

Abstract
Background
Spiroplasma virus, SpV4, is a small, non-enveloped virus that infects the helical mollicute Spiroplasma melliferum. SpV4 exhibits several similarities to the Chlamydia phage, Chp1, and the Coliphages α3, φK, G4 and φX174. All of these viruses are members of the Microviridae. These viruses have isometric capsids with T = 1 icosahedral symmetry, cause lytic infections and are the only icosahedral phages that contain single-stranded circular DNA genomes. The aim of this comparative study on these phages was to understand the role of their capsid proteins during host receptor recognition.
Results
The three-dimensional structure of SpV4 was determined to 27 Å resolution from images of frozen-hydrated particles. Cryo-electron microscopy (cryo-EM) revealed 20, ∼54 Å long, ‘mushroom-like’ protrusions on the surface of the capsid. Each protrusion comprises a trimeric structure that extends radially along the threefold icosahedral axes of the capsid. A 71 amino acid portion of VP1 (the SpV4 capsid protein) was shown, by structural alignment with the atomic structure of the F capsid protein of φX174, to represent an insertion sequence between the E and F strands of the eight-stranded antiparallel β-barrel. Secondary structure prediction of this insertion sequence provided the basis for a probable structural motif, consisting of a six-stranded antiparallel β sheet connected by small turns. Three such motifs form the rigid stable trimeric structures (mushroom-like protrusions) at the threefold axes, with hydrophobic depressions at their distal surface.
Conclusions
Sequence alignment and structural analysis indicate that distinct genera of the Microviridae might have evolved from a common primordial ancestor, with capsid surface variations, such as the SpV4 protrusions, resulting from gene fusion events that have enabled diverse host ranges. The hydrophobic nature of the cavity at the distal surface of the SpV4 protrusions suggests that this region may function as the receptor-recognition site during host infection.
Keywords: cryo-electron microscopy, icosahedral capsid, sequence alignment, spiroplasma virus, SpV4
Introduction
Spiroplasma virus, SpV4, is a small, spherical virus that infects the Spiroplasma melliferum. This non-enveloped, single-stranded DNA virus was discovered in 1980 in strain B63 of S. melliferum cultured from a honey bee in Morocco [1] and has since been propagated in S. melliferum strain G1 [2,3]. Spiroplasmas are helical, wall-free eubacteria belonging to the class Mollicutes, a group of organisms derived by regressive evolution from ancestors of gram-positive bacteria with low guanine-plus-cytosine DNA [4,5].
SpV4 shares several properties with the Chlamydia phage Chp1 and the Coliphages α3, φK, G4 and φX174. All these viruses are members of the Microviridae family; they are lytic and contain single-stranded circular DNA genomes of 4421, 4877, 6087, 6081, 5386 and 5577 nucleotides for SpV4, Chp1, α3, φK, G4 and φX174, respectively [6-10]. The genomes encode similar numbers of proteins: nine for SpV4 and eleven for Chp1, α3, φK, φX174 and G4. The SpV4 and Chp1 capsids, however, only contain 60 copies of a single protein, VP1, whereas the capsids of α3, φK, G4 and φX174 consist of 60 copies each of the major capsid protein, F, the major spike protein G, the DNA packaging protein, J, and 12 copies of the pilot protein, H.
SpV4 virions have a molecular weight of 5.4 × 106 and a density of 1.40 g/cm3 in cesium chloride. VP1 is a 63.9 kDa protein that forms a T = 1 capsid [11] with a mean diameter of ∼270 Å (Figure 1) [12], which is roughly equivalent to a φX174 capsid consisting of only the major capsid protein F, not including the G, H or J proteins [13,14].
Figure 1.

SpV4 particles negatively stained with 1% uranyl acetate on a carbon support film. Arrows highlight protrusions on three particles. The inset, a close-up view of one particle, shows several, stain-excluding (white) ‘dots’ inside the particle. The dots are likely to be end-on views of the protrusions. The scale bar = 500 Å.
The structures of φX174 and G4 virions have been determined by X-ray crystallography to 3 Å resolution [14,15]. These studies revealed the structure and capsid organization of the F, G, and J proteins in atomic detail. Each φX174 or G4 capsid contains 60 copies of the F protein arranged with T = 1 icosahedral symmetry [11]. The F protein has the β barrel tertiary structure common to many of the structural proteins in small, spherical viruses [16]. This canonical, eight β-strand motif is oriented with the strands aligned roughly tangential to the capsid surface and with the B–C, D–E, F–G and H–I corners (where the letters refer to the individual strands of the β barrel) pointed towards the fivefold axes (Figure 2). The β barrel projects into the interior of the virus, where there is little contact between adjacent, symmetry-related β barrels. Two large insertion loops that comprise 65% of the F protein polypeptide chain occur at the corners of the E–F and H–I strands, these loops are 163 and 112 residues in length, respectively. The two insertion loops form the exterior surface topology of the capsid and are responsible for most of the contacts between neighboring F protein subunits (Figure 2).
Figure 2.

Ribbon drawing of the atomic structure of the φX174 capsid protein F. The β-barrel motif is coloured red with the β strands (B, I, D, G, and C, H, E, F) labeled according to standard convention. Arrows highlight the equivalent positions of the capsid protein VP1 of SpV4, where insertion loops (identified as IN1–7) occur relative to the capsid protein F of φX174 (refer to Figure 6). Residue numbering is given for the F protein of φX174. The orientation of the F protein is shown viewed towards the interior of the virus down a twofold axis. An icosahedral asymmetric unit (large open triangle) includes the region bounded by a fivefold axis (filled pentagon) and two adjacent threefold axes (filled triangles). (Figure produced with MOLSCRIPT [53].)
Comparison of the sequences of the structural proteins of four Coliphage members of the Microviridae family, α3, φK, G4, and φX174, showed that the capsid protein F is the most highly conserved structural protein [17]. The VP1 capsid proteins in SpV4 and Chp1 contain 553 and 595 amino acids, respectively [6,7], and are homologous to the 426 amino acid F proteins of φX174 and G4 [8–10].
The SpV4 and Chp1 genomes do not encode a major spike protein like the G protein of the Coliphage virions. Pentamers of the G protein in the Coliphages form 12, characteristic, star-shaped ‘spikes’ (∼70 Å in diameter and extending ∼30 Å above the viral capsid surface) situated at the fivefold vertices of the icosahedron [13–15]. Previously, the SpV4 capsid was postulated to be devoid of such spikes or other projections [12], and hence it was believed that the SpV4 morphology might bear more resemblance to the smoother, spherical capsids of the Parvoviridae [18–20]. The characteristic Coliphage spikes can be removed from capsids with 4 M urea, leaving an intact capsid formed by the F protein [21]. These spikeless particles were thought to be similar in structure to SpV4 or Chp1, the genomes of which do not encode the G, H and J structural proteins found in the Coliphages. Thus, the known atomic structures of the φX174 and G4 F capsid proteins should serve as excellent models of the VP1 capsid protein of SpV4 and Chp1.
We have used cryo-electron microscopy (cryo-EM) and three-dimensional image reconstruction methods to determine the structure of frozen-hydrated SpV4 particles to 27 Å resolution. We also report an amino acid sequence alignment of six members of the Microviridae family which made possible the fit of the atomic coordinates of the F protein of φX174 into the cryo-EM density map of SpV4. Finally, modeling of three, 71 amino acid loops into the cryo-EM density for the ‘mushroom-like’ protrusions at the threefold vertices, and the hydrophobic nature of the cavity at the distal surface of these protrusions, suggest a function as putative receptor recognition sites for SpV4.
Results and discussion
Cryo-EM and three-dimensional image reconstruction
SpV4 for use in the cryo-EM studies was propagated and purified following an established protocol [3] with minor modifications to prevent precipitation of the virus (see Materials and methods section). Cursory inspection of transmission electron micrographs of negatively stained (Figure 1) or vitrified (Figure 3) samples of SpV4 revealed particles with circular profiles, consistent with a spherical structure of diameter ∼270 Å. However, when the three-dimensional structure of SpV4 was determined to 27 Å resolution based on reconstruction analysis of 22 images of vitrified particles, it was immediately apparent that virions contain a prominent protrusion at each of the 20 threefold axes of symmetry (Figure 4). Careful reinspection of the images of stained (Figure 1) and unstained SpV4 particles (Figure 3) revealed features consistent with the presence of such external protrusions. In stain, most particles have small projections or knob-like features at their periphery (Figure 1; arrows) as well as stain-excluding knobs within the particle (e.g. Figure 1 inset). These latter features correspond to end-on views of the protruding structures. Similar features at the particle periphery can sometimes be discerned in the noisy images of unstained SpV4 particles (Figure 3; arrows and inset).
Figure 3.

Unstained SpV4 particles suspended in a layer of vitreous ice over holes in a carbon support film. Arrows highlight several protrusions on two particles. The inset shows an enlarged view of one of the highlighted particles. The scale bar = 500 Å.
Figure 4.

Shaded surface representations of the SpV4 three-dimensional reconstruction. The representations are viewed down the (a) twofold (in stereo), (b) threefold and (c) fivefold axes. The reconstruction was computed to 27 Å resolution from 22 different SpV4 particle images. The scale bar = 100 Å.
The prominent protrusions project radially outwards from the capsid, the surface of which has a ‘rough’ texture (Figure 4). Large, ‘sausage-like’ ridges encircle five smaller ‘bumps’ and an even smaller bump at each fivefold axis. In addition, three small cavities surround each protrusion near its base. Close-up views reveal finer details of the protrusion structure (Figure 5). As depicted in Figure 5, the mushroom-like protrusion extends ∼54 Å above the capsid surface and consists of a globular head (‘bud’) of approximate dimensions 41 × 41 × 29 Å and a neck (‘stalk’) that is ∼25 Å long and ∼13 Å in diameter. The cavities at the base of the protrusion are channels (∼10 Å narrowest diameter) that appear to provide solvent accessibility to the virus interior (Figures 5a,b). These putative channels persist in isosurface rendered views even at much lower density threshold levels. A cross-section through the longitudinal axis of the protrusion reveals a dimple at the outermost tip of the protrusion (Figure 5c; compare with top view in Figure 5a) in the isosurface view. A more realistic rendering of the density distribution in the longitudinal section (Figures 5d,e) reveals a ‘cup-like’ as opposed to solid structure for the bud domain. At 27 Å resolution, however, it is impossible to determine whether the low density central region is a solvent-filled cavity of approximate dimensions 10 Å diameter and 17 Å deep. Alternatively, the average protein density may simply be very low in this region.
Figure 5.

Close-up views of one SpV4 trimeric protrusion. (a) Shaded surface view from above the top of the protrusion. (b) Side view, as from the bottom of (a), with the front of the map (as indicated by the dotted line towards the bottom of (a)) removed to reveal a channel near the base of the stalk. (c) The same view as (b) but with more density removed, to dashed line in center of (a), to reveal a cross-section through the center of the protrusion. (d) Density distribution in the center of the protrusion at the same plane where the map was cut in (c). Highest density features (protein) appear black, whereas lowest densities (solvent) appear white or gray. This representation gives a more realistic rendering of the density fluctuations inside the protrusion and, for example, shows a low density along the vertical axis at the top of the protrusion. (e) The view is the same as in (c) but with four density contour levels shown to illustrate the varying density in the cross-section.
The protrusion appears to be a fairly rigid structure because its densities in the icosahedrally averaged three-dimensional map are in the same range as those attributed to the capsid shell. Our model building experiments (described later) are consistent with this observation.
Alignment of Microviridae capsid proteins
Members of the Microviridae, including the Coliphages α3, φK, G4 and φX174, have been shown to have the same gene order and considerable sequence similarities [22]. An amino acid sequence alignment of the F, G, and J structural proteins [17], together with knowledge of the atomic structures of the φX174 [14] and G4 [15] capsids, showed the F protein to be the most conserved structural protein. Among all four Coliphages, the F proteins showed 63% amino acid sequence identity, as compared to 19% identity for the G spike proteins. Presumably, functions of the F protein, such as involvement in capsid assembly and genome protection, are indispensable enough for evolutionary pressure to conserve them. The sequence alignment and crystallography studies also showed that the F protein residues involved in intersubunit interactions were more conserved than those in other regions. In addition, the residues at a surface depression in the φX174 capsid that runs from a position near the threefold axis to the twofold axis were almost completely conserved [17]. Part of this depression in φX174 forms a putative carbohydrate-binding site [23].
The amino acid sequences of the SpV4 and Chp1 VP1 proteins and of the F proteins of α3, φK, G4 and φX174, were aligned with the use of the program PILEUP (part of the University of Wisconsin Genetics Computer Group [GCG] package). To maximize the alignment agreement of the capsid protein sequences, seven insertion loops were introduced into the SpV4 and Chp1 sequences in relation to the Coliphages (Figures 6a,b). The criteria for scoring insertions required a difference in sequence length of six or more amino acids. By reference to the atomic structure of the φX174 F protein, six insertions were located in the E–F turn of the β barrel and the other insertion was in the H–I turn. Two of the E–F insertions would occur inside the capsid (i.e. at low radius) and all other insertions would occur on the exterior surface of the capsid (Figures 2 and 6a,b). These sequence alignment results suggest that the tertiary structure of VP1 may be quite similar to that for the F protein but with the largest differences occurring at the outer and inner surfaces of the capsid attributed to the VP1 insertions. The longest insertion, IN5, occurs between Thr187 and Thr188 of the F protein of φX174 (Figures 2 and 6), and is 71 and 104 residues in SpV4 and Chp1, respectively. This insertion point occurs in φX174 within the portion of the E–F loop that forms a small bump at the icosahedral threefold axes (Figure 7). On this basis, it is possible to ascribe the 71-residue IN5 insertion of SpV4 to the prominent mushroom-like protrusions at the threefold positions (Figures 4 and 5).
Figure 6.

Comparison of the amino acid sequences of capsid proteins of members of the Microviridae family. (a) Sequence alignment of VP1 of SpV4 and Chp1 and protein F for the Coliphages α3, φK, φX174 and G4. Bold letters indicate residues that are completely conserved in all the phages compared; residues that form part of the β-barrel structure motif elements are colored red and labeled (as shown in Figure 2). The sequence numbers are given for SpV4 (top) and φX174 (bottom). Insertions are labeled and marked by green boxes. (b) Assignment and location of the seven insertion loops (assigned as IN1–7) found in the SpV4 and Chp1 sequences in relation to the Coliphages. (c) The percentage of identical amino acids between the aligned capsid proteins, for all residues (shown in purple) and those involved in the β-barrel motif (shown in red).
Figure 7.
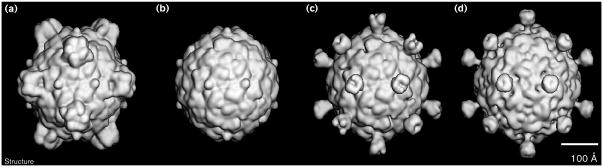
Shaded surface representation comparisons of φX174 and SpV4 structures. (a) The atomic φX174 structure [14]. (b) The φX174 structure, as in (a), but with the prominent spikes (G protein pentamers) removed to reveal just the F capsid. (c) An SpV4–φX174 hybrid model, formed by combining the φX174 F capsid model (b) with the pseudo-atomic model of the SpV4 protrusion. (d) The cryo-EM reconstruction of SpV4. All structures are shown at ∼27 Å resolution and are viewed along a twofold axis of symmetry. The scale bar = 100 Å.
Only 33 (7.7%) of the 426 amino acid residues of the φX174 F protein were identical in all six phages, whereas 268 (63%) of these residues were identical among the four Coliphage members and 142 (33%) were identical between SpV4 and Chp1. The percentage of identical residues for each pairwise comparison of sequences was calculated not only for all residues but also for just those residues of the structurally conserved β-barrel motif identified from the X-ray crystal structures of φX174 and G4 [14,15] (Figure 6c). The residues within the β-barrel motif were observed to be significantly greater in identity in every pair sequence comparison between ‘weakly’ related phages (identity less than 21% for all residues). For example, when comparing the F protein of φX174 with VP1 of SpV4, for all residues the sequence identity is 19%, whereas for residues contained only in the β-barrel motif the percentage identity increases to 30% (Figure 6c). This same general trend holds (with one exception, α3 and φK) for ‘strongly’ related sequences (identity greater than 50% for all residues), although the increase in identity is only slight.
These pairwise sequence comparisons suggest that an evolutionary pressure exists to conserve ‘critical’ residues within the β-barrel motif while the surface insertion loops can accommodate a high mutation rate. Even though there is no obviously conserved specific sequence in the major capsid proteins of the related phages, close inspection of the sequence comparisons (Figure 6a) reveals a tendency for hydrophobic residues in the β-barrel motif to be conserved. Such conservation is consistent with the preservation of a β-barrel structure that is the structural core of each capsid. Rapid evolution within the large insertion loops that decorate this core structure ensures diversity in the exposed surface of each phage.
A phylogenetic tree for the six bacteriophages (Figure 8) was calculated from the data shown in Figure 6c by means of an unweighted pairwise distance matrix method [24]. As expected, the Coliphages cluster closely together, with α3 and φK being most closely related (91% sequence identity) and G4 being the most distantly related member of the Coliphages (∼63% sequence identity to all three other Coliphages). Chp1 and SpV4 cluster together, though they are still quite distant (only 33% sequence identity between VP1s), and they are even more distant from the Coliphages (15% mean sequence identity).
Figure 8.

Phylogenetic tree for six Microviridae capsid proteins.
When pairs of structurally similar protein chains were taken from the CATH structural classification database [25], two distributions of sequence identities were observed. Those sequences which shared a common ancestry had a mean percentage sequence identity of 15%, and those which had a mean percentage sequence identity less than 10% did not [26]. These observations indicate that, although the VP1s of Chp1 and SpV4 are genetically distinct from the F proteins of the Coliphages, there is likely to be a common ancestral link among these proteins.
Modeling the IN5 loop and fit to the cryo-EM density map
The secondary structure of the 71-residue IN5 loop (Figure 6a), that was ascribed to the mushroom-like projections, was predicted with the aid of the PredictProtein Server [27] to be approximately 60% β strand and 40% coil. No α-helical segments were predicted by the program.
A three-dimensional model was built for the 71-residue SpV4 loop that satisfied both the envelope of the cryo-EM reconstruction (Figures 4 and 5) and the secondary structure prediction. The modeling incorporated three, symmetry-related, six-stranded (each strand being six residues long) antiparallel β-sheet motifs connected by small turns of five residues in length (Figure 9). This structure fit extremely well into the distal globular bud domain (41 × 41 × 29 Å) of the trimeric protrusion (Figure 10). This simple model also allowed for intersubunit hydrogen-bonding interactions between antiparallel β strands from the symmetry-related motifs. The lengths of the β strands numbered 1 and 6 (Figure 9) were extended to 12 residues each, to form a stalk that allows the loop to insert at the Thr187–Thr188 position (φX174 sequence designation). In the model, the stalk region consists of the two extended antiparallel strands, which form a β-hexamer structure with the symmetry-related strands. This proposed β-hexamer structure is very similar to that observed on the quasi-sixfold axes of symmetry in cowpea chlorotic mottle virus [28]. The stalk is ∼25 Å long and 13 Å wide and connects the globular bud of the protrusion to the capsid (Figure 10). As modeled, the protruding loop has 48 residues in β-strand structure and 23 residues in loop structures. The motif is a very rigid and stable structure, which is consistent with the observation of the protrusion as a high-density feature in the three-dimensional image reconstruction. This model also includes a 17 Å deep depression at the distal surface of the bud on the threefold axes (Figure 10). Of the 71 amino acids in the loop, 30 are hydrophobic and of the 11 residues, Val237-Pro-Asn-Leu-Ser-Gly-Thr-Pro-Leu-Ile-Phe247, lining the surface of the 17 Å depression, seven are hydrophobic. The model, therefore, predicts the presence of a mainly hydrophobic cavity at the distal surface of the protrusion.
Figure 9.

Proposed structural motif model for residues Gly226–Thr297 of the capsid protein of SpV4. These residues comprise one third of the protrusion at each threefold axis. The residues in the F protein of φX174 at the exit and entry points for the SpV4 loop are indicated in parentheses; β strands are shown as arrows.
Figure 10.

Stereo views of the atomic model of the SpV4 protrusion. (a) The fit of a pseudo-atomic model of an SpV4 VP1 trimer (separate monomers colored blue, green and red) into the cryo-EM reconstruction (gray isodensity contour). Shown also is the viral asymmetric unit, depicted as an open triangle. The view is along a twofold axis. The fit of three symmetry-related SpV4 loop structures (Gly226–Thr297) into the cryo-EM density map, as viewed from (b) the top, along a threefold axis or (c) the side, perpendicular to a threefold axis. Corresponding shaded surface representations are shown to the right of each stereo view. (The stereo views were produced with MacInPlot [54].)
This model is the simplest that satisfies both the cryo-EM density of the protrusion at the threefold axis and the predicted secondary structure profile of the amino acid sequence of the IN5 insertion loop of SpV4. Obviously other more complex models may also satisfy the envelope of the cryo-EM density, but would not satisfy the secondary structure prediction profile or the formation of a hydrophobic cavity at the distal surface of the protrusion. Other structural motif insertions have been observed between strands of the eight-stranded antiparallel β barrel of viruses. For example, an immunoglobulin (Ig) fold domain insertion occurs between the E and F strands of the insect virus nudaurelia ω capensis virus [29], but this motif would be too large for the protrusion in SpV4.
Conclusions
The multisequence alignments for the major capsid proteins of the Microviridae phages demonstrate that the VP1 proteins of Chp1 and SpV4 most likely have a common ancestral link to the F proteins of the Coliphages. The three-dimensional cryo-EM reconstruction of SpV4 revealed a mushroom-like protrusion on the threefold axes of the icosahedral capsid. The sequence alignment results, along with knowledge of the atomic structures of the F proteins for φX174 and G4, lead to the assignment of a 71-residue loop in the SpV4 VP1 sequence (Gly226 to Thr297) to one third of the trimeric protrusion structure. The secondary structure predicted from the sequence of the loop suggested a peptide fold, the main part of which consists of a six-stranded, antiparallel β sheet, that trimerizes and fits well into the globular domain (bud) of the protrusion. The distal globular domain of the trimeric hemagglutinin spike of influenza virus has a similar structure, although hemagglutinin is much larger (∼145 residues) and consists of an eight-stranded, antiparallel β-sheet structure with larger extended loops between the strands [30]. A fragment sequence search of the 71-residue loop was performed, using the BLAST network service, and sequences of influenza hemagglutinin B were among the highest scoring segment pairs [31].
The predicted hydrophobic character of the depression at the distal tip of the SpV4 bud suggests a possible role in cellular receptor binding. The human parvovirus, B19, has a depression at each threefold axis that binds globoside, the cellular receptor [32]. Unlike SpV4, however, B19 does not have long protrusions.
Biological implications
The bacteriophage spiroplasma virus, SpV4, which infects the wall-free prokaryote Spiroplasma melliferum, is a member of the Microviridae family. Other members of this family include the Chlamydia phage, Chp1, and Coliphages, such as φX174. The surface of a virion is one factor that helps control host tropism, because it presents the required specific interactions for host receptor recognition which leads to infection. A detailed description of the SpV4 surface, and comparison with related phages, may help us to further understand the mechanisms underlying host cell recognition and specificity.
We report here the three-dimensional structure of SpV4. We present a detailed description of the SpV4 capsid structure based on a three-dimensional cryo-electron microscopy (cryo-EM) image reconstruction of SpV4 samples and structural sequence alignment with the atomic model of the major capsid protein (protein F) of the related bacteriophage, φX174.
A striking feature of the SpV4 structure, which was not previously seen, is the presence of 20, ‘mushroom-like’ protrusions (∼54 Å long) that extend radially outwards from the capsid surface along the icosahedral threefold axes. The amino acid sequence that folds to form this protrusion was identified as a 71-residue insertion in the SpV4 coat protein, VP1, relative to the φX174 F capsid protein. Sequence alignments of the F capsid proteins of the Coliphages, and the VP1 capsid proteins of Chp1 and SpV4, exhibit an evolutionary relationship. Distinct differences in the surface morphologies of the Microviridae phages are attributable mainly to large variations in protruding structural motifs. The Coliphages have a specialized spike structure (pentamers of the G protein) at each of the 12 fivefold vertices. The G and F proteins both affect host range in Escherichia coli strains [33-35]. Chp1 and SpV4 have no gene for a G-like protein, but instead have an insertion loop (IN5), between the E and F strands of the capsid protein β barrel; these insertions associate at the threefold axes to form 20 protrusions. These protrusions may be utilized as host range determinants in an analogous manner to the G protein spikes of the Coliphages.
Phages of the Microviridae family probably evolved from a common primordial phage gene. Distinct genera might have then evolved by means of genetic fusion events that enabled a diverse range of hosts to be infected. It is probable that Coliphages incorporated a gene, G, to encode the large pentameric spike structure which enables the phages to recognize the outer cell membrane of E. coli. Likewise, Chp1 and SpV4 incorporated a gene that encodes a trimeric structural motif that enables these phage to recognize the chlamydial envelope or the wall-free spiroplasmas, respectively.
Materials and methods
Propagation and purification of SpV4
S. melliferum strain G1 [36] was grown in SP4 medium [37] at 32°C, and used for the propagation of SpV4 [3]. SpV4-infected Spiroplasma broth cultures were prepared by inoculation of an early log phase culture of S. melliferum (108–109 cfu/ml) with an SpV4 inoculum at a multiplicity of infection of approximately 0.2.
For virus purification, SpV4-infected cultures (6 × 150 ml) were harvested 24h after inoculation. Spiroplasma cells were pelleted by centrifugation at 22,000 × g for 45 min. The supernatant was adjusted to 0.5 M NaCl and the virions were precipitated by adding 8% polyethylene glycol 6000. After overnight incubation at 4°C, the precipitate was collected by centrifugation (22,000 × g for 45 min), and the pellets were suspended in 50 mM sodium tetraborate buffer at pH 9.2 containing 0.5 M NaCl. The suspension was treated with 10 μg/ml DNAse in the presence of 25 mM MgCl2 for 1 h at 37°C, with 0.2% Triton X100 for an additional 1 h, and then clarified three times by vigorous shaking with an equal volume of chloroform. The virions were sedimented from the final aqueous phase by centrifugation (130,000 × g for 7 h in a Beckman SW27 rotor) through a 30% sucrose cushion (5 ml). The pellets were resuspended in 50 mM sodium tetraborate buffer at pH 9.2 containing 0.6 g/ml CsCl and centrifuged to equilibrium at 150,000 × g for 48 h. The fraction containing virions was collected and dialyzed against 40 mM sodium tetraborate buffer at pH 9.2. The virus concentration was adjusted to 1 mg/ml assuming an extinction coefficient of 7.5 mg/ml per cm at 260 nm. This protocol yielded between 1.0 to 1.5 mg of purified virions per liter of SpV4 infected culture.
Cryo-EM and three-dimensional image reconstruction
Small aliquots (3.5 μl) of SpV4 sample were placed on holey carbon grids, blotted with filter paper, and plunged into liquid ethane to suspend the SpV4 particles in a thin layer of vitreous ice as described [38–40]. Grids were inserted into a precooled Gatan 626 cryotransfer holder (Gatan Inc., Warrendale, PA, USA) that maintained a constant temperature of −175°C. The sample was examined in a Philips EM420 transmission electron microscope (Philips Electronics Instruments, Mahwah, NJ, USA) and images were recorded on Kodak SO-163 film (Eastman Kodak Company, Rochester, NY, USA) under low dose conditions (∼18 e–/Å2) at a nominal magnification of × 49,000 and a defocus value of ∼0.8 m m. A micrograph that displayed uniform specimen ice thickness and with minimal drift and astigmatism, as determined by eye, was digitized at 25 m m intervals (5.1 Å step size) and processed as described [40]. The orientations and origins of different particles imaged in the micrograph were determined and a subset of 22 SpV4 images were combined to compute a three-dimensional reconstruction which was used for subsequent model-based refinement of the view angle and origin parameters [41]. The resolution of the final reconstructed density map was determined to be at least 27 Å as measured by structure-factor comparisons [42] and Fourier ring correlation measurements [43].
All computations were performed with FORTRAN programs [41, 44–46] on VAX/VMS 8550 and AlphaStation 500/400 computers (Digital Equipment Corp., Maynard, MA, USA). Digitized images and shaded-surface representations were viewed on a raster graphics device (Model 3400; Lexidata Corp., Billerica, MA, USA) and final figures were prepared on a MacIntosh computer (Apple Computer, Inc., Cupertino, CA, USA) with Adobe Photoshop 3.0 (Adobe Systems Inc., Mountain View, CA, USA) and ClarisDraw (Claris Corporation, Santa Clara, CA, USA) programs. The threshold level for rendering iso-surface views (Figures 4, 5, 9 and 10) was selected to give a molecular volume consistent with the expected volume of the SpV4 capsid. Though the image and reconstruction data were not compensated for the effects of the contrast transfer function of the microscope [47], the chosen threshold gave a molecular envelope that fit quite well the X-ray structure of the φX174 F protein capsid (Figure 10).
Alignment of Microviridae capsid proteins
The programs used for the manipulation and alignment of sequences were those of the University of Wisconsin GCG [48]. The representative sequences of the F capsid proteins of α3, φK, φX174 and G4, and the VP1 proteins of SpV4 and Chp1 were obtained from the GenBank database [49]. Residues Gly35, Gly36, Pro68 and Arg281 of the F protein of G4 were changed to Val35, Val36, Ser68 and Gly281, according to sequencing performed by Bentley Fane and is in agreement with the interpretation of the high-resolution electron-density map of G4 [15]. The program PILEUP was used to align each sequence to a profile of all other sequences [50] with gap penalties of Gi = 3.0 and G1 = 0.1. The known secondary structural elements of φX174 and G4 [14,15] were used to make a few minor adjustments to the final sequence alignment. The alignment procedure was carried out on a Silicon graphics Indigo 2 workstation (Silicon Graphics Computer Systems, Mountain View, CA, USA).
Modeling the IN5 loop and fit to the cryo-EM density map
Three copies of the 71-residue loop (IN5) of SpV4 were built and fitted interactively into the cryo-EM density map with the program O [51] on a Silicon graphics Indigo 2 workstation. The β strand structural elements were modeled using segments from the β-barrel domain of the previously determined atomic structure of φX174 [14]. The turn regions were inserted between the β strands to complete the connectivity of the loop. The model was constrained using the structural geometry library of the program O.
Acknowledgments
We thank Norm Olson, Tom Smith and Chris Storey for useful discussions and Sybille Duret for excellent technical assistance. This work was supported by National Institutes of Health grant GM33050 to TSB, Program Project grant AI35212 to the Purdue University Structural Virology group, and a University of Warwick Research and Teaching Initiative grant 0952 to RM.
Footnotes
Accession numbers The Cα atomic coordinates of the SpV4 model have been deposited with the Brookhaven Protein Data Base [52], with the accession code 1KVP.
References
- 1.Ricard B, Garnier M, Bove JM. Characterization of SPV3 from spiroplasmas and discovery of a new spiroplasma virus (SPV4) Rev Infect Dis. 1982;4:S275. [Google Scholar]
- 2.Renaudin J, Pascarel MC, Garnier M, Carle P, Bove JM. Characterization of spiroplasma virus group 4 (SPV4) Isr J Med Sci. 1984;20:797–799. [PubMed] [Google Scholar]
- 3.Renaudin J, Pascarel MC, Garnier M, Junca-Carle P, Bove JM. SpV4, a new spiroplasma virus with circular, single stranded DNA. Ann Virol. 1984;135E:343–361. [Google Scholar]
- 4.Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisburg WG, et al. Woese CR. A phylogenetic analysis of the mycoplasmas: basis for their classification. J Bacteriol. 1989;171:6455–6467. doi: 10.1128/jb.171.12.6455-6467.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renaudin J, Pacarel MC, Bove JM. Spiroplasma virus 4: nucleotide sequence of the viral DNA, regulatory signals and proposed genome organisation. J Bacteriol. 1987;169:4950–4961. doi: 10.1128/jb.169.11.4950-4961.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storey CC, Lusher M, Richmond SJ. Analysis of the complete nucleotide sequence of Chp1, a phage which infects avian Chlamydia psittaci. J Gen Virol. 1989;70:3381–3390. doi: 10.1099/0022-1317-70-12-3381. [DOI] [PubMed] [Google Scholar]
- 8.Sanger F, et al. Smith M. Nucleotide sequence of bacteriophage φX174 DNA. Nature. 1977;265:687. doi: 10.1038/265687a0. [DOI] [PubMed] [Google Scholar]
- 9.Sanger F, et al. Smith M. The nucleotide sequence of bacteriophage φX174. J Mol Biol. 1978;125:225. doi: 10.1016/0022-2836(78)90346-7. [DOI] [PubMed] [Google Scholar]
- 10.Godson GN, Barrell BC, Staden R, Fiddes JC. Nucleotide sequence of bacteriophage G4 DNA. Nature. 1978;276:236. doi: 10.1038/276236a0. [DOI] [PubMed] [Google Scholar]
- 11.Caspar DLD, Klug A. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp Quant Biol; 1962. pp. 1–24. [DOI] [PubMed] [Google Scholar]
- 12.Renaudin J, Bove JM. Advances in Virus Research. Vol. 44. Academic Press; London, UK: 1995. SpV1 and SpV4, spiroplasma viruses with circular, single-stranded DNA genomes, and their contribution to the molecular biology of spiroplasmas; pp. 429–463. I. [DOI] [PubMed] [Google Scholar]
- 13.Olson NH, Baker TS, Willingmann P, Incardona NL. The three-dimensional structure of frozen-hydrated bacteriophage φX174. J Struct Biol. 1992;108:168–175. doi: 10.1016/1047-8477(92)90016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna R, et al. Incardona NL. Atomic structure of single-stranded DNA bacteriophage φX174 and its functional implications. Nature. 1992;355:137–143. doi: 10.1038/355137a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna R, Bowman BR, Ilag LL, Rossmann MG, Fane BA. Atomic structure of the degraded procapsid particle of the bacteriophage G4: induced structural changes in the presence of calcium ions and functional implications. J Mol Biol. 1996;256:736–750. doi: 10.1006/jmbi.1996.0121. [DOI] [PubMed] [Google Scholar]
- 16.Rossmann MG, Johnson JE. Icosahedral RNA virus structure. Annu Rev Biochem. 1989;58:533–573. doi: 10.1146/annurev.bi.58.070189.002533. [DOI] [PubMed] [Google Scholar]
- 17.McKenna R, Ilag LL, Rossmann MG. Analysis of the single-stranded DNA bacteriophage φX174, refined at a resolution of 3.0 Å. J Mol Biol. 1994;237:517–543. doi: 10.1006/jmbi.1994.1253. [DOI] [PubMed] [Google Scholar]
- 18.Tsao J, et al. Parrish CR. The three-dimensional structure of canine parvovirus and its functional implications. Science. 1991;251:1456–1464. doi: 10.1126/science.2006420. [DOI] [PubMed] [Google Scholar]
- 19.Agbandje M, McKenna R, Rossmann MG, Strassheim ML, Parish CR. Structure determination of feline panleukopenia virus empty particles. Proteins. 1993;16:155–171. doi: 10.1002/prot.340160204. [DOI] [PubMed] [Google Scholar]
- 20.Llamas-Saiz AL, et al. Rossmann MG. Structure determination of the minute virus of mice. Acta Cryst D. 1997;53:93–102. doi: 10.1107/S0907444996010566. [DOI] [PubMed] [Google Scholar]
- 21.Edgell MH, Hutchinson CA, III, Sinsheimer RL. The process of infection with bacteriophage φX174. XXVIII. Removal of the spike proteins from the phage capsid. J Mol Biol. 1969;42:547–557. doi: 10.1016/0022-2836(69)90242-3. [DOI] [PubMed] [Google Scholar]
- 22.Kodaira KI, Nakano K, Okada S, Taketo A. Nucleotide sequence of the genome of the bacteriophage α3: interrelationship of the genome structure and the gene products with those of the phages φX174, G4 and φK. Biochim Biophys Acta. 1992;1130:277–288. doi: 10.1016/0167-4781(92)90440-b. [DOI] [PubMed] [Google Scholar]
- 23.Ilag LL, McKenna R, Yadav MP, BeMiller JN, Incardona NL, Rossmann MG. Calcium ion-induced structural changes in phage φX174. J Mol Biol. 1994;244:291–300. doi: 10.1006/jmbi.1994.1730. [DOI] [PubMed] [Google Scholar]
- 24.Fitch WM, Margoliash E. Construction of phylogenetic trees. Science. 1967;155:279–284. doi: 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- 25.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH – a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 26.Jones DT. Progress in protein structure prediction. Curr Opin in Struct Biol. 1997;7:377–387. doi: 10.1016/s0959-440x(97)80055-3. [DOI] [PubMed] [Google Scholar]
- 27.Rost B, Sander C. Prediction of protein structure at better than 70% accuracy. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 28.Speir JA, Munshi S, Wang G, Baker TS, Johnson JE. Structures of the native and swollen forms of cowpea chlorotic mottle virus determined by X-ray crystallography and cryo-electron microscopy. Structure. 1995;3:63–78. doi: 10.1016/s0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munshi S, et al. Johnson JE. The 2.8 Å structure of a T=4 animal virus and its implication for membrane translocation of RNA. J Mol Biol. 1996;261:1–10. doi: 10.1006/jmbi.1996.0437. [DOI] [PubMed] [Google Scholar]
- 30.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1991;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Millar W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Chipman PR, et al. Rossmann MG. Cryo-electron microscopy studies of empty capsids of human parvovirus B19 complexed with its cellular receptor. Proc Natl Acad Sci USA. 1996;93:7502–7506. doi: 10.1073/pnas.93.15.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinsheimer RL. Bacteriophage φX174 and related viruses. Prog Nucleic Acids Res Molec Biol. 1968;8:115–169. [PubMed] [Google Scholar]
- 34.Newbold JE, Sinsheimer RL. Process of infection with bacteriophage φX174. XXXIV. Kinetics of the attachment and eclipse steps of the infection. J Virol. 1970;5:427–431. doi: 10.1128/jvi.5.4.427-431.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dowell CE, Jansz HS, Zandberg J. Infection of Escherichia coli K-12 by bacteriophage φX174. Virology. 1981;114:252–255. doi: 10.1016/0042-6822(81)90271-3. [DOI] [PubMed] [Google Scholar]
- 36.Davies RE. Spiroplasmas from flowers of Bidens pilosa L. and honey bees in Florida: relationship to honey bee spiroplasma AS576 from Maryland. Phytopatho News. 1978;12:7. [Google Scholar]
- 37.Whitcomb RF. Culture media for spiroplasmas. In: Razin S, Tully JG, editors. Methods in Mycoplasmology. I. Academic Press; London: 1983. pp. 147–159. [Google Scholar]
- 38.Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- 39.Dubochet J, et al. Schultz P. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 40.Olson NH, Baker TS, Johnson JE, Hendry DA. The three-dimensional structure of frozen-hydrated Nudaurelia capensis β virus, a T=4 insect virus. J Struct Biol. 1990;105:111–122. doi: 10.1016/1047-8477(90)90105-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker TS, Cheng RH. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 42.Baker TS, Newcomb WW, Olson NH, Cowsert LM, Olson C, Brown JC. Structures of bovine and human papillomaviruses: analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys J. 1991;60:1445–1456. doi: 10.1016/S0006-3495(91)82181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conway JF, Trus BL, Booy FP, Newcomb WW, Brown JC, Steven AC. Visualization of three-dimensional density maps reconstructed from cryoelectron micrographs of viral capsids. J Struct Biol. 1996;116:200–208. doi: 10.1006/jsbi.1996.0031. [DOI] [PubMed] [Google Scholar]
- 44.Fuller SD, Butcher SJ, Cheng RH, Baker TS. Three-dimensional reconstruction of icosahedral particles – the uncommon line. J Struct Biol. 1996;116:48–55. doi: 10.1006/jsbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 45.Baker TS, Drak J, Bina M. Reconstruction of the three-dimensional structure of simian virus 40 and visualization of the chromatin core. Proc Natl Acad Sci USA. 1988;85:422–426. doi: 10.1073/pnas.85.2.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baker TS, Drak J, Bina M. The capsid of small papova viruses contains 72 pentameric capsomeres: direct evidence from cryo-electron microscopy of simian virus 40. Biophys J. 1989;55:243–253. doi: 10.1016/S0006-3495(89)82799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyoshima C, Unwin N. Contrast transfer for frozen-hydrated specimens: determination from pairs of defocused images. Ultramicroscopy. 1988;25:279–292. doi: 10.1016/0304-3991(88)90003-4. [DOI] [PubMed] [Google Scholar]
- 48.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bilofsky HS, et al. Tung CS. The GenBank genetic sequence databank. Nucleic Acids Res. 1986;14:1–4. doi: 10.1093/nar/14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gribskov M, McLachlan AD, Eisenberg D. Profile analysis: detection of distantly related proteins. Proc Natl Acad Sci USA. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Cryst A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 52.Bernstein FC, et al. Tasumi M. The protein data bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 53.Kraulis P. MOLSCRIPT: a program to produce both detail and schematic plots of proteins structures 1991 [Google Scholar]
- 54.Smith TJ. MacInPlot: a program to display electron density and atomic models on the Macintosh personal computer. J Appl Cryst. 1990;23:141–142. [Google Scholar]