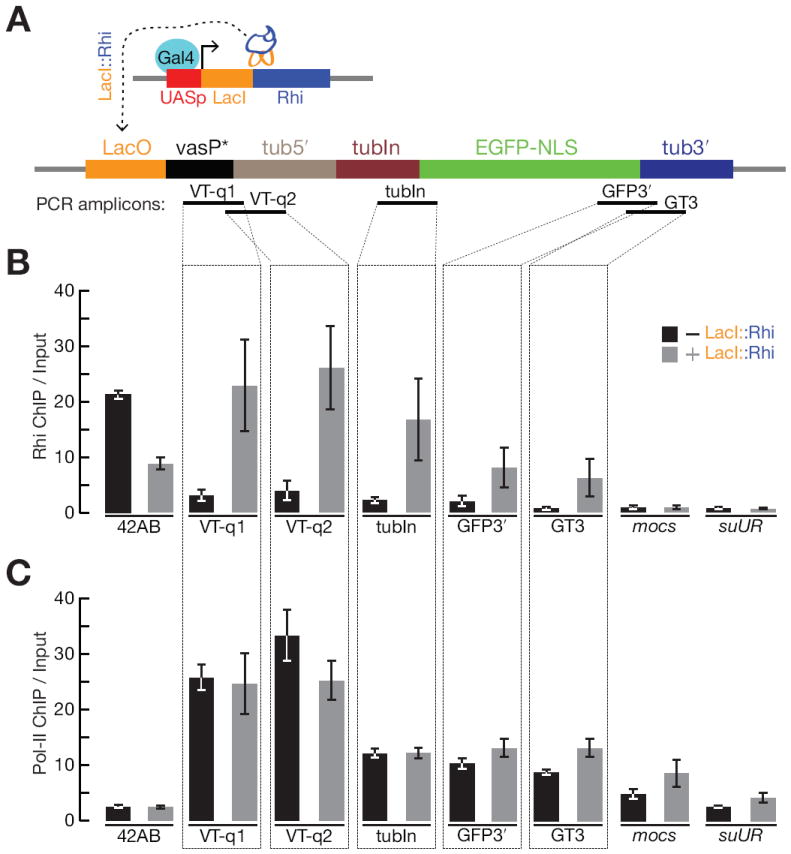
(A) Schematic diagram of the experimental design. An inducible UASp promoter was used to drive expression of the DNA binding protein LacI fused to full length Rhi, in the presence of a reporter gene containing LacI binding sites (LacO) upstream of a promoter (truncated vasa promoter) that drives expression of the 84B alpha tubulin 5′UTR and first intron fused to EGFP with nuclear localization signal (NLS). PCR amplicons indicates positions assayed for Rhi and Pol-II binding in panel B and C.
(B) Fold enrichment by Rhi ChIP across the reporter in the absence (grey) or presence of LacI∷Rhi. The LacI∷Rhi lead to Rhi binding through the transcription unit.
(C) Fold enrichment by RNA pol-II ChIP across the reporter, bars as indicated for panel B. RNA polymerase binding across the transcription unit is not altered in the presence of the LacI∷Rhi. The 42AB locus is used as a positive control for Rhi binding. The mocs and suUR loci are located downstream of reporter construct in the genome. We do not detect Rhi spreading or changes in RNA Pol-II at these loci.
Data are mean ± standard deviation for 3 independent biological samples.
Figure 5. Tethering Rhi suppresses EGFP expression and splicing.
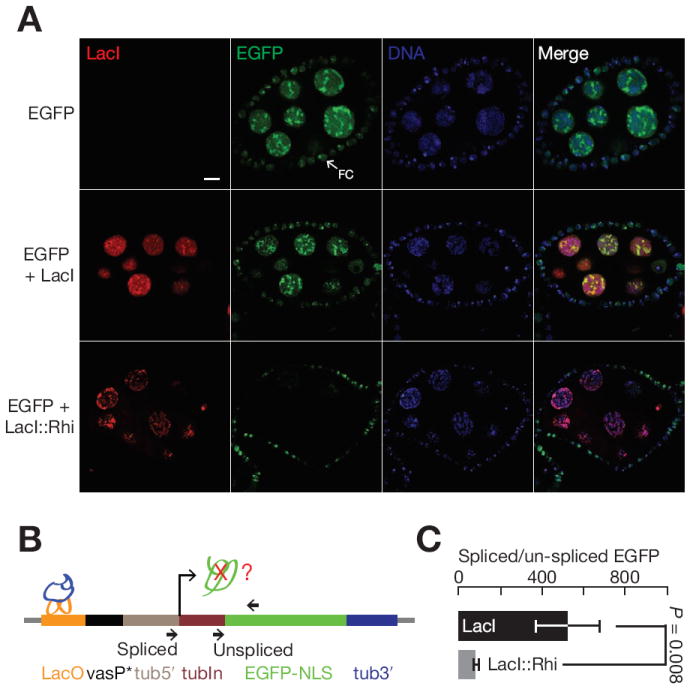
(A) Germline expression of LacI (red) does not suppress EGFP expression (green). By contrast, expression of LacI∷Rhi (red) suppresses EGFP accumulation in the nurse cell nuclei (green). Note that the EGFP reporter is expressed in both the germline and surrounding somatic follicle cells (arrows, FC). The fusion does not expressed in the follicle cells, and EGFP expression in these cells is not reduced. The bar in the up right panel is 10 μm, and applies to all panels.
(B) Splicing at the target locus. The diagram shows the target transgene and indicates that position of LacI or LacI∷Rhi binding (LacO) and the primers used to assay both spliced and unspliced transcripts by qRT-PCR.
(C) Bar graph showing the ratio of spliced to unspliced target in the presence of LacI (black) or LacI∷Rhi (grey). LacI∷Rhi binding lead to a significant reduction in splicing efficiency (p = 0.008), data are mean ± standard deviation for 3 independent biological samples.
See also Figure S6.
HP1a recruits the methyl transferase that modifies Histone H3 to generate HP1a binding sites, which leads to heterochromatin spreading (Danzer and Wallrath, 2004). Rhi is an HP1a homolog, and we speculated that LacI∷Rhi binding upstream of the reporter gene promoter may lead to Rhi spreading into the downstream transcription unit. We therefore assayed Rhi binding at sites through the transgene reporter using ChIP and qPCR. In the absence of LacI∷Rhi fusion protein, we observed background binding of Rhi through the transgene. However, expression of the LacI∷Rhi fusion was linked to significant Rhi binding through the transcription unit, with the highest levels near the LacO binding sites at 5′ end of the gene (Figure 4B). To determine if LacI∷Rhi binding and Rhi spreading reduces transcription, we used ChIP-qPCR to measure total RNA Polymerase II (Pol II) binding at the target transgene (Figure 4C). Essentially identical levels of Pol II were observed in the absence or presence the LacI∷Rhi fusion, suggesting that silencing is post-transcriptional. Consistent with this hypothesis, qPCR revealed a 6 fold reduction in the ratio of spliced to unspliced reporter transcripts in the presence of the LacI∷Rhi fusion (Figure 5B and 5C). Rhi binding thus appears to suppress pre-mRNA splicing.
To assay piRNA production, we sequenced small RNAs from ovaries carrying the target transgene and expressing either the LacI control or the LacI∷Rhi fusion. With both combinations, we detected only very low levels of 23 to 30 nt putative piRNAs mapping to the reporter transgene (Figure 6 and S7). Rhi is specifically required for piRNA production from dual-strand clusters, and the target transgene is transcribed from only one strand. We therefore constructed a second transgene with a promoter driving expression of antisense target sequences, integrated this gene into the same chromosomal locus as the sense strand reporter, and generated females carrying a trans combination of sense and anti-sense reporters. Small RNA sequencing showed that expression of the LacI∷Rhi fusion, but not the LacI control, triggered production of 23 to 30nt small RNAs from both strands of the reporter construct, including the intron, EGFP and LacO binding sites (Figure S7).
Figure 6. Rhi binding to complementary transcription units triggers piRNA production.
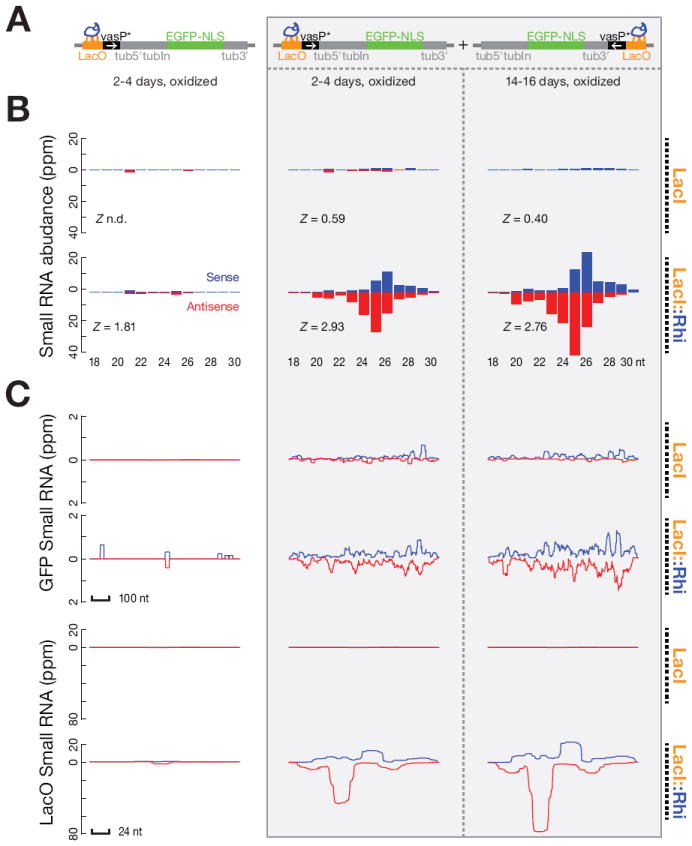
(A) Diagrams show sense strand reporter and combination of sense and antisense reporters, indicating positions of LacI or LacI∷Rhi binding and position and orientation of promoters (vasP).
(B) Length distribution of the small RNAs mapping to the reporter constructs. Blue indicates sense strand species and red indicates antisense species. Z scores indicate the significance of the 10 nt overlap between sense and antisense piRNAs (Ping-Pong signature). Z-score = 1.96 corresponds to p-value = 0.05. Too few piRNAs were detected with the LacI control for the Z score to be determined (indicated as n.d.). piRNAs carry a 3′ end modified and therefore are resistant to oxidation. Both un-oxidized (Figure S7) and oxidized RNAs (shown) were used to prepare libraries for sequencing. LacI∷Rhi binding to the sense strand reporter did not lead to significant production of oxidation resistant species between 23 and 30 nt. By contrast, LacI∷Rhi binding to the combination of sense and anti-sense reporters trigger production of oxidation resistant species showing length distributions characteristic of mature piRNAs. piRNAs from opposite strands showed a weak bias toward a 10 nt overlap, which is typical of primary piRNAs produced by a ping-pong independent mechanism.
(C) Distribution of Small RNA reads over EGFP and the LacO repeats in the presence of LacI or LacI∷Rhi. Sense signal is in blue and anti-sense signal is in red.
See also Figure S7.
piRNAs and endo-siRNAs bear a 2′-O-methyl group at their 3’ termini, which protects them from oxidation (Horwich et al., 2007; Saito et al., 2007). miRNAs and non-specific RNA degradation products, by contrast, don’t carry this modification and are rendered unclonable by oxidation. We therefore performed deep sequencing on oxidized RNA samples, which are enriched for 21 nt siRNAs and 23-30 nt piRNAs. As shown in Figure 6, LacI∷Rhi tethering to the trans-combination of reporter genes triggered production of oxidation resistant small RNA with a size distribution typical of endogenous piRNAs. During the P-M hybrid dysgenesis, de novo production of P-element piRNAs increase with adult female age (Khurana et al., 2011). Reporter specific piRNAs also increased as flies carrying LacI∷Rhi were aged from 2-4 days to 12-14 days (Figure 6). Total endogenous ovary piRNAs, by contrast, did not change (Table S2b). LacI∷Rhi binding is therefore sufficient to drive de novo production of primary piRNAs from a trans combination of reporters producing complementary RNAs.
DISCUSSION
Primary piRNA production from dual-strand and uni-strand piRNA clusters
The piRNA pathway has an evolutionarily conserved role in transposon control during germline development, and is essential for transmission of the inherited genetic complement. In the Drosophila ovary, unique piRNAs are concentrated in “ clusters” composed of complex arrays of nested transposon fragments that are generally localized to pericentromeric or subtelomeric heterochromatin (Brennecke et al., 2007). These loci fall into two classes, based on strand bias. Clusters that produce piRNAs from both genomic strands (dual-strand clusters) are dominant in the germline, while clusters that are expressed on only one genomic strand (uni-strand clusters) produce most of the piRNAs in somatic follicle cells that surround the germline (Brennecke et al., 2007; Malone et al., 2009). Primary piRNAs from dual-strand clusters, bound to PIWI proteins, appear to drive a ping-pong cycle that amplifies the silencing RNA pool (Brennecke et al., 2007; Gunawardane et al., 2007). Primary piRNAs that initiate the amplification cycle, by definition, are produced by a ping-pong independent mechanism. Similarly, ping-pong amplification is not required for production of piRNAs from uni-strand clusters. These observations suggest a simple model in which primary piRNAs from uni-strand and dual-strand clusters are produced by a common mechanism, and dual-strand clusters are equivalent to convergently transcribed uni-strand clusters. However, uni-strand cluster piRNA production is independent of rhi, uap56 and cuff, which are essential for production of piRNAs that map uniquely to dual-strand clusters (Klattenhoff et al., 2009; Pane et al., 2011; Zhang et al., 2012a). In addition, we show that Rhi-dependent piRNA production from an ectopic locus requires a combination of transgenes expressing complementary transcripts (Figure 6 and S7). Primary piRNA production by dual-strand and uni-strand clusters thus appear to proceed by distinct mechanisms. These findings also suggest that piRNA production by dual-strand clusters requires complementary precursors. The role of complementary RNAs in the germline piRNA biogenesis pathway, however, remains to be determined.
Does stalled splicing distinguish piRNA precursors from mRNAs?
piRNA pathway mutations increase expression of a subset of transposons by over 200 fold, but do not alter germline gene expression (Klattenhoff et al., 2009; Zhang et al., 2012a; Li et al., 2009; Czech et al., 2013; Handler et al., 2013). This remarkable specificity is almost certainly essential to gamete production, but how piRNA precursors are differentiated from mRNAs is not understood. The vast majority of protein coding pre-mRNAs are efficiently spliced, exported from the nucleus, and translated in the cytoplasm. By contrast, splicing is suppressed at a transgene inserted into the Drosohila X-TAS piRNA cluster, (Muerdter et al., 2012), and our transcriptome wide studies indicate that rapidly evolving HP1 homolog Rhi, the Rai1 related protein Cuff, and the DEAD box protein UAP56 suppress slicing at resident consensus donor and acceptor sites in germline clusters. This is most clearly illustrated at the sox102F locus, which produces efficiently spliced pre-mRNAs in the soma, but is the source of piRNAs from unspliced primary transcripts in the germline (Figure S5D). Significantly, accumulation of both unspliced transcripts and piRNAs requires rhi, cuff and uap56, and tethering a LacI∷Rhi fusion to a intron-containing reporter transgene suppresses splicing, and is sufficient to trigger de novo piRNA production from a trans combination of sense and anti-sense transgenes (Figure 6 and S7). We therefore propose that Rhi functions with Cuff and UAP56 to suppress cluster transcript splicing, and that the stalled splicing intermediates are the precursors for primary piRNAs.
Cuff is a homolog of Rai1/DXO, which binds and degrades mRNAs carrying incomplete cap structures (Jiao et al., 2013). This would appear to support a role for Cuff in destabilizing spliced cluster transcripts, but the residues required for the 5′ to 3′ exonuclease activity of Rai1/DXO are not conserved in Cuff (Chen et al., 2007; Pane et al., 2011). Murine DXO has been co-crystallized with an uncapped RNA, and with a cap analog. The crystal structures reveal the protein residues that interact with the RNA backbone, and indicate that the cap is bound in a pocket in the interior of the protein (Jiao et al., 2013). We aligned the sequences of Drosophila Cuff with the murine DXO. Twenty one percent of the positions are identical, and conserved amino acids are present throughout the entire alignment. Overall protein fold of Cuff is therefore highly likely to resemble DXO. We therefore used the Modeller and I-TASSER algorithms to build a homology model of Cuff based on the Murine DXO structures (Roy et al., 2010; Zhang, 2008; Sali and Blundell, 1993). Essentially all of the RNA binding interactions are preserved in the homology model of Cuff (Figure 7A). We therefore propose that Cuff binds to capped RNAs, but is not a catalytically active nuclease.
Figure 7. Predicted Cuff structure and a model for Rhi complex function in piRNA biogenesis.
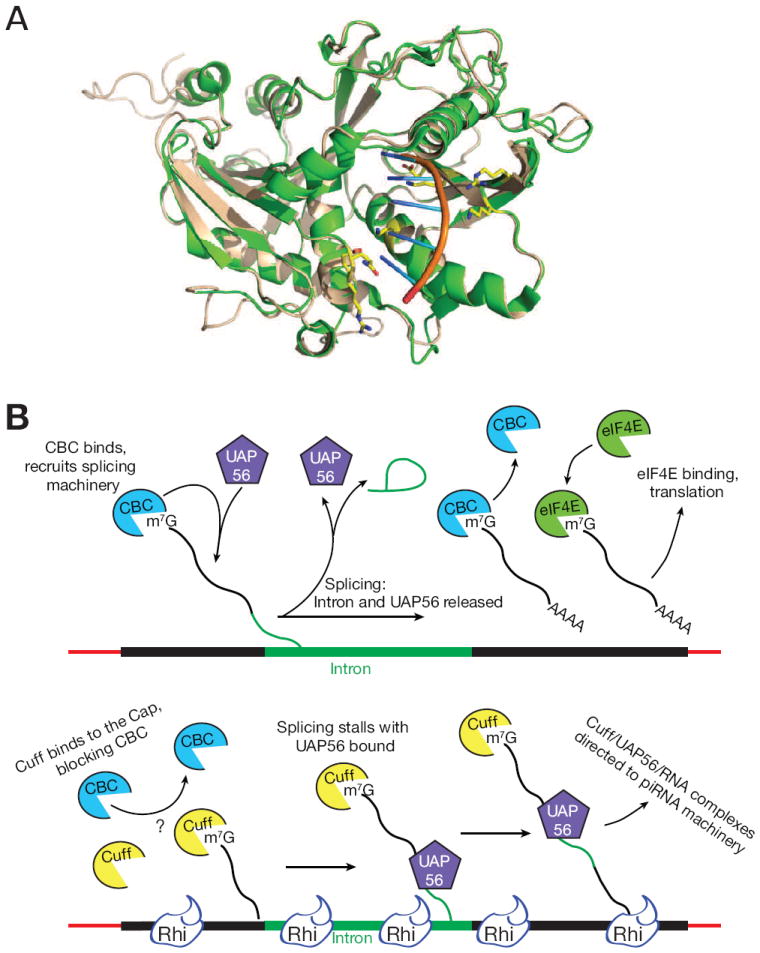
(A) Predicted overlap of homology modeled Drosophila Cuff (wheat) and mouse DXO (Green), a murine mRNA decapping enzyme (Jiao et al., 2013). Catalytic residues are not conserved in Cuff, but residues that interact with the RNA backbone and bases are preserved (yellow).
(B) Top: Co-transcriptional binding by the cap binding complex (CBC) promotes efficient pre-mRNA splicing, through a process that requires transient binding by UAP56. Subsequent cap binding by EIF4e promotes translation. Bottom: Cuff localizes to clusters through Rhi, and we speculate that co-transcriptional loading on capped cluster transcripts competes with CPC and stalls splicing with UAP56 bound. Stable cluster transcript-UAP56 complexes are directed to the piRNA biogenesis machinery.
Pre-mRNAs are co-transcriptionally capped, and cap binding by the nuclear Cap Binding Complex (CBC) is required for splicing and polyadenylation (Izaurralde et al., 1994; Topisirovic et al., 2011). We therefore propose that Cuff binds to Rhi and co-transcriptionally associates with cluster transcripts, which prevents recognition by the CBC and thus blocks splicing (Figure 7B). We previously showed that UAP56 immunoprecipitation significantly enriches for cluster transcripts, not for mRNAs, and that RNAs mapping to the major 42AB cluster are the most highly enriched species in the immunoprecipitated pool (Zhang et al., 2012a). The point mutation in uap56 that specifically blocks piRNA biogenesis disrupts a salt bridge predicted to stabilize the ATP and RNA bound form of the protein (Zhang et al., 2012a). These observations suggest that stable cluster transcript binding by UAP56 is required for piRNA biogenesis. Mutations in the yeast cap binding complex lead to arrest at an early step in the splicing pathway, with the U2 snRNP bound to primary transcripts (Gornemann et al., 2005). UAP56 was identified as a binding partner of the U2 snRNP protein U2AF65 (Fleckner et al., 1997). Cuff binding to capped cluster transcripts may therefore prevent cap recognition by the CBC, arresting splicing with UAP56 stably bound. This aberrant stable complex could differentiate piRNA precursors from pre-mRNAs (Figure 7). While this model is highly speculative, it makes several clear predictions and should therefore serve as a useful starting point to additional studies.
Adaptation to transposon invasion by the piRNA pathway appears to be initiated by insertion of the invading element into a cluster (Khurana et al., 2011). This speculate model, with the observation that Rhi can spread from anchor sites, suggest an adaption model in which Rhi spreads into active transposons that insert into clusters, leading to Cuff binding to capped transcripts from transposon promoters. This would block processing and promote production of new piRNAs, thus coordinately silence the inserted element and produce the trans-silence species that control dispersed active elements.
Studies in the pathogenic yeast Crypotoccous provide evidence for a direct link between stalled splicing and transposon silencing by the siRNA pathway (Dumesic et al., 2013). Dumesic et al., (2013) showed that splicing factors associate with the Crypotoccous siRNA biogenesis machinery and siRNAs are produced from unspliced transposon transcripts. In addition, intron removal reduces siRNA production, and splice site mutations that reduce splicing efficiency increase siRNA production (Dumesic et al., 2013). Furthermore, recent genome wide screens have implicated splicing factors in transposon silencing, and the splicing and small RNA silencing pathways appears to be co-evolving (Czech et al., 2013; Handler et al., 2013; Muerdter et al., 2013; Tabach et al., 2013). These findings, with the studies reported here, suggest that stalled splicing generates a conserved molecular signature for potentially deleterious RNAs, which directs these transcripts to small silencing RNA biogenesis pathways. Retrotransposons and retroviruses encode essential spliced transcripts, but splicing must be suppressed to produce full length genomic RNAs. This novel feature of the retroviral life cycle may have driven evolution of silencing systems that use stalled splicing as a hallmark of pathogenic RNAs.
EXPERIMENTAL PROCEDURES
General Methods
RNA isolation, small RNA library construction and sequencing data analysis, immunoblotting, immunostaining and quantitative RT-PCR were performed as described (Zhang et al., 2011). Figures were generated using Excel (Microsoft, Redmond, WA, USA), IgorPro (WaveMetrics, Lake Oswego, OR, USA), Adobe Photoshop and Illustrator (Adobe systems, San Jose CA, USA). Table S2 reports the statistics for the ChIP-Seq, RNA-Seq and small RNA-Seq data generated in this study. Table S3 reports primer sequences for ChIP-qPCR and qRT-PCR. PCR primers used to clone the LacI binding domain, Rhi open reading frame and LacO repeats are detailed along with the supplemental text. The sources of the published deep sequencing data used in this study are summarized in Table S5. Table S6 lists the antibody information. Unless otherwise specified, p-values were calculated from at least three independent biological replicates using a two-tailed, two-sample unequal variance t-test (Excel, Microsoft).
Drosophila stocks
All flies were raised at 25°C. Table S4 summarizes the published fly alleles used in this study. Transgenic flies for tethering Rhi to EGFP locus and for rescuing suppress splicing were made as described in supplemental information.
ChIP-Seq
Detailed protocols are provided in Supplemental information. Briefly, ovaries were crosslinked and sonicated to generate ~150 bp fragments. After immunoprecipitation with antibodies to Rhi or control serum, crosslinking was reversed and enriched DNA was purified and subjected to library cloning by End-Repair, A-tailing, Adapter ligation and PCR amplification (Zhang et al., 2012b). Antibodies to Rhi are described in (Klattenhoff et al., 2009).
Bioinformatics analysis of splicing
Strand-specific RNA-Seq libraries were made as described (Zhang et al., 2012b). RNA-seq reads were aligned to the genome and the transcriptome (Flybase r5.50) using TopHat 2.0.8 (Trapnell et al., 2009) with the parameters “-x 1000 -g 1000 --read-mismatches 2 --read-edit-dist 2 --read-realign-edit-dist 0 --segment-length 50 --segment-mismatches 2”. Only reads mapping uniquely were considered in the downstream analysis. BEDTools (Quinlan and Hall, 2010)was used to count the fragments within a transcript or piRNA cluster, and the number of reads per transcript were normalized by the sequencing depth and transcript length. We collapsed introns detected by TopHat from six libraries (three control strains: Ore. R, cn,bw, w1; three mutants: rhi2/KG, cuffwm25, uap56sz/28), then we counted the spliced reads and the unspliced reads across the donor/acceptor sites. The introns with fewer than 10 spliced reads in all six libraries were discarded in the analysis. Splicing efficacy was calculated as the ratio reads mapping to mature splice junctions multiplied by two over the sum of reads to the corresponding donor and acceptor sites, a pseudo count 10 was used.
Supplementary Material
highlights.
Rhino is a piRNA cluster specific HP1.
Rhino binding correlates with piRNA production.
Rhino “tethering” to complementary transgenes triggers piRNA production.
Rhino functions with the Cuff and UAP56 to suppress piRNA precursor splicing.
Acknowledgments
We thank members of the Theurkauf and Weng labs and the UMass RNA biology community for critical feedback and encouragement. This work was supported by a grant R01HD049116 from the National Institute of Child Health and Human Development, NIH, to WET, ZW and PDZ, and in part by National Institutes of Health grant GM62862 to PDZ.
Footnotes
Accession number.
Sequence data generated in this study are available via the NCBI trace archives (http://www.ncbi.nlm.nih.gov/Traces/) using accession number SRP030460.
SUPPLEMENTAL INFORMATION
Supplemental information includes 7 figures and 6 tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, Chien M, Russo JJ, Ju J, Sheridan R, Sander C, Zavolan M, Tuschl T. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–1170. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL. Transposable element contributions to plant gene and genome evolution. Plant Mol Biol. 2000;42:251–269. [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Chen Y, Pane A, Schüpbach T. Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Current biology CB. 2007;17:637–642. doi: 10.1016/j.cub.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Preall JB, McGinn J, Hannon GJ. A Transcriptomewide RNAi Screen in the Drosophila Ovary Reveals Factors of the Germline piRNA Pathway. Molecular cell. 2013;50:749–761. doi: 10.1016/j.molcel.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer JR, Wallrath LL. Mechanisms of HP1-mediated gene silencing in Drosophila. Development. 2004;131:3571–3580. doi: 10.1242/dev.01223. [DOI] [PubMed] [Google Scholar]
- Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR, Bartel DP, Madhani HD. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell. 2013;152:957–968. doi: 10.1016/j.cell.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckner J, Zhang M, Valcarcel J, Green MR. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 1997;11:1864–1872. doi: 10.1101/gad.11.14.1864. [DOI] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- Gornemann J, Kotovic KM, Hujer K, Neugebauer KM. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol Cell. 2005;19:53–63. doi: 10.1016/j.molcel.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, Brown JB, Cherbas L, Davis CA, Dobin A, Li R, Lin W, Malone JH, Mattiuzzo NR, Miller D, Sturgill D, Tuch BB, Zaleski C, Zhang D, Blanchette M, Dudoit S, Eads B, Green RE, Hammonds A, Jiang L, Kapranov P, Langton L, Perrimon N, Sandler JE, Wan KH, Willingham A, Zhang Y, Zou Y, Andrews J, Bickel PJ, Brenner SE, Brent MR, Cherbas P, Gingeras TR, Hoskins RA, Kaufman TC, Oliver B, Celniker SE. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI. RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin Genet Dev. 2010;20:134–141. doi: 10.1016/j.gde.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivna ST, Beyret E, Wang Z, Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–1714. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC. A slicer-mediated mechanism for repeat-associated siRNA 5’ end formation in Drosophila. Science. 2007;315:1587–1590. doi: 10.1126/science.1140494. [DOI] [PubMed] [Google Scholar]
- Guzzardo PM, Muerdter F, Hannon GJ. The piRNA pathway in flies: highlights and future directions. Curr Opin Genet Dev. 2013;23:44–52. doi: 10.1016/j.gde.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler D, Meixner K, Pizka M, Lauss K, Schmied C, Gruber FS, Brennecke J. The genetic makeup of the Drosophila piRNA pathway. Mol Cell. 2013;50:762–777. doi: 10.1016/j.molcel.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich MD, Li C, Matranga C, Vagin V, Farley G, Wang P, Zamore PD. The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Current biology : CB. 2007;17:1265–1272. doi: 10.1016/j.cub.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Iida T, Nakayama J, Moazed D. siRNA-mediated heterochromatin establishment requires HP1 and is associated with antisense transcription. Mol Cell. 2008;31:178–189. doi: 10.1016/j.molcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M. A mammalian pre-mRNA 5’ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell. 2013;50:104–115. doi: 10.1016/j.molcel.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Adaixo R, Stunnenberg R, Woolcock KJ, Hiller S, Buhler M. HP1(Swi6) mediates the recognition and destruction of heterochromatic RNA transcripts. Mol Cell. 2012;47:215–227. doi: 10.1016/j.molcel.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Khurana JS, Theurkauf W. piRNAs, transposon silencing, and Drosophila germline development. J Cell Biol. 2010;191:9. doi: 10.1083/jcb.201006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana JS, Wang J, Xu J, Koppetsch BS, Thomson TC, Nowosielska A, Li C, Zamore PD, Weng Z, Theurkauf WE. Adaptation to P element transposon invasion in Drosophila melanogaster. Cell. 2011;147:1551–1563. doi: 10.1016/j.cell.2011.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C, Xi H, Li C, Lee S, Xu J, Khurana JS, Zhang F, Schultz N, Koppetsch BS, Nowosielska A, Seitz H, Zamore PD, Weng Z, Theurkauf WE. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell. 2009;138:1137–1149. doi: 10.1016/j.cell.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–367. doi: 10.1126/science.1130164. [DOI] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, Kittler ELW, Zapp ML, Klattenhoff C, Schulz N, Theurkauf WE, Weng Z, Zamore PD. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell. 2009;137:509–521. doi: 10.1016/j.cell.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137:522–535. doi: 10.1016/j.cell.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. The origin and behavior of mutable loci in maize. Proc Natl Acad Sci U S A. 1950;36:344–355. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Guzzardo PM, Gillis J, Luo Y, Yu Y, Chen C, Fekete R, Hannon GJ. A Genome-wide RNAi Screen Draws a Genetic Framework for Transposon Control and Primary piRNA Biogenesis in Drosophila. Molecular cell. 2013;50:736–748. doi: 10.1016/j.molcel.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Olovnikov I, Molaro A, Rozhkov NV, Czech B, Gordon A, Hannon GJ, Aravin AA. Production of artificial piRNAs in flies and mice. Vol. 18. RNA; New York, N.Y.: 2012. pp. 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pane A, Jiang P, Zhao DY, Singh M, Schüpbach T. The Cutoff protein regulates piRNA cluster expression and piRNA production in the Drosophila germline. The EMBO journal. 2011;30:4601–4615. doi: 10.1038/emboj.2011.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Sakaguchi Y, Suzuki T, Suzuki T, Siomi H, Siomi MC. Pimet, the Drosophila homolog of HEN1, mediates 2’-O-methylation of Piwi- interacting RNAs at their 3’ ends. Genes Dev. 2007;21:1603–1608. doi: 10.1101/gad.1563607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 2011;12:246–258. doi: 10.1038/nrm3089. [DOI] [PubMed] [Google Scholar]
- Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, Borowsky M, Kim JK, Ruvkun G. Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature. 2013;493:694–698. doi: 10.1038/nature11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA. 2011;2:277–298. doi: 10.1002/wrna.52. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang J, Xu J, Zhang Z, Koppetsch BS, Schultz N, Vreven T, Meignin C, Davis I, Zamore PD, Weng Z, Theurkauf WE. UAP56 Couples piRNA Clusters to the Perinuclear Transposon Silencing Machinery. Cell. 2012a;151:871–884. doi: 10.1016/j.cell.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, Zamore PD. Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence. 2012b;3:9. doi: 10.1186/1758-907X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Xu J, Koppetsch BS, Wang J, Tipping C, Ma S, Weng Z, Theurkauf WE, Zamore PD. Heterotypic piRNA Ping-Pong requires qin, a protein with both E3 ligase and Tudor domains. Molecular cell. 2011;44:572–584. doi: 10.1016/j.molcel.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.