

Abstract
Endocannabinoids (EC), particularly anandamide (AEA), released constitutively in pain pathways might be accountable for the inhibitory effect on nociceptors. Pathogenesis of neuropathic pain may reflect complex remodeling of the dorsal root ganglia (DRGs) and spinal cord EC system. Multiple pathways involved both in the biosynthesis and degradation of AEA have been suggested. We investigated the local synthesis and degradation features of AEA in DRGs and spinal cord during the development and maintenance of pain in a model of chronic constriction injury (CCI). All AEA synthesis and degradation enzymes are present on the mRNA level in DRGs and lumbar spinal cord of intact as well as CCI-treated animals. Deregulation of EC system components was consistent with development of pain phenotype at days 3, 7, and 14 after CCI. The expression levels of enzymes involved in AEA degradation was significantly upregulated ipsilateral in DRGs and spinal cord at different time points. Expression of enzymes of the alternative, sPLA2-dependent and PLC-dependent, AEA synthesis pathways was elevated in both of the analyzed structures at all time points. Our data have shown an alteration of alternative AEA synthesis and degradation pathways, which might contribute to the variation of AEA levels and neuropathic pain development.
1. Background
The development of neuropathic pain after nerve injury occurs when peripheral nerve fibers are damaged or dysfunctional, which results in incorrect signals being sent to the brain and loss of afferent sensory function with typical features such as allodynia and hyperalgesia [1, 2]. Chronic pain serves no protective biological function unlike a symptom of a disease process, and there is a strong need to identify novel therapeutic targets [3]. Among the many suggested strategies to treat neuropathic pain, cannabinoids have the potential to become analgesic targets for drug development. Indeed cannabinoid agonists suppress neuropathic symptoms in animal models of neuropathic pain evoked by chronic constriction injury (CCI) to the sciatic nerve [4–6] or spinal nerve ligation [7–10]. Yet, this therapeutic intervention is also associated with a number of adverse effects, including sedation, motor impairment, and cognitive impairment. Therefore, an alternative approach to target endocannabinoid (EC) signaling has been proposed [11], which may provide a more effective strategy in relieving neuropathic pain [12–14]. Anandamide (AEA), the first discovered and best studied EC, acts via cannabinoid 1 (CB1) and cannabinoid 2 (CB2) receptors in a manner similar to naturally derived and synthetic cannabinoid agonists, but it may also modulate nociception via other receptors, that is, transient receptor potential vanilloid 1 (TRPV1) [15–18].
ECs are present in multiple pain-modulating regions throughout the central nervous system (CNS), including the dorsal horn of the spinal cord and the dorsal root ganglia (DRGs), where their levels are modified by acute nociceptive stimuli and stress [19–23]. Tissue concentrations of AEA in the spinal cord become altered [14, 23–25] as an adaptive response to neuropathic pain, which further confirms the significant role of the AEA in chronic pain development.
Previous reports suggest that AEA is synthesized “on demand” [26] in regions of cellular stress (such as injured tissues or nerves). Although AEA is mainly generated from phospholipid precursor N-arachidonoylphosphatidylethanolamine (NAPE) through hydrolysis by a N-arachidonoylphosphatidylethanolamine phospholipase D (NAPE-PLD) [27] in a Ca2+-sensitive manner, recent evidence [28] indicates the existence of two parallel, additional, phospholipase C (PLC) and secreted phospholipase A2 (sPLA2)—catalyzed, Ca2+-independent pathways. The PLC pathway involves PLC itself and two other enzymes with parallel activity: protein tyrosine phosphatase non-receptor type 22 (PTPN22) and phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1 (INPP5D) [29, 30]. The sPLA2 pathway also includes the α/β hydrolase domain containing protein 4 (ABHD4) and glycerophosphodiesterphosphodiesterase 1 (GDE1) [31] (Figure 1). Similarly, multiple pathways involved in the degradation of AEA have been suggested [27, 32, 33]. Due to the efficient enzymatic degradation mainly by fatty acid amide hydrolase (FAAH) and also cyclooxygenase 2 (COX-2) as well as arachidonate lipoxygenases 12 and 15 (LOX-12/15), locally released ECs have a short half-life [34, 35]; thus, corrective relevance is limited.
Figure 1.
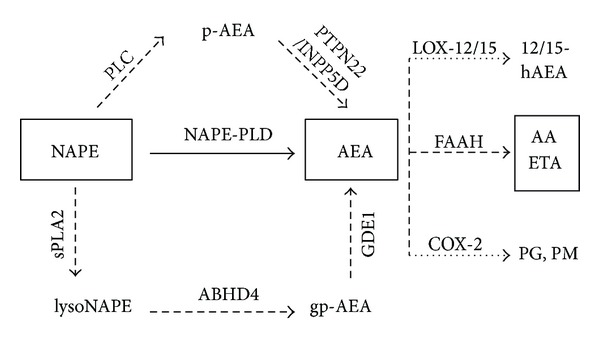
Schematic illustration of parallel AEA synthesis and degradation pathways. NAPE-PLD represents a Ca2+-dependent route of AEA formation (—), while other enzymes (- - -) act in a Ca2+-insensitive manner. Main route of AEA degradation is hydrolysis by FAAH (- - -). High AEA tissue concentration triggers parallel catabolic pathways through LOX-12/15 and COX-2 enzymes (⋯).AEA: anandamide, NAPE: N-acylphosphatidylethanolamine, NAPE-PLD: N-acylphosphatidylethanolamine phospholipase D, GDE1: glycerophosphodiester phosphodiesterase 1, ABHD4: α/β hydrolase domain containing protein 4, PTPN22: protein tyrosine phosphatase nonreceptor type 22, sPLA2: soluble phospholipase A2, INPP5D: inositol 5-phosphatase, PLC: phospholipase C, FAAH: fatty acid amide hydrolase, COX-2: cycloxygenase 2, LOX-12: arachidonate 12-lipoxygenase, LOX-15: arachidonate 15-lipoxygenase, gp-AEA: glycerophosphoanandamide, p-AEA: phospho-anandamide, lysoNAPE: lyso-N-acylphosphatidylethanolamine, AA: arachidonic acid, ETA: ethanolamine, PG: prostaglandins, PM: prostamides, and 12/15-hAEA: 12/15-hydroxyanandamide.
Deregulation of the EC system underlies several neurological disorders including chronic pain; thus, there is a strong need for detailed characterization of the changes in the EC system during the development of neuropathic pain. Therefore, the aim of our studies was to investigate the role of multiple AEA production (both in Ca2+-dependent and Ca2+-independent manners) and degradation pathways as well as the possible consequences of altering its signaling during the development of neuropathic pain. We examined mRNA expression of EC system elements in DRGs and lumbar spinal cord, as both of these structures play a critical role in the integration and modulation of nociceptive signals from the peripheral nervous system. Additionally, changes at the site of nerve injury and in DRGs may give rise to the perception of pain in conditions such as neuropathy, which modifies the transmission of pain from peripheral tissues through the spinal cord to higher centers of the brain.
2. Results
2.1. Rats Subjected to CCI Showed Signs of Allodynia on the Operated Paw at 3, 7, and 14 Days after Induction of Injury
Presurgery thresholds reached the cut-off values for both thermal and mechanical allodynia, for both animals which undergo CCI procedure and intact. Cut-off values were also reached for intact animals in all tested time points. Neuropathic animals developed thermal allodynia as indicated by the cold plate test thermal withdrawal latency observed at day 3 after CCI (18.88 ± 2.88 s; ∗P < 0.01; Figure 2(a)) and progressed at days 7 and 14 (12.85 ± 1.61 s, ∗P < 0.001 and 16.63 ± 1.64 s, ∗P < 0.001, resp.; Figure 2(a)). Mechanical allodynia was observed as decreased mechanical withdrawal threshold ipsilateral to injury in all time points tested (18.18 ± 1.88 g, ∗P < 0.001; 11.68 ± 1.54, ∗P < 0.001; 12.75 ± 1.40, ∗P < 0.001; 3, 7, and 14 days after sciatic nerve injury, resp.; Figure 2(b)). In both tests, allodynia was maximal at day 7. The thresholds to mechanical stimulation were unaffected in contralateral paws and sham-operated rats (data not shown).
Figure 2.
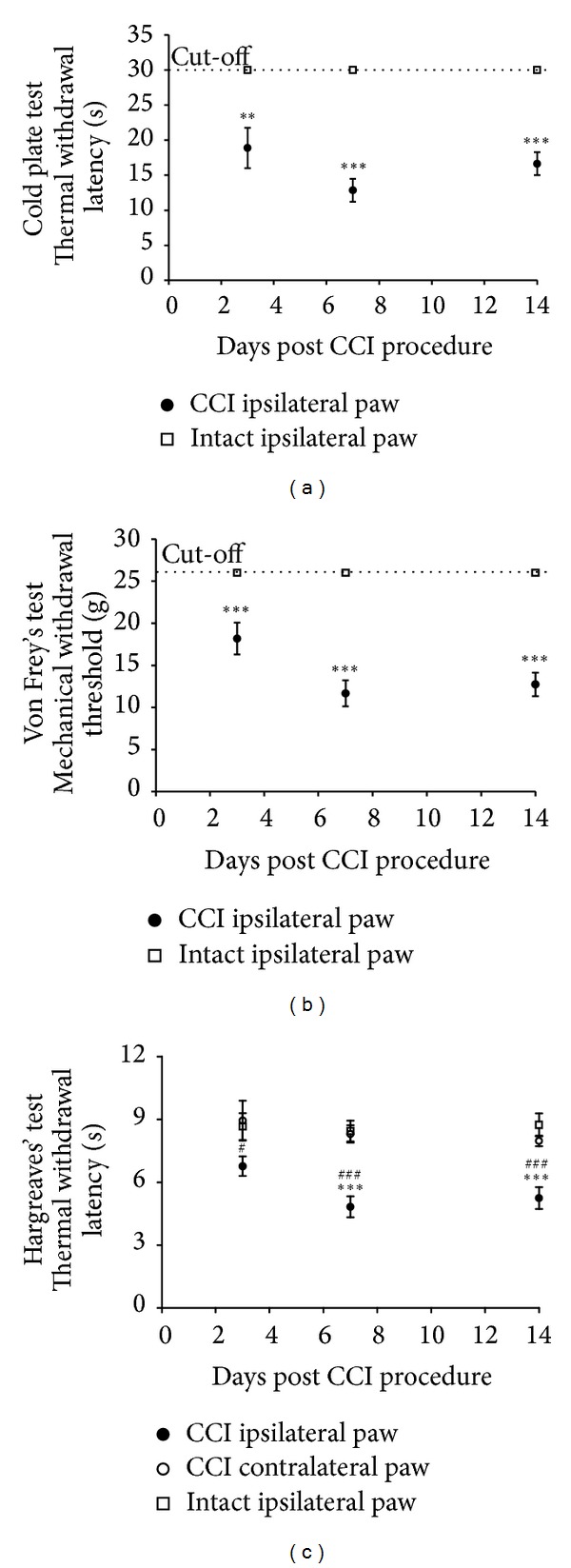
Thermal (a) and mechanical (b) allodynia and thermal hyperalgesia (c) in intact or CCI animals at 3, 7, and 14 days after CCI of the sciatic nerve. Thermal allodynia and hyperalgesia were measured as thermal withdrawal latency in seconds (mean ± SEM) and mechanical allodynia as mechanical withdrawal threshold in grams (mean ± SEM). Cut-off values were 30 s, 26 g, and 20 s for the cold plate, von Frey's and Hargreaves' tests, respectively. Statistical analysis was performed using a one-way ANOVA followed by Bonferroni post hoc tests. Values with P < 0.05 were considered significant. ∗ denotes significant difference versus intact and # versus contralateral paw.
2.2. CCI Rats Exhibited a Significant Decrease in Thermal Hyperalgesia Thresholds after Nerve Ligation
Thermal ipsi-/contralateral withdrawal latency did not significantly change in intact animals or in the contralateral side of the CCI operated rats in all time points tested (Figure 2(c)). CCI to the sciatic nerve reduced ipsilateral thermal withdrawal latency compared with both the intact animals and the contralateral paw of CCI animals at all-time points tested. It reached the lowest values at day 7 after the procedure (6.77 ± 0.46 s, # P < 0.05; 4.83 ± 0.50 s, ∗# P < 0.001; 5.25 ± 0.52 s, ∗# P < 0.001; at day 3, 7 and 14 after CCI, resp.; Figure 2(c)).
2.3. Alteration of CB2 Expression Was Observed in DRGs (L4-L6) and Lumbar Spinal Cord during the Development of Neuropathic Pain, While No Changes Were Observed in the Expression of CB1 and TRPV1
CB1, CB2, and TRPV1 receptor transcripts were detected both in the DRGs and in the lumbar spinal cord of intact and neuropathic rats. Higher levels of Cnr1 (CB1) receptor transcripts were detected contralateral to the injury in the DRGs throughout the development of neuropathic pain in comparison with the ipsilateral side (1.31 ± 0.11, # P < 0.05; 1.78 ± 0.14, ∗# P < 0.001; 1.35 ± 0.09, # P < 0.01; fold change at days 3, 7, and 14 in CCI rats, resp.; Figure 3(a)). Although there were significant differences in Cnr1 expression in ipsilateral versus contralateral DRGs at respective days, only day 7 was characterized by a significant upregulation in the contralateral versus intact. No significant changes in Cnr1 mRNA levels were observed in the lumbar spinal cord during development of neuropathic pain (Figure 3(b)). Cnr2 (CB2) mRNA expression levels were altered in both structures (Figures 3(c) and 3(d)). In DRGs, an elevated level of Cnr2 transcript was observed ipsilateral at day 7 after CCI procedure (1.78 ± 0.11, ∗P < 0.01; fold change at day 7 in CCI rats). Expression of Cnr2 contralateral to the injury decreased significantly 14 days after sciatic nerve injury in comparison to earlier time points ($ P < 0.05). The strongest upregulation of Cnr2 transcript was observed, exclusively ipsilateral to the injury, in the lumbar spinal cord at all-time points tested (6.53 ± 1.42, ∗P < 0.01; 7.43 ± 1.49 ∗P < 0.01# P < 0.05; 6.30 ± 1.93 ∗P < 0.05; fold change ipsilateral at days 3, 7, and 14 in CCI rats, resp.; Figure 3(d)). Expression of Trpv1 mRNA was not altered in the examined structures during the development of neuropathic pain at days 3, 7, or 14 in comparison to intact animals (Figures 3(e) and 3(f)). However, an alterations in the DRGs at day 7 versus day 3 was observed ($ P < 0.01).
Figure 3.
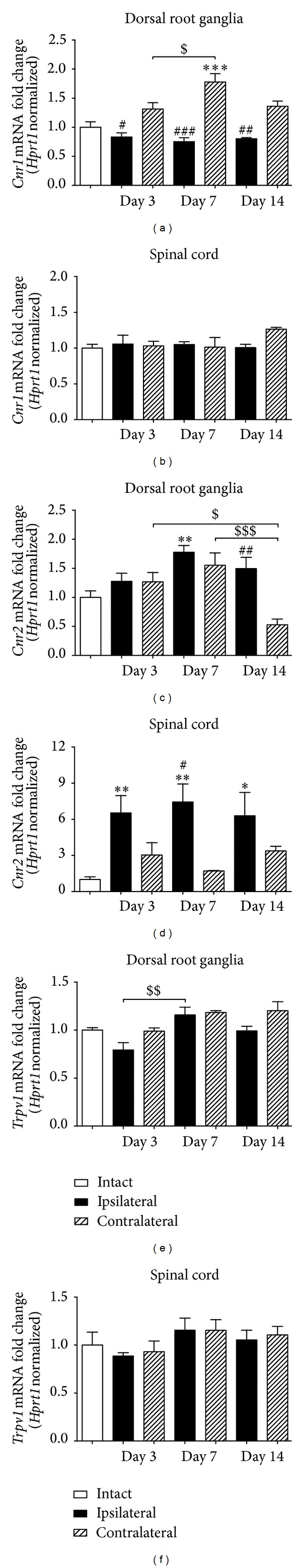
Results of qPCR analysis of Cnr1, Cnr2, and Trpv1 gene expression levels in the L4-L6 dorsal root ganglia and in the dorsal part of the lumbar spinal cord during the development of neuropathic pain in CCI rats. Samples were collected at 3, 7, and 14 days after CCI procedure. Data are presented as the mean ± SEM and represent normalized averages derived from 4–6 samples for each group. Results are presented as a fold change normalized to the expression of a reference gene Hprt1, compared to the intact animals. Statistical analysis was performed using a one-way ANOVA followed by Bonferroni post hoc tests. Values with P < 0.05 were considered significant. ∗ denotes significant differences versus intact, # versus contralateral side, and $ versus indicated bar.
2.4. Upregulation of Alternative Synthesis Enzymes of AEA in DRGs and Lumbar Spinal Cord as a Consequence of Sciatic Nerve Injury
There were no significant changes in the expression levels of Napepld (NAPE-PLD) mRNA, the main AEA synthesizing enzyme, in DRGs or in the lumbar spinal cord (Figures 4(a) and 4(b)). Neuropathic pain led to an upregulation of mRNA encoding enzymes of alternative synthesis pathways in L4-L6 DRGs and the lumbar spinal cord (Figure 5). Pla2g2a (sPLA2) transcript levels were elevated in DRGs ipsilateral to the injury 7 days after CCI compared with the intact animals (1.40 ± 0.17, # P < 0.01; 1.69 ± 0.07, ∗P < 0.01# P < 0.001; 1.40 ± 0.06, # P < 0.01; fold change ipsilateral at days 3, 7, and 14 in CCI rats, resp.; Figure 5(a)) and in all-time points in the lumbar spinal cord (2.15 ± 0.14, ∗# P < 0.001; 1.94 ± 0.06, ∗# P < 0.001; 1.87 ± 0.04, ∗P < 0.001# P < 0.01; fold change at days 3, 7 and 14 after sciatic nerve ligation, resp.; Figure 5(g)). The abundance of Inpp5d (INPP5D) mRNA was increased ipsilateral to the injury in all tested time points in the lumbar spinal cord exclusively (2.83 ± 0.18, ∗# P < 0.001; 2.36 ± 0.10, ∗# P < 0.001; 2.51 ± 0.05, ∗# P < 0.001; fold change ipsilateral at days 3, 7, and 14 in CCI rats, resp.; Figure 5(l)). The mRNA levels of other enzymes involved in AEA synthesis did not significantly change in the measured time points after CCI of the sciatic nerve.
Figure 4.
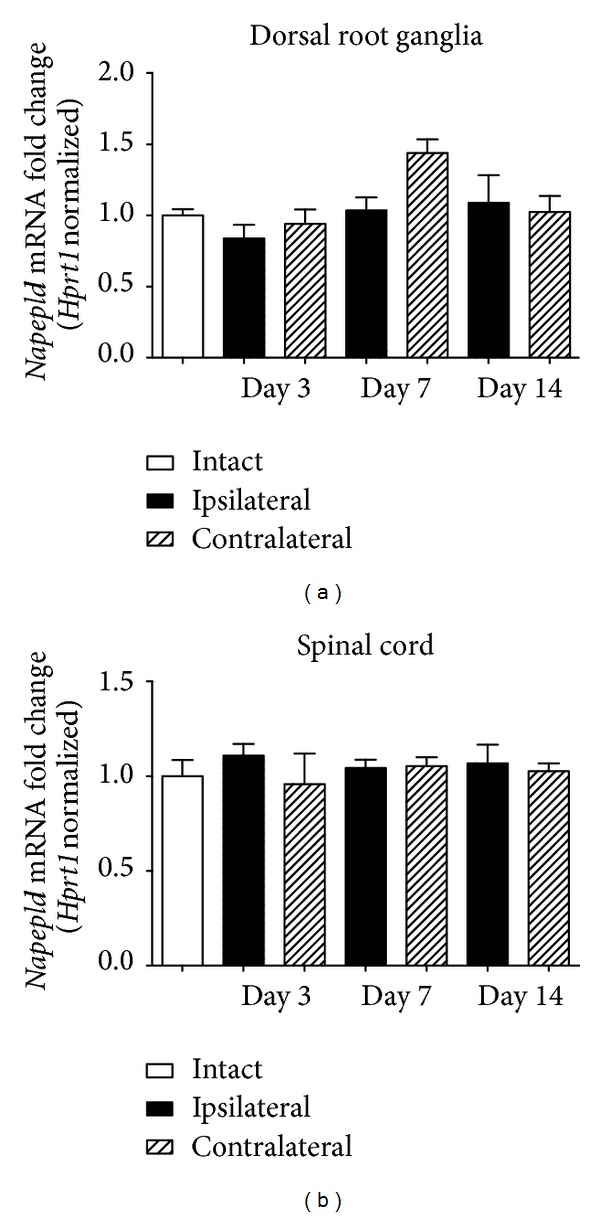
Expression of Napepld mRNA in L4-L6 dorsal root ganglia and in the dorsal part of the lumbar spinal cord during the development of neuropathic pain in CCI rats. Samples were collected at 3, 7, and 14 days after CCI procedure. Data are presented as the mean ± SEM and represent normalized averages derived from 4–6 samples for each group. Results are presented as a fold change normalized to the expression of a reference gene Hprt1, compared to the intact animals. Statistical analysis was performed using a one-way ANOVA followed by Bonferroni post hoc tests. Values with P < 0.05 were considered significant. ∗ denotes significant differences versus intact, # versus contralateral side, and $ versus indicated bar.
Figure 5.

Gene expression analysis of enzymes involved in alternative pathways of anandamide synthesis—Pla2g2a, Abdh4, Gde1, Plcb1, Ptpn22, and Inpp5d in L4-L6 dorsal root ganglia and in the dorsal part of the lumbar spinal cord during the development of neuropathic pain in CCI rats. Samples were collected at 3, 7, and 14 days after CCI procedure. Data are presented as the mean ± SEM and represent normalized averages derived from 4–6 samples for each group. Results are presented as a fold change normalized to the expression of a reference gene Hprt1, compared to the intact animals. Statistical analysis was performed using a one-way ANOVA followed by Bonferroni post hoc tests. Values with P < 0.05 were considered significant. ∗ denotes significant differences versus intact, # versus contralateral side, and $ versus indicated bar.
2.5. Alteration in the Expression of Main and Alternative Enzymes for AEA Degradation in Tested Tissues of CCI Rats during the Development of Neuropathic Pain
Analysis of Faah (FAAH) transcript levels revealed that the main AEA degradation enzyme showed no significant changes in expression in the L4-L6 DRGs during the development of neuropathic pain (Figure 6(a)). Alterations of Faah mRNA levels were limited to the ipsilateral side of the lumbar spinal cord at days 3, 7, and 14 after CCI (1.73 ± 0.23, ∗P < 0.001; 2.54 ± 0.36, ∗P < 0.001; 2.57 ± 0.20, ∗P < 0.001# P < 0.05; fold change at respective days; Figure 6(b)). Ptgs2 (COX2) transcript levels were altered in DRGs both at the ipsilateral and contralateral side of the injury at different time points. The highest levels of transcript were observed at day 7 after injury (3.50 ± 0.25, ∗P < 0.001; 3.40 ± 0.42, ∗P < 0.001; Figure 6(c)). Ptgs2 expression declined to baseline at day 14 after CCI surgery. We observed no appreciable changes in the abundance of Ptgs2 in lumbar spinal cord, except for the Ptgs2 ipsilateral upregulation at day 3 (1.86 ± 0.21, ∗P < 0.001; fold change ipsilateral at day 3 in CCI rats; Figure 6(d)). Similar patterns of gene expression levels of the major lipoxygenases (Alox12, Alox15) in neuropathic rats were observed in the DRGs and the lumbar spinal cord (Figures 6(e)–6(h)). Alox12 (LOX-12) mRNA levels were significantly upregulated ipsilateral to the injury in DRGs from day 7 after CCI (1.87 ± 0.17, ∗P < 0.001# P < 0.01$ P < 0.001; 1.63 ± 0.09, ∗P < 0.001$ P < 0.01; fold change at days 7 and 14 in CCI rats, resp.; Figure 6(e)). Elevation of Alox12 transcript levels in lumbar spinal cord was observed solely ipsilateral to the injury at all-time points measured (1.65 ± 0.06, ∗P < 0.001# P < 0.01; 1.52 ± 0.08, ∗P < 0.01; 1.74 ± 0.18, ∗P < 0.001# P < 0.05; fold change ipsilateral at days 3, 7, and 14 in CCI rats, resp.; Figure 6(f)). Elevated levels of Alox15 (LOX-15) mRNA were observed ipsilateral at days 7 and 14 after sciatic nerve injury in both of the assayed tissues (2.04 ± 0.28, ∗P < 0.01$ P < 0.05; 2.64 ± 0.27, ∗P < 0.001# P < 0.001$ P < 0.001; fold change DRGs ipsilateral at day 7 and 14 in CCI rats, resp., and 2.26 ± 0.36, ∗P < 0.001# P < 0.05$ P < 0.001; 2.49 ± 0.25, ∗P < 0.001$ P < 0.001; fold change lumbar spinal cord ipsilateral at day 7 and 14 in CCI rats, resp; Figures 6(g) and 6(h)).
Figure 6.
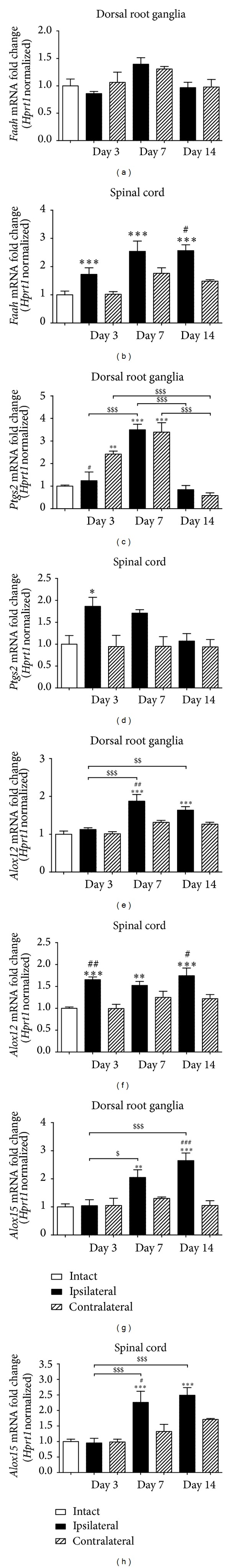
Expression of main anandamide degradation enzymes—Faah, Ptgs2, Alox12, and Alox15 in L4-L6 dorsal root ganglia and in the dorsal part of the lumbar spinal cord during the development of neuropathic pain in CCI rats. Samples were collected at 3, 7, and 14 days after CCI procedure. Data are presented as the mean ± SEM and represent normalized averages derived from 4–6 samples for each group. Results are presented as a fold change normalized to the expression of a reference gene Hprt1, compared to the intact animals. Statistical analysis was performed using a one-way ANOVA followed by Bonferroni post hoc tests. Values with P < 0.05 were considered significant. ∗ denotes significant differences versus intact, # versus contralateral side, and $ versus indicated bar.
3. Discussion
A large number of research articles have demonstrated the efficacy of cannabinoids and modulators of the EC system in the alleviation of neuropathic pain in various animal models of surgically induced trauma, such as chronic constriction injury, partial sciatic nerve ligation, or spinal nerve ligation [14, 36]. Recent studies highlight the importance of alterations in the spinal and supraspinal EC levels in neuropathic rats [24] as well as the involvement of peripheral CB1/CB2 receptors in the antinociceptive effects of EC system modulation [37]. However, the exact mechanisms involved in the dynamic changes of EC concentrations in nervous tissues have never been investigated. Given the importance of the first-order neurons located in the DRGs and the spinal cord for pain sensation, in the present study, we investigated the putative AEA synthesizing and degradation enzymatic pathways in those structures. We reported for the first time changes in the expression of AEA metabolic enzymes at the DRG and spinal cord levels in a rat model of neuropathic pain.
In the rat CCI model of neuropathic pain, we evaluated pain behavior in three independent tests (Figure 2). We have determined that the nocifensive behavior in neuropathic animals: allodynia and hyperalgesia are accompanied by multiple changes in the expression of receptors and metabolic enzymes for AEA. ECs are produced on demand in regions of cellular stress, for example, in injured tissues during the development of neuropathic pain. Unfortunately, locally released ECs are rapidly broken down in the tissue, so their physiological effectiveness is limited. Therefore, the endogenous control of the EC system during chronic pain remains an important issue to study, and it might provide new insight into the possibilities of EC modulation. The present investigation has expanded the knowledge of endogenous control mechanisms by showing that parallel pathways of synthesis and degradation of AEA become activated in response to the development of neuropathic pain and in consequence may influence levels of AEA in effected tissues. Studies on the endogenous levels of AEA in neuronal tissues during the development of chronic pain have yielded conflicting results in this regard. It was reported that the development of chronic pain was accompanied by a significant elevation of AEA levels at the spinal cord level [6, 24, 33, 38], although other studies showed no changes or even decreases in AEA concentration in different models of chronic pain [12, 22, 23, 25, 39, 40]. Several parallel pathways are suggested to contribute to the synthesis of AEA, among which the main occurs from its membrane precursor through cleavage by NAPE-PLD. It was reported that tissues from NAPE-PLD knockout mice exhibited enzymatic activity converting NAPE to AEA in a calcium-independent manner [41], suggesting the involvement of parallel biosynthetic pathways and supporting the theory that NAPE-PLD only makes a partial contribution to the biosynthesis of AEA [42]. Although some data showed the expression of enzymes of alternative pathways in neuronal tissues [43], comparison of expression profiles between control and neuropathic pain animals has never been performed. The present investigation has supplemented these observations by showing the upregulation of AEA synthetic enzymes in parallel pathways in a rat model of neuropathic pain (Figure 5). We confirmed that NAPE-PLD shows no alteration during the induction of pain (Figure 4) (as previously reported [40]). Therefore, we hypothesized that the variations in AEA levels are derived from disparities in the activity of alternative synthesis pathways. As a matter of fact, parallel pathways for AEA synthesis involving Ca2+-insensitive enzymes were elevated ipsilateral to the injury in both of the tissues examined in our studies (Figure 5). Additionally, PLC-dependent pathway activity was altered in the site of injury in the lumbar spinal cord exclusively (Figure 5). Our findings stress the importance of the activation of these pathways in the endogenous control of AEA levels during the development of chronic disorders.
Similar to the synthetic pathways, there is more than one degradation route of AEA. It has been assumed that AEA undergoes mainly FAAH-mediated hydrolysis. In the present study, we report strong upregulation of FAAH transcripts ipsilateral to the injury on the spinal cord level (Figure 6). This finding supports our previous studies, which were focused on investigating the role of FAAH inhibition in the alleviation of pain behavior through the endogenous elevation of AEA levels (for details see [13]). Yet, diminishing or eliminating the hydrolysis of AEA by FAAH would increase the probability that AEA might undergo alternative routes of metabolism, such as oxidation by fatty acid oxygenases that are known to act on endogenous arachidonic acid, namely, the members of the lipoxygenase (LOX) and cyclooxygenase (COX) [33, 44, 45] families. Herein, we also examined mRNA levels of LOX-12 and LOX-15 and showed an ipsilateral alteration of these enzymes in both the DRGs and lumbar spinal cord during the development of neuropathic pain (Figure 6). This result suggests that changes in LOX expression, as well metabolism of AEA via this pathway, may influence nociceptive processing. Moreover, LOX catabolism may lead to the production of active AEA metabolites, for example, 12/15-hydroxy-AEA, which may act via TRPV1 and/or PPARalfa receptors, which contribute to the modification of pain behavior [33, 46, 47]. AEA was also shown to serve as a substrate for COX-2 [48]. As a result, it is a precursor of prostaglandins and prostamides, which can induce neuroinflammation and can result in the attenuation of therapeutic benefits of FAAH inhibitors [49, 50]. Moreover, some oxidized forms of AEA might serve as FAAH inhibitors [51]. Because they can be formed in vivo, they might also play an important role in controlling AEA degradation and, as a consequence, its levels in tissues. As in our studies, both LOX-12/15 and COX-2 levels were elevated, the effects of AEA metabolites should be considered based on the estimation of the benefits of pharmacological FAAH enzyme inhibition.
The analgesic effects produced by the activation of CB1 receptors have been well described and extensively reviewed [14, 52]. However, the broad distribution of CB1 receptors in the central nervous system emphasizes both their therapeutic effects, such as analgesia, as well as their side effects. Although it was reported by many authors that CB1 receptor expression is increased in the chronic pain conditions [53, 54], there is evidence showing no effect of pain development on CB1 receptor level alterations [55, 56], which is consistent with our studies. Due to the side effects mediated by CB1 receptor, it is clinically relevant to focus on the peripherally restricted CB1 agonists [57] as well as on signaling through the CB2 receptor. In our studies on neuropathic pain development, the CB2 receptor rather than the CB1 receptor showed significant upregulation, which is consistent with results obtained by other authors [58–60] and this finding might contribute to the hypothesis of the involvement of CB2 receptors in the attenuation of nociceptive response in models of neuropathic pain [61, 62]. Moreover, studies showed that the administration of CB1/CB2 agonist can attenuate pain response, although no change in expression of those receptors was observed [63]. This result might suggest more complex interactions between cannabinoid receptors during the development chronic pain dependent on the various features of the animal model used. Because AEA might act on different molecular targets, we examined the expression of the TRPV1 receptor at the transcript level as well. Although we observed no changes in the transcript levels of this receptor, its activity depends on phosphorylation and dephosphorylation processes, which are crucial for its function and act to decrease or increase channel activity, respectively [64].
4. Conclusions
The present investigation has expanded the knowledge of EC system modulation by showing that all AEA synthesis and degradation enzymes are present in DRGs and lumbar spinal cord of intact as well as neuropathic animals. Alterations in a variety of synthesis and degradation enzymes of AEA illustrate the flexibility of the EC system. This may explain why genetic ablation or pharmacological inhibition of only one of its metabolic pathways does not cause a substantial change in the cellular levels of AEA and may lead to unexpected behavioral effects. By combining behavioral tests and measuring the transcript levels of metabolic enzymes of AEA, we provide new insight into the involvement of the EC system in the development of neuropathic pain. Because therapies using ECs hold substantial promise, an understanding of the plasticity of the EC system is crucial and should be further investigated.
5. Methods
5.1. Animals
Male Wistar rats (Charles River, Hamburg, Germany), initially weighing 225–250 g, were used for all experiments. Animals were housed five per cage under a standard 12/12 h light/dark cycle (lights on at 08:00 h) with food and water available ad libitum. All animals were allowed to acclimatize to their holding cages for 3 to 4 days before any behavioral or surgical procedures were carried out. All experiments were conducted during the light cycle between 8:00 and 13:00. All experiments were performed according to the NIH Guide for the Care and Use of Laboratory Animals with recommendations by IASP [65] and were approved by the Local Bioethics Committee. Care was taken to implement the 3 Rs rule (replacement, reduction, and refinement) both to reduce the number of animals used and the suffering during the experiments. Different sets of animals were used for behavioral and biochemical studies to avoid changes in expression levels caused by thermal and mechanical stimulation. Results obtained in our research group [66] as well as those reported by others [67] showed no significant differences between sham operated group and intact (naive) animals in allodynia and hyperalgesia thresholds in development of neuropathic pain. Moreover Paszcuk et al. reported no significant differences in expression of EC system components in sham versus intact animals [54]. Therefore, respecting 3 R policy in laboratory animals use, we decided to compare only intact (naive) and neuropathic pain groups in our biochemical experiments.
5.2. Sciatic Nerve Surgery
Peripheral neuropathy was induced by chronic constriction injury (CCI) as described by Bennett and Xie [68]. The sciatic nerve injury was performed under sodium pentobarbital anesthesia (60 mg/kg, i.p.). The biceps femoris and the gluteus superficialis were separated, and the right sciatic nerve was exposed. Proximal to the sciatic trifurcation, approximately 7 mm of nerve was freed from the adhering tissue, and the injury was produced by tying four loose ligatures (4/0 silk, 1 mm spacing) around the sciatic nerve until they elicited a brief twitch in the respective hind limbs. This twitch prevented us from applying a ligation that was too strong. The total length of nerve affected was 5-6 mm. No procedure was conducted on the control animals.
5.3. Nociceptive Behavior
All experiments were conducted 3, 7, and 14 days after the sciatic nerve injury to determine thermal and mechanical withdrawal thresholds during the development of neuropathic pain. Thermal allodynia was assessed using the cold plate test (Cold/Hot Plate Analgesia Meter No. 05044 Columbus Instruments, USA). The temperature of the cold plate was kept at 5°C, and the cut-off latency was 30 s. The rats were placed on the cold plate, and the time until the hind paw was lifted was recorded. The injured paw exhibited lower reaction latency. For the assessment of mechanical allodynia (von Frey's test), rats were tested for their foot withdrawal threshold in response to an automatic von Frey apparatus (Dynamic Plantar Aesthesiometer Cat. No. 37400, Ugo Basile Italy). Rats were placed in plastic cages with a wire net floor 5 min before the experiment. The von Frey's filament was applied to the midplantar surface of the ipsilateral hind paw, and the measurements of applied mechanical force were taken automatically. The strength of the von Frey's stimuli in our experiments ranged from 0.5 to 26 g. Thermal hyperalgesia (Hargreaves' test). For the assessment of paw withdrawal latency (PWD) to a noxious thermal stimulus the Analgesia Meter (mod 33, IITC INC., Landing, NJ) was used. On the day of the experiment, each animal was placed in a plastic cage with a heated glass floor. After 5 min of habituation, a noxious thermal stimulus, a light beam, was focused onto the plantar aspect of a hind paw until the animal lifted the paw away from the heat source. The paw withdrawal latency was automatically rounded to the nearest 0.1 s. A cut-off latency of 20 s was used to avoid tissue damage.
5.4. Sample Preparation & RNA Isolation
Animals were sacrificed at either the 3, 7, or 14 day after nerve ligation. A group of naive animals was used as a reference. The L4-L6 dorsal root ganglia (DRGs) and dorsal lumbar spinal cord were collected from both ipsilateral and contralateral side to the injury. Tissue samples were placed in individual tubes with the tissue storage reagent RNAlater (Qiagen Inc., Valencia, CA, USA), frozen on dry ice, and stored at −80°C until RNA isolation. Samples were homogenized in 1 mL of Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA isolation was performed according to Chomczynski's method [69]. RNA concentration was measured using a NanoDrop ND-1000 Spectrometer (Thermo Scientific, Wilmington, USA).
5.5. qPCR Analysis of Gene Expression
Reverse transcription of total RNA (1 μg per sample) was performed using Omniscript reverse transcriptase (Qiagen Inc., Valencia, CA, USA) at 37°C for 60 minutes. For quantitative PCR, 45 ng of cDNA was used as a template. Reactions were performed using Assay-On-Demand TaqMan probes and TaqMan Universal PCR Master Mix (Applied Biosystems, Foster, CA, USA) according to the manufacturer's protocol. The following assays were used: Rn02758689_s1 (Cnr1), Rn03993699_s1 (Cnr2), Rn00583117_m1 (Trpv1), Rn01786262_m1 (Napepld), Rn00668379_g1 (Pla2g2a), Rn01488539_m1 (Abhd4), Rn00583529_m1 (Gde1), Rn01514511_m1 (Plcb1), Rn01533758_m1 (Ptpn22), Rn01400935_m1 (Inpp5d), Rn00577086_m1 (Faah), Rn00568225_m1 (Ptpgs2), Rn01461082_m1 (Alox12), Rn00696151_m1 (Alox15), Rn01527840_m1 (Hprt1). Cycle threshold values (Ct) were calculated automatically by the iCycler IQ 3.0 software. Expression levels were normalized with the Ct for a reference gene, which was hypoxanthine phosphoribosyltransferase 1 (Hprt1). The abundance of RNA was calculated as 2−(normalized threshold cycle).
5.6. Statistics
All data are presented as the mean S.E.M. The results of behavioral experiments and RT-qPCR were evaluated by the analysis of variance (ANOVA) followed by Bonferroni tests. Groups included 8–10 animals for behavioral tests or 4–6 animals for RT-qPCR experiments. A value of P < 0.05 was considered to be statistically significant.
Acknowledgments
This work was supported by Grant LIDER/29/60/L- 2/10/NCBiR/2011 and statutory funds. Natalia Malek is a recipient of a scholarship from the KNOW sponsored by the Ministry of Science and Higher Education, Poland. The authors acknowledge Wioletta Makuch for technical assistance.
List of Abbreviations
- AA:
Arachidonic acid
- ABHD4:
α/β Hydrolase domain containing protein 4
- AEA:
Anandamide
- 12/15-hAEA:
12/15-Hydroxyanandamide
- gp-AEA:
Glycerophosphoanandamide
- p-AEA:
Phosphoanandamide
- LOX-12:
Arachidonate 12-lipoxygenase
- LOX-15:
Arachidonate 15-lipoxygenase
- CB1:
Cannabinoid receptor type 1
- CB2:
Cannabinoid receptor type 2
- CNS:
Central nervous system
- CCI:
Chronic constriction injury to the sciatic nerve
- COX-2:
Cyclooxygenase 2
- DRGs:
Dorsal root ganglia
- EC:
Endocannabinoid
- ETA:
Ethanolamine
- FAAH:
Fatty acid amide hydrolase
- GDE1:
Glycerophosphodiesterphosphodiesterase 1
- INPP5D:
Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1
- NAPE:
N-Arachidonoylphosphatidylethanolamine
- lysoNAPE:
Lyso-N-acylphosphatidylethanolamine
- NAPE-PLD:
N-Arachidonoylphosphatidylethanolamine phospholipase D
- PG:
Prostaglandins
- PLC:
Phospholipase C
- sPLA2:
Secreted phospholipase A2
- PM:
Prostamides
- PTPN22:
Protein tyrosine phosphatase nonreceptor type 22
- TRPV1:
Transient receptor potential vanilloid 1.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Natalia Malek conducted the experiments design, acquisition of data, analysis and interpretation of data, and the writing of the paper. Mateusz Kucharczyk performed the analysis and interpretation of data and the writing of paper. Katarzyna Starowicz also performed study conception, experiments design, analysis and interpretation of data, and the writing of the paper. All authors read and approved the final paper.
References
- 1.Ali Z, Raja SN, Wesselmann U, Fuchs PN, Meyer RA, Campbell JN. Intradermal injection of norepinephrine evokes pain in patients with sympathetically maintained pain. Pain. 2000;88(2):161–168. doi: 10.1016/S0304-3959(00)00327-4. [DOI] [PubMed] [Google Scholar]
- 2.Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nature Medicine. 2004;10(7):685–692. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg DS, McGee SJ. Pain as a global public health priority. BMC Public Health. 2011;11(1, article 770) doi: 10.1186/1471-2458-11-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costa B, Colleoni M, Conti S, et al. Repeated treatment with the synthetic cannabinoid WIN 55,212-2 reduces both hyperalgesia and production of pronociceptive mediators in a rat model of neuropathic pain. British Journal of Pharmacology. 2004;141(1):4–8. doi: 10.1038/sj.bjp.0705587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa B, Trovato AE, Comelli F, Giagnoni G, Colleoni M. The non-psychoactive cannabis constituent cannabidiol is an orally effective therapeutic agent in rat chronic inflammatory and neuropathic pain. European Journal of Pharmacology. 2007;556(1–3):75–83. doi: 10.1016/j.ejphar.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Palazzo E, de Novellis V, Petrosino S, et al. Neuropathic pain and the endocannabinoid system in the dorsal raphe: pharmacological treatment and interactions with the serotonergic system. European Journal of Neuroscience. 2006;24(7):2011–2020. doi: 10.1111/j.1460-9568.2006.05086.x. [DOI] [PubMed] [Google Scholar]
- 7.Ibrahim MM, Deng H, Zvonok A, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guindon J, Beaulieu P. Antihyperalgesic effects of local injections of anandamide, ibuprofen, rofecoxib and their combinations in a model of neuropathic pain. Neuropharmacology. 2006;50(7):814–823. doi: 10.1016/j.neuropharm.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. British Journal of Pharmacology. 2006;147(3):281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fox A, Kesingland A, Gentry C, et al. The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92(1-2):91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- 11.Petrosino S, Di Marzo V. FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Current Opinion in Investigational Drugs. 2010;11(1):51–62. [PubMed] [Google Scholar]
- 12.Kinsey SG, Long JZ, O'Neal ST, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. Journal of Pharmacology and Experimental Therapeutics. 2009;330(3):902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starowicz K, Makuch W, Osikowicz M, et al. Spinal anandamide produces analgesia in neuropathic rats: possible CB1- and TRPV1-mediated mechanisms. Neuropharmacology. 2012;62(4):1746–1755. doi: 10.1016/j.neuropharm.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 14.Starowicz K, Przewlocka B. Modulation of neuropathic-pain-related behaviour by the spinal endocannabinoid/endovanilloid system. Philosophical Transactions of the Royal Society B: Biological Sciences. 2012;367(1607):3286–3299. doi: 10.1098/rstb.2011.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Marzo V, De Petrocellis L. Why do cannabinoid receptors have more than one endogenous ligand? Philosophical Transactions of the Royal Society B. 2012;367(1607):3216–3228. doi: 10.1098/rstb.2011.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sousa-Valente J, Varga A, Ananthan K, Khajuria A, Nagy I. Anandamide in primary sensory neurons: too much of a good thing? European Journal of Neuroscience. 2014;39(3):409–418. doi: 10.1111/ejn.12467. [DOI] [PubMed] [Google Scholar]
- 17.Zygmunt PM, Petersson J, Andersson DA, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400(6743):452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 18.Movahed P, Jönsson BAG, Birnir B, et al. Endogenous unsaturated C18 N-acylethanolamines are vanilloid receptor (TRPV1) agonists. The Journal of Biological Chemistry. 2005;280(46):38496–38504. doi: 10.1074/jbc.M507429200. [DOI] [PubMed] [Google Scholar]
- 19.Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM. Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Research. 1999;822(1-2):237–242. doi: 10.1016/s0006-8993(98)01368-7. [DOI] [PubMed] [Google Scholar]
- 20.Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(21):12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohmann AG, Suplita RL, Bolton NM, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435(7045):1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 22.Bishay P, Schmidt H, Marian C, et al. R-flurbiprofen reduces neuropathic pain in rodents by restoring endogenous cannabinoids. PLoS ONE. 2010;5(5) doi: 10.1371/journal.pone.0010628.e10628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bishay P, Häussler A, Lim H-Y, et al. Anandamide deficiency and heightened neuropathic pain in aged mice. Neuropharmacology. 2013;71:204–215. doi: 10.1016/j.neuropharm.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 24.Petrosino S, Palazzo E, de Novellis V, et al. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology. 2007;52(2):415–422. doi: 10.1016/j.neuropharm.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. The Journal of Neuroscience. 2006;26(51):13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Der Stelt M, Trevisani M, Vellani V, et al. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. The EMBO Journal. 2005;24(17):3026–3037. doi: 10.1038/sj.emboj.7600784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muccioli GG. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discovery Today. 2010;15(11-12):474–483. doi: 10.1016/j.drudis.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 28.Vellani V, Petrosino S, de Petrocellis L, et al. Functional lipidomics. Calcium-independent activation of endocannabinoid/endovanilloid lipid signalling in sensory neurons by protein kinases C and A and thrombin. Neuropharmacology. 2008;55(8):1274–1279. doi: 10.1016/j.neuropharm.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Wang L, Harvey-White J, et al. A biosynthetic pathway for anandamide. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(36):13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Wang L, Harvey-White J, et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology. 2008;54(1):1–7. doi: 10.1016/j.neuropharm.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Y, Tsuboi K, Okamoto Y, et al. Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochemical Journal. 2004;380, part 3:749–756. doi: 10.1042/BJ20040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maccarrone M, Attinà M, Bari M, Cartoni A, Ledent C, Finazzi-Agrò A. Anandamide degradation and N-acylethanolamines level in wild-type and CB1 cannabinoid receptor knockout mice of different ages. Journal of Neurochemistry. 2001;78(2):339–348. doi: 10.1046/j.1471-4159.2001.00413.x. [DOI] [PubMed] [Google Scholar]
- 33.Starowicz K, Makuch W, Korostynski M, et al. Full inhibition of spinal FAAH leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0060040.e60040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rouzer CA, Marnett LJ. Non-redundant functions of cyclooxygenases: oxygenation of endocannabinoids. The Journal of Biological Chemistry. 2008;283(13):8065–8069. doi: 10.1074/jbc.R800005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veldhuis WB, van der Stelt M, Wadman MW, et al. Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. Journal of Neuroscience. 2003;23(10):4127–4133. doi: 10.1523/JNEUROSCI.23-10-04127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahn EJ, Hohmann AG. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 2009;6(4):713–737. doi: 10.1016/j.nurt.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sagar DR, Kelly S, Millns PJ, O'Shaughnessey CT, Kendall DA, Chapman V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. European Journal of Neuroscience. 2005;22(2):371–379. doi: 10.1111/j.1460-9568.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- 38.Guasti L, Richardson D, Jhaveri M, et al. Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Molecular Pain. 2009;5(article 35) doi: 10.1186/1744-8069-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khasabova IA, Khasabov SG, Harding-Rose C, et al. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. The Journal of Neuroscience. 2008;28(44):11141–11152. doi: 10.1523/JNEUROSCI.2847-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okine BN, Norris LM, Woodhams S, et al. Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. British Journal of Pharmacology. 2012;167(3):627–640. doi: 10.1111/j.1476-5381.2012.02028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-Acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45(15):4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simon GM, Cravatt BF. Characterization of mice lacking candidate N-acyl ethanolamine biosynthetic enzymes provides evidence for multiple pathways that contribute to endocannabinoid production in vivo . Molecular BioSystems. 2010;6(8):1411–1418. doi: 10.1039/c000237b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varga A, Jenes A, Marczylo TH, et al. Anandamide produced by Ca(2+)-insensitive enzymes induces excitation in primary sensory neurons. Pflügers Archiv. 2013;466(7):1421–1435. doi: 10.1007/s00424-013-1360-7. [DOI] [PubMed] [Google Scholar]
- 44.Kozak KR, Prusakiewicz JJ, Marnett LJ. Oxidative metabolism of endocannabinoids by COX-2. Current Pharmaceutical Design. 2004;10(6):659–667. doi: 10.2174/1381612043453081. [DOI] [PubMed] [Google Scholar]
- 45.Jhaveri MD, Richardson D, Robinson I, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology. 2008;55(1):85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 46.Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB1, TRPV1 and PPARγ receptors and neurotrophic factors. Pain. 2008;139(3):541–550. doi: 10.1016/j.pain.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Kelly S, Chapman RJ, Woodhams S, et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Annals of the Rheumatic Diseases. 2013 doi: 10.1136/annrheumdis-2013-203413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu M, Ives D, Ramesha CS. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. The Journal of Biological Chemistry. 1997;272(34):21181–21186. doi: 10.1074/jbc.272.34.21181. [DOI] [PubMed] [Google Scholar]
- 49.Nomura DK, Morrison BE, Blankman JL, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334(6057):809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ligresti A, Martos J, Wang J, et al. Prostamide F(2) α receptor antagonism combined with inhibition of FAAH may block the pro-inflammatory mediators formed following selective FAAH inhibition. British Journal of Pharmacology. 2014;171(6):1408–1419. doi: 10.1111/bph.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Stelt M, van Kuik JA, Bari M, et al. Oxygenated metabolites of anandamide and 2-arachidonoylglycerol: conformational analysis and interaction with cannabinoid receptors, membrane transporter, and fatty acid amide hydrolase. Journal of Medicinal Chemistry. 2002;45(17):3709–3720. doi: 10.1021/jm020818q. [DOI] [PubMed] [Google Scholar]
- 52.Pertwee RG. Cannabinoid receptors and pain. Progress in Neurobiology. 2001;63(5):569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 53.Bridges D, Ahmad K, Rice ASC. The synthetic cannabinoid WIN55,212-2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. British Journal of Pharmacology. 2001;133(4):586–594. doi: 10.1038/sj.bjp.0704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paszcuk AF, Dutra RC, da Silva KABS, Quintão NLM, Campos MM, Calixto JB. Cannabinoid agonists inhibit neuropathic pain induced by brachial plexus avulsion in mice by affecting glial cells and MAP kinases. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0024034.e24034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Furuse S, Kawamata T, Yamamoto J, et al. Reduction of bone cancer pain by activation of spinal cannabinoid receptor 1 and its expression in the superficial dorsal horn of the spinal cord in a murine model of bone cancer pain. Anesthesiology. 2009;111(1):173–186. doi: 10.1097/ALN.0b013e3181a51e0d. [DOI] [PubMed] [Google Scholar]
- 56.Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacological Research. 2013;67(1):94–109. doi: 10.1016/j.phrs.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agarwal N, Pacher P, Tegeder I, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neuroscience. 2007;10(7):870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O'Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. European Journal of Neuroscience. 2003;17(12):2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- 59.Racz I, Nadal X, Alferink J, et al. Crucial role of CB2 cannabinoid receptor in the regulation of central immune responses during neuropathic pain. Journal of Neuroscience. 2008;28(46):12125–12135. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beltramo M, Bernardini N, Bertorelli R, et al. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. European Journal of Neuroscience. 2006;23(6):1530–1538. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- 61.Romero-Sandoval A, Nutile-Mcmenemy N, Deleo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008;108(4):722–734. doi: 10.1097/ALN.0b013e318167af74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rahn EJ, Zvonok AM, Thakur GA, Khanolkar AD, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB2 receptors suppresses neuropathic nociception induced by treatment with the chemotherapeutic agent paclitaxel in rats. The Journal of Pharmacology and Experimental Therapeutics. 2008;327(2):584–591. doi: 10.1124/jpet.108.141994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rahn EJ, Makriyannis A, Hohmann AG. Activation of cannabinoid CB 1 and CB 2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. The British Journal of Pharmacology. 2007;152(5):765–777. doi: 10.1038/sj.bjp.0707333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winter Z, Buhala A, Ötvös F, et al. Functionally important amino acid residues in the transient receptor potential vanilloid 1 (TRPV1) ion channel—an overview of the current mutational data. Molecular Pain. 2013;9(1, article 30) doi: 10.1186/1744-8069-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 66.Osikowicz M, Mika J, Makuch W, Przewlocka B. Glutamate receptor ligands attenuate allodynia and hyperalgesia and potentiate morphine effects in a mouse model of neuropathic pain. Pain. 2008;139(1):117–126. doi: 10.1016/j.pain.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 67.Elmes SJR, Jhaveri MD, Smart D, Kendall DA, Chapman V. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naïve rats and in rat models of inflammatory and neuropathic pain. European Journal of Neuroscience. 2004;20(9):2311–2320. doi: 10.1111/j.1460-9568.2004.03690.x. [DOI] [PubMed] [Google Scholar]
- 68.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33(1):87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 69.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]