

Abstract
Pulmonary hypertension (PH) is a progressive lung disease characterized by elevated pressure in the lung vasculature, resulting in right-sided heart failure and premature death. The pathogenesis of PH is complex and multifactorial, involving a dysregulated autonomic nervous system and immune response. Inflammatory mechanisms have been linked to the development and progression of PH; however, these are usually restricted to systemic and/or local lung tissue. Inflammation within the CNS, often referred to as neuroinflammation involves activation of the microglia, the innate immune cells that are found specifically in the brain and spinal cord. Microglial activation results in the release of several cytokines and chemokines that trigger neuroinflammation, and has been implicated in the pathogenesis of several disease conditions such as Alzheimer’s, Parkinson’s, hypertension, atherosclerosis, and metabolic disorders. In this review, we introduce the concept of neuroinflammation in the context of PH, and discuss possible strategies that could be developed for PH therapy based on this concept.
Keywords: Pulmonary hypertension, Neuroinflammation, Microglia
Introduction
Pulmonary hypertension (PH) is a devastating disease of diverse etiology that is characterized by elevated blood pressure in the lung vasculature. The mean pulmonary arterial pressure in normal individuals at rest measures about 14 mmHg. However, when this mean pressure exceeds 25 mmHg at rest, or is greater than 30 mmHg with exercise, the hemodynamic condition is referred to as PH [1•]. In most cases, the precise cause of this disease is unknown, and is believed to be of idiopathic origin. However, genetic predisposition has been noted in certain individuals with mutations in bone morphogenetic protein receptor-2 or activin-like-kinase-1 [2, 3]. Epigenetic silencing of superoxide dismutase-2 [4] or underlying disease conditions such as connective tissue disorders, portal hypertension, and human immunodeficiency virus (HIV) infection can also give rise to PH [5]. Regardless of the cause, PH is associated with endothelial dysfunction, vasoconstriction, smooth muscle proliferation, thrombosis, and inflammation [6, 7]. These aforementioned factors, alone or in combination, contribute to structural alterations of the lung vessels, leading to increased pulmonary vascular resistance. Persistent elevation in vascular resistance increases workload on the right ventricle to cause maladaptive remodeling, eventually leading to dysfunction and end-organ failure. In fact, right-sided heart failure is the primary cause of death in patients with PH [8].
The mechanisms involved in the pathogenesis of PH include imbalances in the levels of vasoconstrictive and vasodilatory mediators, excessive production of growth factors, and altered secretions of neurohumoral factors [9]. In addition, inflammation also plays a key role in PH pathobiology. This is evident from the fact that heightened circulating levels of cytokines, and infiltration of inflammatory cells into the lungs has been observed in patients with PH [7, 10]. However, the focus on inflammatory processes has involved systemic and/or local lung tissue. The role of inflammation within the central nervous system (CNS) has not been studied so far in diseases like PH.
Our focus in this review is to discuss the new concept of integration between neuroinflammation and the development/progression of PH. We aim to assess recent progress in PH physiopathology, and introduce the hypothesis of a neuroinflammatory role in PH and right-sided heart failure. This disorder is marked by elevated pressure in the lung vasculature, inflammation, and increased sympathetic nerve activity, which suggests a strong neurogenic component. The economic, medical, and social impact of PH in terms of illness and mortality is substantial. Potential new and less invasive therapies that target autonomic imbalance and neuroinflammation should significantly improve therapeutic outcomes for patients with PH.
What is Neuroinflammation?
Inflammation is a basic host defense response to injury, ischemia, toxins, or infectious agents. Historically, the Roman, Celsus, defined inflammation as a condition marked by four cardinal signs - redness, swelling, heat, and pain [11]. However, since then, this terminology has undergone considerable change. Inflammation now encompasses the body’s innate and adaptive immune system, along with invasion of cells (lymphocytes and macrophages) to the site of injury, and induction of inflammatory mediators such as cytokines, and chemokines [12]. When inflammation is specific to the central nervous system (CNS), it is termed neuroinflammation. Over the past few years, the field of neuroinflammation has developed into a very active branch of neuroscience. One of the primary reasons for this is that neuroinflammation is viewed as a causative/contributing factor to the pathogenesis of several neurological and/or peripheral diseases [13–15]. It would be pertinent to clarify that the term "neuroinflammation" generally refers to a state of chronic and sustained CNS injury that contributes to pathological effects, thus maintaining or worsening the disease process. Microglia, the nervous system’s own innate immune cells, play an important role in mediating neuroinflammation.
Microglia, Resident Immune Cells of the CNS
The CNS consists of neurons and three different kinds of glial cells: astrocytes, oligodendrocytes, and microglia. All these central cells are insulated from the peripheral tissues by a tight blood brain barrier. Because of the impermeable nature of the blood brain barrier, circulating immunoglobulins and leukocytes fail to access the brain regions under normal conditions. As a result, the immunological mechanisms of the CNS are quite different from that of peripheral tissues. Microglia, which account for 5 – 20 % of all cells in an adult human brain are considered to be the key players in mediating immune responses through secretion of various factors [16]. In fact, they constitute the first line of defense against invading pathogens, infections and tissue injury. Moreover, microglial cells remove cellular debris and/or toxic substances by phagocytosis, thereby maintaining homeostasis. Under physiological conditions, these cells are characterized by a small cell body and many fine processes which send multiple branches extending in all directions. This “ramified” form of microglia is fairly motionless, but its branches constantly move to survey the surrounding areas for signs of brain lesions or nervous system dysfunction. However, the ramified microglia undergoes structural changes to assume an amoeboid form, a process that is generally referred to as ‘microglial activation’ in response to a brain insult [17•]. This amoeboid form enables the entire microglial cell to migrate through the brain tissue, and translocate to the site of injury for performing immunological functions. Thus, microglial cells exhibit extreme plasticity to change shapes and transform to an activated state. While short-term microglial activation might be beneficial, chronic activation is considered detrimental, and has been implicated as a potential mechanism in neurodegeneration [18]. Activated microglia release a host of neurotoxins that include pro-inflammatory cytokines, nitric oxide and reactive oxygen species, all of which contribute to the degenerative process [19]. It is relevant to point out that astrocytes, another type of glial cell also generate pro-inflammatory cytokines to mediate neurodegeneration.
Origin of Microglia During Development and Diseased Conditions
The origin and cell lineage of microglia have been topics of debate for a long time. Though microglia were initially thought to originate from neuroectodermal matrix cells, the general consensus today is that these cells are derived from progenitors, originating from the bone marrow [20]. Studies in rodents have shown that the bone marrow-derived progenitors immigrate into the brain until postnatal day 10 to give rise to microglia [17•].
Recently, Kierdorf et al., have demonstrated that microglia in the mouse brain emerge from c-kit+ erythromyeloid yolk sac precursor cells that invade the developing nervous tissue [21]. It is well accepted that in adult animals there seems to be very little exchange between blood and brain parenchyma. However, on injury, microglial progenitors that are produced and mobilized from the bone marrow penetrate the brain tissue. The mobilization of microglial progenitors is regulated by the sympathetic nervous system (SNS) [22]. Once mobilized, these progenitors infiltrate specific injured brain areas. Furthermore, chemokines and their receptors play an important role in the recruitment process. Monocyte chemotactic protein-1 or CC-chemokine ligand-2 (MCP/CCL-2), along with its receptor CCR2, recruit resident microglia as well as microglial progenitors to the lesion area [23]. It is worth mentioning that MCP/CCL-2 is densely expressed in the paraventricular nucleus (PVN) and other hypothalamic nuclei, which happen to be cardiovascular regulatory regions of the brain [24].
Microglia-Derived Cytokines and Chemokines
Several immuno- and neuroregulatory factors are secreted by activated microglia, which mediate neuroinflammation. Cytokines, chemokines, and nitric oxide constitute a substantial fraction of these factors. Cytokines released by activated microglia consist of interleukins (IL-1 and IL-6), IFN-γ, and tumor necrosis factor-alpha (TNF-α), while chemokines include MCP-1, macrophage inflammatory protein-1 (MIP-1), and RANTES [25]. IL-1, IL-6, and TNF-α are pro-inflammatory cytokines that actively participate in the initiation and coordination of inflammatory processes following microglial activation [26]. Excessive production of these cytokines can exhibit toxic and cell death-inducing potential on the surrounding neurons and oligodendrocytes [27]. Activated microglia can also trigger or increase the release of glutamate from adjacent astrocytes to cause potentially toxic and neuromodulatory effects [28]. On the other hand, chemokines of microglial origin assist in the process of leukocyte invasion via enhanced expression of endothelial adhesion molecules to impact neuroinflammation. Microglial cells are known to express several chemokine receptors that include IL-8R, CXCR2, CXCR3, CXCR4, CCR3, CCR5, and CX3CR1 [29]. The most prominent receptors through which microglia detect infection and tissue damage are the Toll-like receptors (TLRs). Among the different subtypes, Toll-like receptor 4 (TLR-4) mediates microglial activation and production of inflammatory mediators [17•]. Of particular relevance to PH is that TLR4-deficient mice were less susceptible to the development of hypoxia-induced PH and showed decreased pulmonary vascular inflammatory response [30, 31]. Though the contribution of microglial activation on hypoxia-induced PH was not evaluated in this particular study, we speculate that TLR-4 deletion results in decreased microglia-mediated neuroinflammation, and thus renders protection against PH pathophysiology.
Inflammation in Pulmonary Hypertension
Inflammation has long been recognized as an important hallmark of PH. Overwhelming evidence from experimental models and clinical studies have shown elevated levels of circulating inflammatory markers, and accumulation of inflammatory cells in the pulmonary hypertensive lungs [7, 32]. Accumulation of T cells, B cells, and macrophages in the lungs leads to increased production of pro-inflammatory cytokines, along with subsequent activation of inflammatory pathways, which perpetuates PH, and results in end-organ damage [33, 34].
An important initiator of inflammation is the nuclear factor of activated T cells (NFAT), a well-known transcription factor that promotes cytokine gene expression. NFAT is found to be severely upregulated in PH [35]. Animal studies have shown that activation of macrophages results in the release of several pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α, all of which contribute to inflammatory process [36]. In fact, circulating levels of IL-1 and IL-6 have been reported to be considerably elevated in patients with PH [37]. Recently IL-18, a pro-inflammatory cytokine was also found to be elevated in pulmonary hypertensive subjects [38]. This cytokine has been shown to exert proliferative effects on the pulmonary artery smooth muscle cells, which contributes to vessel wall thickening. Interestingly, an increased prevalence of PH has been observed in some patients with autoimmune disorders such as HIV, connective-tissue diseases, and thyroiditis [39, 40], which underscores the importance of inflammation in PH.
The role of inflammatory cytokines in the development of PH is also supported by studies in animal models. Levels of IL-6 are consistently elevated in animal models of PH [41]. This elevation may be causal as deletion of IL-6 protects from hypoxia induced-PH [42] and overexpression of IL-6 causes severe PH [43]. Therefore, it is well recognized that the circulating and pulmonary levels of pro-inflammatory cytokines are elevated in PH; however, the induction and involvement of neuroinflammation with its manifestation of glial activation and cytokine overexpression is so far unknown in PH. Preliminary studies from our lab suggest that pro-inflammatory cytokine TNF-α is elevated in the PVN of pulmonary hypertensive animals, an indication of neuroinflammation.
An exceptional recent study suggested a causal role for hematopoietic myeloid progenitors in PH. It was found that transplantation of bone marrow-derived CD133+ cells from PH patients induced vascular injury, thromboses, and right ventricular hypertrophy in immunodeficient mice, suggesting a detimental role of these cells in cardiopulmonary remodeling [44•]. These findings also indicate that progenitor cells themselves may be dysfunctional in the disease state. Along similar lines, we have demonstrated that the migratory capacity of CD34+ cells harvested from patients with PH is impaired as compared with CD34+ cells from healthy individuals. However, treatment of these dysfunctional cells with diminazene, an activator of angiotensin converting enzyme-2 (ACE2) resulted in a significant increase in SDF-1α–induced migration [45•]. The migratory ability of these cells is also dependent on the bioavailability of nitric oxide (NO), and is adversely disrupted by oxidative stress. Our preliminary data suggests that cells from PH patients produce less NO and have greater ROS production than similar cells harvested from control subjects.
We have previously demonstrated that the ACE2 activator, diminazene can prevent and reverse MCT-induced PH, which is associated with reduction in the levels of pro-inflammatory cytokines [45•]. Preliminary data, using adipose-derived stem cells (ASCs) from normal rats have also been effective in attenuating MCT-induced PH, along with decreasing pro-inflammatory, and elevating anti-inflammatory cytokines. Further studies will be required to investigate if these treatments mediate their effects via central mechanisms.
Brain RAS and Neuroinflammation
Traditionally, the renin angiotensin system (RAS) has been viewed as a circulating system that plays a central role in the regulation of blood pressure and body fluid homeostasis. However, it is now well established that an intrinsic local RAS exists in many tissues, including the brain, where it mediates tissue-specific physiological and pathological effects [46]. The classical RAS comprises angiotensin converting enzyme (ACE), whose metabolic action leads to the formation of angiotensin II (Ang II), a vasoactive peptide. Ang II exerts its biological actions via stimulation of two different subtypes of G-protein-coupled angiotensin receptors, the AT1- and the AT2-receptor (AT1R and AT2R). However, since the expression of AT2R is very low in adult tissues, most of the classical actions of Ang II are mediated by AT1R. In addition to these classical RAS components, a counter-regulatory system comprising angiotensin converting enzyme2 (ACE2), its metabolic product, angiotensin-(1-7) [Ang-(1-7)], and the receptor Mas has been recently discovered, which together forms the ACE2/Ang-(1-7)/Mas axis, and opposes the detrimental effects of Ang II [47, 48]. All the above-mentioned RAS components (both classical and newly discovered) are expressed in the brain [49]. AT1R activation in the brain leads to regulation of immune cell infiltration, modulation of antigen-presenting cells, suppression of regulatory T cells, and induction of T helper cells (TH1 and TH17), all of which are involved in inflammation [50]. In vitro studies have confirmed the presence of AT1R on microglial cells, which on stimulation triggers the release of pro-inflammatory cytokines and NO, key factors that participate in neuroinflammation [17•]. Conversely, blockade of AT1R on the cultured microglia suppressed its activation, resulting in reduced production of cytokines and NF-κB, a potent cytokine transcription factor [51]. Similarly, in distinct experimental models of brain inflammation involving LPS administration [52], stroke [53], or cerebral hemorrhage [54], blockade of AT1R protected the brain parenchyma.
Experimental Evidence for Neuroinflammation in PH
In the well-studied monocrotaline (MCT)-model of lung injury, we observed that development of PH, identified by an increase in right ventricular systolic pressure (RVSP, a surrogate marker), was associated with microglial activation in the PVN of the brain. Additionally, these changes were accompanied by significant increases in the expression of inflammatory cytokine and RAS components, suggesting for the first time that neuroinflammation and a dysregulated brain RAS could be involved in the pathogenesis of PH. Overwhelming evidence from both experimental and clinical studies have shown the importance of RAS, particularly that of Ang II in the development and progression of PH [55–57]. Circulating levels of Ang II have been found to be considerably increased during PH [55]. We speculate that this increase in Ang II levels might contribute to neuroinflammation and other changes in the MCT-challenged brain. This speculation is based on our previous rodent experiments, wherein chronic Ang II infusion resulted in microglial activation and neuroinflammation in the PVN [58•]. Likewise, neuroinflammation was accompanied by changes in the brain RAS components in this study. Conversely, all these detrimental changes in the PVN were reversed on treatment with minocycline, an antibiotic that is known to inhibit microglial activation [58•]. We have also established a direct neuronal connection between the PVN and the right-ventricle of the heart using retrograde tracing with pseudo rabies virus (PRV). This is an important finding, which suggests that modulation of the PVN can affect right-sided heart function.
Possible Therapeutic Strategies
It is possible that treatments which directly target the neuroinflammatory pathways in the brain can protect against PH and associated pathology. In this regard, TNF-α-neutralizing antibodies (infliximab or etanercept) can be of potential therapeutic use. As indicated earlier, administration of minocycline, an inhibitor of microglial activation can also render beneficial effects against PH. All these drugs have been previously shown to be effective in reducing neuroinflammation in several disease models, and are currently being evaluated in the clinics. We believe that these drugs have the potential to treat PH as well, and must be evaluated in relevant experimental models of PH.
Conclusion
We are at a very nascent, but exciting stage of identifying the role and involvement of microglial activation/neuroinflammation in the pathogenesis of PH. Much of the historical PH research has focused primarily on understanding endothelial dysfunction, particularly the importance of an imbalance between endothelial-derived vasoconstrictors and vasodilators. Certainly, these endeavors have been fruitful in terms of target identification and validation, with subsequent development of several drugs that successfully modify the prostacyclin, NO, and endothelin signaling pathways. Despite these currently available therapies, PH still remains an incurable disease with high mortality rates, underscoring the need for identifying novel targets and drugs. A better understanding of the disease pathology is fundamental towards achieving this goal. The significance of systemic and pulmonary inflammation has been well recognized. However, the role of neuroinflammation in the PH pathogenesis has yet to be dissected. Learning about how inflammatory processes are induced within the brain and the mechanisms by which these responses ultimately contribute to the development and progression of PH will help us develop novel therapeutics. Though we have provided some preliminary data in support of neuroinflammation in PH, and propose an interaction between lungs, brain, and right ventricle of the heart (Fig. 1), much more research needs to be done to exploit this concept for the development of an effective therapy. Although several questions need to be addressed, some of the more pressing ones are the following: i) is neuroinflammation a cause or consequence of PH; ii) are there specific regions of the brain associated with microglial activation and neuroinflammation; iii) what are the mechanisms that regulate neuroinflammation; iv) do these mechanisms involve the SNS and RAS or are they neurogenic independent; and v) can therapeutic targets of microglial activation/migration attenuate or restore pathological alterations associated with PH?
Fig. 1.
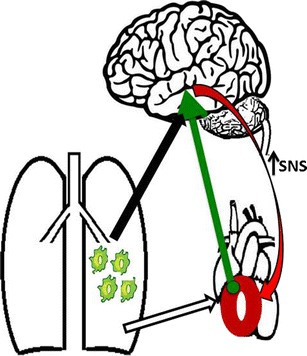
Working model: Interaction between lung, brain, and right ventricle of the heart in PH. Augmented inflammatory cells in the lung may affect the brain PVN (black arrow) and activate microglia. Activated microglia increase sympathetic nerve activity, contributing to PH pathology. Importantly, PVN neurons project onto the right ventricle of the heart (green arrow) and increase sympatho-excitation (red arrow), which could lead to right-sided heart hypertrophy and heart failure. Part of right ventricle hypertrophy is due to elevated pulmonary arterial pressure, which increases right ventricle workload (white arrow). Together, microglia activation leads to increased SNS activity to the right ventricle of the heart, inducing right heart hypertrophy, deteriorating cardiac function, resulting in heart failure and ultimately death
Acknowledgement
This work was supported by N.I.H Grants HL 102033 and HL 056921 (awarded to MKR and MJK), and SDG from the AHA (awarded to VS).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
None
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of the Topical Collection on Hypertension and the Brain
Aline M. Hilzendeger and Vinayak Shenoy contributed equally to this work.
References
Papers of particular interest, published recently, have been highlighted as: • Of major importance
- 1.•.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6):1219–1263. doi: 10.1183/09031936.00139009. [DOI] [PubMed] [Google Scholar]
- 2.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26(1):81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 3.Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, Imamura S, et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J. 2008;72(1):127–133. doi: 10.1253/circj.72.127. [DOI] [PubMed] [Google Scholar]
- 4.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121(24):2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dellegrottaglie S, Garcia-Alvarez A, Guarini P, Perrone-Filardi P, Fuster V, Sanz J. Prevalence and severity of ventricular dysfunction in patients with HIV-related pulmonary arterial hypertension. Heart Lung. 2014;43(3):256–261. doi: 10.1016/j.hrtlng.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Beck WC. Hospital asepsis and the advance of medical instrumentation. Med Instrum. 1978;12(3):148. [PubMed] [Google Scholar]
- 7.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122(9):920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 8.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114(17):1883–1891. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 9.Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D4–12. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jasiewicz M, Kowal K, Kowal-Bielecka O, Knapp M, Skiepko R, Bodzenta-Lukaszyk A, et al. Serum levels of CD163 and TWEAK in patients with pulmonary arterial hypertension. Cytokine. 2014;66(1):40–45. doi: 10.1016/j.cyto.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 11.Tedgui A. Focus on inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):958–959. doi: 10.1161/ATVBAHA.111.227355. [DOI] [PubMed] [Google Scholar]
- 12.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer's disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis. 2010;19(1):355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 15.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.•.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 18.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- 19.Qian L, Hong JS, Flood PM. Role of microglia in inflammation-mediated degeneration of dopaminergic neurons: neuroprotective effect of interleukin 10. J Neural Transm Suppl. 2006;70:367–371. doi: 10.1007/978-3-211-45295-0_56. [DOI] [PubMed] [Google Scholar]
- 20.Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1(1):9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 22.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 23.Old EA, Malcangio M. Chemokine mediated neuron-glia communication and aberrant signalling in neuropathic pain states. Curr Opin Pharmacol. 2012;12(1):67–73. doi: 10.1016/j.coph.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 24.de Kloet AD, Krause EG, Shi PD, Zubcevic J, Raizada MK, Sumners C. Neuroimmune communication in hypertension and obesity: a new therapeutic angle? Pharmacol Ther. 2013;138(3):428–440. doi: 10.1016/j.pharmthera.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87(1):10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura Y, Si QS, Kataoka K. Lipopolysaccharide-induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci Res. 1999;35(2):95–100. doi: 10.1016/S0168-0102(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 27.Graeber MB, Li W, Rodriguez ML. Role of microglia in CNS inflammation. FEBS Lett. 2011;585(23):3798–3805. doi: 10.1016/j.febslet.2011.08.033. [DOI] [PubMed] [Google Scholar]
- 28.Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77(1):10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 29.Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013;2013:480739. doi: 10.1155/2013/480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young KC, Hussein SM, Dadiz R, de Mello D, Devia C, Hehre D, et al. Toll-like receptor 4-deficient mice are resistant to chronic hypoxia-induced pulmonary hypertension. Exp Lung Res. 2010;36(2):111–119. doi: 10.3109/01902140903171610. [DOI] [PubMed] [Google Scholar]
- 31.Bauer EM, Chanthaphavong RS, Sodhi CP, Hackam DJ, Billiar TR, Bauer PM. Genetic deletion of toll-like receptor 4 on platelets attenuates experimental pulmonary hypertension. Circ Res. 2014;114(10):1596–1600. doi: 10.1161/CIRCRESAHA.114.303662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC. Inflammatory cytokines in pulmonary hypertension. Respir Res. 2014;15:47. doi: 10.1186/1465-9921-15-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(9):897–908. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 34.Gerasimovskaya E, Kratzer A, Sidiakova A, Salys J, Zamora M, Taraseviciene-Stewart L. Interplay of macrophages and T cells in the lung vasculature. Am J Physiol Lung Cell Mol Physiol. 2012;302(10):L1014–1022. doi: 10.1152/ajplung.00357.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104(27):11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y, et al. Distinct contribution of IL-6, TNF-alpha, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest. 2004;114(4):582–588. doi: 10.1172/JCI200421795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151(5):1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 38.Ross DJ, Strieter RM, Fishbein MC, Ardehali A, Belperio JA. Type I immune response cytokine-chemokine cascade is associated with pulmonary arterial hypertension. J Heart Lung Transplant. 2012;31(8):865–873. doi: 10.1016/j.healun.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Chu JW, Kao PN, Faul JL, Doyle RL. High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension. Chest. 2002;122(5):1668–1673. doi: 10.1378/chest.122.5.1668. [DOI] [PubMed] [Google Scholar]
- 40.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26(6):1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 41.Bhargava A, Kumar A, Yuan N, Gewitz MH, Mathew R. Monocrotaline induces interleukin-6 mRNA expression in rat lungs. Heart Dis. 1999;1(3):126–132. [PubMed] [Google Scholar]
- 42.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6. doi: 10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104(2):236–244. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.•.Asosingh K, Farha S, Lichtin A, Graham B, George D, Aldred M, et al. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood. 2012;120(6):1218–1227. doi: 10.1182/blood-2012-03-419275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.•.Shenoy V, Gjymishka A, Jarajapu YP, Qi Y, Afzal A, Rigatto K, et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am J Respir Crit Care Med. 2013;187(6):648–657. doi: 10.1164/rccm.201205-0880OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Callaghan EL, Choong YT, Jancovski N, Allen AM. Central angiotensinergic mechanisms associated with hypertension. Auton Neurosci. 2013;175(1–2):85–92. doi: 10.1016/j.autneu.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 47.Ferreira AJ, Santos RA, Bradford CN, Mecca AP, Sumners C, Katovich MJ, et al. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension. 2010;55(2):207–213. doi: 10.1161/HYPERTENSIONAHA.109.140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216(2):R1–R17. doi: 10.1530/JOE-12-0341. [DOI] [PubMed] [Google Scholar]
- 49.Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 2008;23:187–193. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platten M, Youssef S, Hur EM, Ho PP, Han MH, Lanz TV, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U S A. 2009;106(35):14948–14953. doi: 10.1073/pnas.0903958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyoshi M, Miyano K, Moriyama N, Taniguchi M, Watanabe T. Angiotensin type 1 receptor antagonist inhibits lipopolysaccharide-induced stimulation of rat microglial cells by suppressing nuclear factor kappaB and activator protein-1 activation. Eur J Neurosci. 2008;27(2):343–351. doi: 10.1111/j.1460-9568.2007.06014.x. [DOI] [PubMed] [Google Scholar]
- 52.Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36(4):857–870. doi: 10.1038/npp.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sironi L, Gelosa P, Guerrini U, Banfi C, Crippa V, Brioschi M, et al. Anti-inflammatory effects of AT1 receptor blockade provide end-organ protection in stroke-prone rats independently from blood pressure fall. J Pharmacol Exp Ther. 2004;311(3):989–995. doi: 10.1124/jpet.104.072066. [DOI] [PubMed] [Google Scholar]
- 54.Jung KH, Chu K, Lee ST, Kim SJ, Song EC, Kim EH, et al. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral hemorrhage. J Pharmacol Exp Ther. 2007;322(3):1051–1058. doi: 10.1124/jpet.107.120097. [DOI] [PubMed] [Google Scholar]
- 55.de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, Francois C, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(8):780–789. doi: 10.1164/rccm.201203-0411OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abraham WT, Raynolds MV, Badesch DB, Wynne KM, Groves BM, Roden RL, et al. Angiotensin-converting enzyme DD genotype in patients with primary pulmonary hypertension: increased frequency and association with preserved haemodynamics. J Renin Angiotensin Aldosterone Syst. 2003;4(1):27–30. doi: 10.3317/jraas.2003.003. [DOI] [PubMed] [Google Scholar]
- 57.Chung WK, Deng L, Carroll JS, Mallory N, Diamond B, Rosenzweig EB, et al. Polymorphism in the angiotensin II type 1 receptor (AGTR1) is associated with age at diagnosis in pulmonary arterial hypertension. J Heart Lung Transplant. 2009;28(4):373–379. doi: 10.1016/j.healun.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.•.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56(2):297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]