

Abstract
Recent technological advances led to an appreciation of the genetic complexity of human acute myeloid leukemia (AML) but underlying progenitor cells remain poorly understood because their rarity precludes direct study. We developed a co-culture method integrating hypoxia, aryl hydrocarbon receptor inhibition, and micro-environmental support via human endothelial cells to isolate these cells. X-chromosome inactivation studies of the least mature precursors derived following prolonged culture of CD34+/CD33− cells revealed polyclonal growth in highly curable AMLs, suggesting mutations necessary for clonal expansion were acquired in more mature progenitors. Consistently, in core-binding factor (CBF) leukemias with known complementing mutations, immature precursors derived following prolonged culture of CD34+/CD33− cells harbored neither mutation or the CBF mutation alone, whereas more mature precursors often carried both mutations. These results were in contrast to those with leukemias with poor prognosis that showed clonal dominance in the least mature precursors. These data indicate heterogeneity among progenitors in human AML that may have prognostic and therapeutic implications.
Keywords: Acute myeloid leukemia (AML), Core-binding factor, Leukemic stem/progenitor cell, Long-term culture, Prognosis, X chromosome inactivation
INTRODUCTION
Recent technological advances led to an appreciation of the genetic complexity of human acute myeloid leukemias (AMLs).1 Still, their cellular origin remains unclear, with ongoing controversy as to whether these leukemias arise from transformed hematopoietic stem cells or emerge as a result of genetic events occurring in more mature CD33+ myeloid progenitor cells.2–7 Heterogeneity among AML stem/progenitor cells was suggested several decades ago in studies on X chromosome inactivation patterns. Studying a small cohort of women with AML and heterozygosity for glucose-6-phosphate dehydrogenase (G6PD) isoenzymes, Fialkow et al. found the clonal process to be dominant in multiple cell lineages in some cases, suggesting AML origination and disease expansion at the level of pluripotent stem/progenitor cells.8 In others, clonal dominance was limited to granulocytes and monocytes,8 suggesting that expansion of the malignant clone could occur at the level of committed myeloid precursors. Indeed, in some of the latter cases, removal of CD33+ cells in vitro via CD33-directed complement-mediated lysis or fluorescence-activated cell sorting (FACS) followed by placement of the remaining CD33− cells in long-term culture together with irradiated allogeneic stroma yielded colony-forming cells (CFCs) with X chromosome inactivation patterns consistent with predominantly or completely non-clonal hematopoiesis.9, 10 While it is conceivable that the nature of the cells giving rise to AML may have important implications, the clinical significance of the apparent stem cell heterogeneity was not studied in these classic investigations.
Next-generation sequencing now allows the genetic mapping of distinct human AML clones and can even identify relatively minor disease subclones.11–14 However, the estimated frequency of leukemia-initiating cells in unsorted human AML specimens15, 16 is several orders of magnitude lower than the current resolution of 1–2% achieved with genomic techniques. Thus, these cells escape study without prior enrichment. Yet, enrichment strategies for AML stem/progenitor cells are challenging because of their antigenic heterogeneity,15, 17–19 and because stem cells share phenotypes with cells that have more limited functional capabilities, as suggested by the observation that phenotypically enriched “leukemic stem cell populations” engraft immunodeficient mice only at low frequency.15, 19–22 Therefore, it is generally accepted that indirect strategies based on functional stem/progenitor cell characteristics are required to study these key cells, with prominent examples being xenotransplantation assays and prolonged in vitro culture on feeder layers.
Xenotransplantation assays are nowadays widely used to interrogate AML stem/progenitor cells,23 although their success rate in unselected patient specimens can be relatively low, and the testing of patient cohorts large enough to provide clinically relevant analyses is costly and time consuming. However, engraftment in immunodeficient mice may be a non-random, inherent AML cell property indicative of poor outcome,24–30 offering the possibility that xenotransplantation models underestimate AML stem cell diversity and provide a skewed, incomplete assessment of these cells.7 Furthermore, some observations have raised the concern that cells without bona fide human-relevant stem cell properties may be transplantable in such model systems.7 This possibility was suggested by the finding that CD33+ cord blood cells could engraft with multilineage hematopoiesis.31 This was unexpected because in vitro studies on normal bone marrow indicated that CD33 was not expressed on pluripotent hematopoietic stem cells32–35 and because clinical studies demonstrated delayed but durable multilineage engraftment after transplantation of CD33-depleted autografts in patients with AML.36, 37
As an alternative to xenotransplantation assays, prolonged in vitro culture methodologies have been established that enable the study of highly immature hematopoietic cells on a functional rather than phenotypic level;38 yet, their utility for human AML was so far limited because of the infrequent in vitro growth of patient specimens. To overcome this hurdle and functionally assess progenitor cells involved in the leukemogenic process, we have developed a novel long-term culture method to isolate highly immature normal, preleukemic, and leukemic precursors contained in specimens from patients with AML. Through their increased proliferative potential after extended in vitro culture, such rare precursors and immediate CFC progeny can be separated from the large number of blasts, and then become amenable to clonal and molecular analysis. Using this approach, we are able to demonstrate distinct clinical outcomes associated with clonal expansion among the least mature progenitor cells. To further assess the precursor populations with respect to acquisition of mutations, we also molecularly studied phenotypically less and more mature progenitors of selected leukemias, focusing on core-binding factor (CBF) leukemias with known complementing mutations. We show distinct molecular profiles of precursors at distinct developmental stages, indicative of a multistep, maturation-linked mutation acquisition, including cells with a profile consistent with that of preleukemic progenitor cells in the least mature cell population in some cases.
MATERIALS AND METHODS
Primary AML specimens
For our studies, frozen aliquots of Ficoll-isolated mononuclear cells (containing normal, preleukemic, and leukemic progenitor cells) from pretreatment bone marrow or peripheral blood specimens from AML patients were used from the Children’s Oncology Group (COG) AML Reference Laboratory, the SWOG Tumor Bank, and a Fred Hutchinson Cancer Research Center (FHCRC) sample repository. The FHCRC Institutional Review Board, the COG Myeloid Disease Biology Committee, and the SWOG Leukemia Committee approved the research.
Fluorescence-activated cell sorting (FACS) of primary AML specimens
After thawing and staining with directly labeled antibodies, CD34+/CD33− and CD34+/CD33+ cells were separated using a FACSAria flow cytometer (BD Biosciences, San Jose, CA). CD34+/CD33− cells were defined as CD34+ cells that included the bottom 5% of CD33-stained cells as long as staining did not exceed 85% of the isotype control staining, whereas CD34+/CD33+ cells were defined as the top 50% of CD33+ cells that were also CD34+.
In vitro culture of primary hematopoietic cells
For liquid cultures, unsorted and FACS-isolated cells were cultured in Iscoves modified Dulbecco medium (IMDM; Invitrogen, Carlsbad, CA) with 20% fetal bovine serum (FBS; HyClone, Thermo Scientific, Logan, UT) containing the recombinant human cytokines SCF, IL-6, FLT3 ligand, and TPO (all from Invitrogen), StemRegenin-1 (SR1, Cellagen Technology, San Diego, CA), and penicillin-streptomycin. For co-cultures of primary normal and abnormal hematopoietic cells, human umbilical cord endothelial cells (ECs) transduced with a lentiviral construct encoding the open reading from 1 of the early region 4 of adenovirus (E4ORF1) were plated in 48-well plates at 30,000 cells per well in growth medium (Medium199 with Earle’s balanced salt solution, L-glutamine, HEPES, NaHCO3; BioWhittaker, Lonza, Walkersville, MD) supplemented with 20% FBS, ECGS endothelial mitogen (Biomedical Technologies, Stoughton, MA), heparin, HEPES, L-glutamine, penicillin-streptomycin, and Fungizone. 18–24 hours later, medium was removed and aliquots of unsorted cells and FACS-isolated immature myeloid cell populations were added in StemSpan (Stemcell Technologies, Vancouver, Canada) containing human recombinant SCF, SR1, and penicillin-streptomycin. Cultures were replenished with fresh medium weekly and kept at 37°C in 3% O2 and 5% CO2 for up to 8 weeks. Defined fractions of the cultures were removed biweekly and subjected to CFC assays.
CFC assays
Aliquots of uncultured and cultured hematopoietic cells were subjected to CFC assays using standard semi-solid methylcellulose-based media.39, 40 After 10–14 days at 37°C in 3% O2 and 5% CO2, colony forming units-granulocyte and/or monocyte (CFU-GM) of at least 30–50 cells were harvested.40
Determination of X chromosome inactivation
Clonality analyses of unsorted AML cells and individual CFU-GMs were performed using a methylation-specific polymerase chain reaction (PCR), which identifies the patterns of methylated and unmethylated alleles of the polymorphic CAG repeat in the human androgen receptor alpha (HUMARA) gene by bisulfite modification, essentially as described previously.41 A representative example of this assessment for clonality is depicted in Supplemental Figure 1. Specimen growth was considered sufficient if it yielded ≥5 CFU-GMs. A clonal growth pattern was assumed if the specimen yielded >75% disease-specific CFU-GMs.
Molecular analyses of patient-specific somatic mutations
Unsorted and sorted cell subpopulations as well as resulting individual CFU-GMs were analyzed for the presence of the patient-specific somatic abnormalities using PCR- or reverse transcriptase (RT)-PCR-based assays.
Statistical analyses
The Kaplan-Meier method was used to estimate overall survival (defined as time from study entry to death) and relapse-free survival (defined as time from remission entry to relapse or death), and compared between patient groups with the log-rank statistic. Cox proportional hazards models were used to estimate the hazard ratio (HR) for defined groups of patients in univariate analyses.
RESULTS
Characterization of co-culture assay to study human hematopoietic progenitor cells
To address the limitation of infrequent in vitro growth of primary AML patient samples, we developed an optimized culture method to support normal, preleukemic, and leukemic progenitor cells contained in such specimens. Modeled after the classic long-term culture initiating cell assay, this assay detects primitive cells capable of giving rise to CFCs after prolonged culture on competent feeder layers.38 We integrated 3 novel components – hypoxia, inhibition of aryl hydrocarbon receptors (AHRs),42 and micro-environmental support via primary human endothelial cells (ECs) transduced with the adenoviral E4ORF1 gene43 – each uniquely supporting AML-associated human progenitor cells. Specifically, as detailed in the Data Supplement, we found that hypoxia better supported growth of transformed CFCs derived from uncultured CD34+/CD33− cells than normoxia. Furthermore, the AHR antagonist, SR1, supported CD34+/CD33− cells contained in human AML specimens and maintained their immature phenotype when cultured over 8 weeks in cytokine-containing liquid medium in the presence of serum, and enhanced growth of derived CFU-GMs (Supplemental Figure 2). Finally, E4ORF1+ EC co-cultures, unlike cytokine-containing liquid medium cultures, expanded progenitor cells with CD34+/CD33− phenotype over 8 weeks (Supplemental Figure 3). Importantly, the supporting effects of SR1 and E4ORF1+ ECs on leukemic or preleukemic hematopoietic progenitor cells were complimentary, with the combined use providing superior support of progenitor cells harboring AML-associated somatic mutations compared to either component alone (Supplemental Figure 4), and only in SR1 containing EC co-cultures did we typically observe leukemic or preleukemic CFU-GMs after long-term culture. Taken together, the combined use of hypoxia, SR1, and E4ORF1+ ECs appeared most effective and was utilized in subsequent studies. Among 51 AML specimens (38 pediatric, 13 adult) across the entire cytogenetic/molecular spectrum, 39 (76%) and 31 (61%) yielded CFC growth after short-term (2 weeks) and long-term (4–8 weeks) co-culture, respectively, when starting from 50,000–300,000 unsorted cells and/or FACS-isolated CD34+/CD33− and CD34+/CD33+ cells from 1 frozen cell aliquot. Of note, during the 8 weeks of co-cultures, CD34+/CD33− cells displayed greater proliferative potential consistent with their immaturity by yielding a higher number of cells and CFU-GMs than CD34+/CD33+ cells (Supplemental Figure 5). Furthermore, while E4ORF1+ EC co-cultures with SR1 in hypoxia supported phenotypically immature CD34+/CD33− cells after long-term culture of CD34+/CD33− cells, no CD34+/CD33− cells were found after long-term culture of CD34+/CD33+ cells (Supplemental Figure 6). Together, these data were consistent with the more limited proliferative potential of the more mature CD34+/CD33+ (relative to the CD34+/CD33−) cell population, supporting the notion of stable CD33 expression in the appropriate context of hematopoietic cell maturation, and arguing against the inappropriate “loss” of expression of this antigen in culture. Relevant to the studies presented herein, paired analyses of different culture methodologies in a set of 25 core-binding factor (CBF) AMLs revealed that E4ORF1+ EC/SR1-based co-cultures most successfully supported long-term growth, with 16/25 specimens (64%) resulting in CFC growth and 10 out of these 16 specimens (63%) yielding CFU-GMs harboring the CBF translocation (Table 1). In contrast, following cytokine-containing liquid medium cultures kept under normoxic conditions, only 8/25 (32%) yielded some but limited CFU-GM growth after short- or long-term culture in cytokine-containing liquid medium, and CFU-GMs from only 1/8 specimens (13%) carried the CBF translocation in some colonies after long-term culture, indicating that E4ORF1+ EC/SR1-based co-cultures are significantly superior in supporting leukemic or preleukemic progenitor cells in vitro long-term.
TABLE 1.
Growth and molecular analysis of CFU-GMs derived from various culture methods of primary hematopoietic cells from 25 patients with CBF leukemias
| Culture Condition | Growth after 2 weeks | Specimens with mutation+ CFU-GMs after growth for 2 weeks | Growth after ≥4 weeks | Specimens with mutation+ CFU-GMs after growth for ≥4 weeks |
|---|---|---|---|---|
| Cytokine-containing liquid medium, normoxia | 8/25 (32%) | 3/8 (37.5%) | 8/25 (32%) | 1/8 (25%) |
| Cytokine-containing liquid medium, hypoxia | 12/25 (48%) | 6/12 (50.0%) | 6/25 (24%) | 2/6 (33%) |
| Cytokine-containing liquid medium, hypoxia+SR1 | 20/25 (80%) | 12/20 (60.0%) | 18/25 (72%) | 5/18 (28%) |
| E4ORF1+ ECs, Hypoxia+SR1 | 16/25 (64%) | 13/16 (81.3%) | 16/25 (64%) | 10/16 (63%) |
Heterogeneous clonal expansion of human AML amongst CD34+/CD33− cells
Utilizing this co-culture assay, we studied the stage of progenitor cell maturation at which clonal expansion occurred in human AML. To accomplish this, we isolated phenotypically less mature CD34+/CD33− and more mature CD34+/CD33+ cells from AML specimens and subjected these cell aliquots to direct CFC assays (i.e. without prior culture) or CFC assays after short-term (2 weeks) or long-term (4, 6, and 8 weeks) co-culture; the experimental strategy pursued is depicted in Figure 1A, and 3 representative examples of CD34+/CD33− and CD34+/CD33+ cell isolation from primary human AML specimens are shown in Figure 1B. Individual CFU-GMs derived from uncultured cells as well as cells that were cultured short- and long-term were then individually analyzed for their X chromosome inactivation pattern; results from 18 informative bone marrow specimens are summarized in Table 2. After long-term co-culture, CFU-GMs derived from CD34+/CD33− cells yielded colonies consistent with a polyclonal growth pattern in 5 specimens, whereas such CFU-GMs yielded colonies consistent with a clonal growth pattern in the other 13 specimens. These data indicated heterogeneity amongst the immature progenitor cells, with existence of a subset of leukemias that did not predominantly involve CD34+/CD33− progenitors. Importantly, we found examples of AMLs with similar cytogenetic abnormalities (e.g. t(8;21), see Table 2) that showed polyclonal growth in long-term cultures of CD34+/CD33− cells in specimens from some patients but clonal growth in samples from other patients, indicating progenitor cell heterogeneity even within cytogenetically well-defined AML subsets.
Figure 1. Experimental Strategy.

(A) Diagram depicting the experimental strategy pursued with primary human AML specimens to assess bulk AML cells for the disease-associated X chromosome inactivation pattern and presence of somatic mutations, to isolate phenotypically immature cell populations based on expression of CD34 and CD33 with subsequent direct clonal/molecular analysis, and to subject these isolated cell populations to short- and long-term in vitro E4ORF1+ EC co-culture with SR1 in the presence of hypoxia, with subsequent CFC assays and clonal/molecular analyses of individual CFU-GMs. (B) Scatter and histogram plots from 3 representative primary AML specimens illustrating the sorting strategy pursued to isolate CD34+/CD33− and CD34+/CD33+ cells. Fluorescence intensity of CD34 and CD33 is shown in black/bold, whereas staining with isotype control antibodies is shown in grey/dashed. FSC, forward scatter; SSC, side scatter.
TABLE 2.
Clonal analysis of CFU-GMs derived after direct plating (i.e. without culture) or from short-term (2 weeks) or long-term (4–8 weeks) co-culture of primary hematopoietic cells from bone marrow specimens from patients with AML
| Direct Plating | Short-Term Culture | ||||
|---|---|---|---|---|---|
| Sample | Cytogenetics | CD34+/CD33− Cells |
CD34+/CD33+/ Unsorted Cells |
CD34+/CD33− Cells |
CD34+/CD33+/ Unsorted Cells |
| 205377 | 46,XX,t(8;21)(q22;q22)[19]/46,XX[1] | Polyclonal | Polyclonal | Polyclonal | Polyclonal |
| 213865 | 46,XX,inv(16)(p13.1q22)[21] | Clonal | Clonal | -- | -- |
| 208567 | 46,XX,inv(16)(p13.1q22)[17]/47,idem,+22[3] | -- | -- | -- | Clonal |
| 767487 | 46,XX,t(6;11)(q27;q23)[17]/46,XX[4] | -- | -- | -- | -- |
| 774353 | 46,XX,t(8;21)(q22;q22)[13]/47,idem,+6[7] | Clonal | Clonal | Polyclonal | Clonal |
| 795324 | 45,XX,add(2)(q37),−10,der(11)del(11)(q13q22)trp(11)(q23q23) t(10;11)(q11.2;q23)[14]/45,idem,− der(2),dup(7)(p15p22)[4]/46,XX[2] | -- | Clonal | -- | Clonal |
| 790790 | 46,XX[21] | Clonal | Clonal | Clonal | Clonal |
| 765911 | 46,XX,t(6;11)(q27;q23)[20] | Clonal | Clonal | Clonal | Clonal |
| 768832 | 46,XX,t(6;9)(p23;q34) | Clonal | Clonal | Clonal | Clonal |
| 211571 | 46,XX,t(8;21)(q22;q22)[20] | -- | Clonal | Clonal | Clonal |
| 153112 | 45,X,−X,t(8;20;21)(q22;p11.2;q22)[19] | Clonal | Clonal | -- | Clonal |
| 126865 | 46,X,t(X;21)(p11.2;q22)[20] | -- | Polyclonal | Clonal | Clonal |
| 212496 | 46,XX,inv(5)(q2?2q3?3),del(12)(q13q24.1)[19]/46,XX[1] | Polyclonal | Polyclonal | Clonal | Clonal |
| 212565 | 46,XX[20] | Clonal | Clonal | Clonal | Clonal |
| 206869 | 47,XX,+1,der(1;18)(q10;q10),+8[18] | Clonal | Clonal | -- | Clonal |
| 162772 | 45,XX,add(3)(q29),−7[13]/48,XX,+X,add(3)(q29),−7,+10,+21[8] | -- | Clonal | Clonal | Clonal |
| 117802 | 46,XX[21] | -- | Clonal | -- | -- |
| 159278 | 45,XX,der(14;21)(q10;q10)?c[18]/46,XX,der(14;21)(q10;q10)?c,+i(21)(q10)[2] | Clonal | Clonal | Clonal | Clonal |
--denotes no/insufficient growth
Clinical significance of AML progenitor cell heterogeneity
To determine the prognostic significance of this AML progenitor cell heterogeneity, we studied pre-treatment specimens from 50 women with newly diagnosed AML who received standard induction therapies without the CD33-targeting immunoconjugate, gemtuzumab ozogamicin (GO), on recent SWOG trials. To avoid any potential bias introduced by the source of hematopoietic progenitor cells, only bone marrow specimens were used for this analysis. Forty-five of the 50 specimens (90%) were informative with regard to HUMARA length polymorphism (i.e. allowed clonal assessment of isolated cell progeny), and 16 of these (36%) yielded CFU-GMs derived from CD34+/CD33− cells after long-term (≥4 weeks) co-culture sufficient for clonal analyses. In 10 of the 16 specimens (including 1 CBF AML), such CFU-GMs yielded a growth pattern consistent with clonal growth, while a growth pattern consistent with polyclonal growth was found in such CFU-GMs in 6 specimens. Of note, among the 10 specimens with clonal growth, there were 2 cases of CBF AML, 1 normal karyotype AML with NPM1 mutation but without FLT3/ITD, 2 normal karyotype AMLs without NPM1 or FLT3 mutation, 4 other intermediate-risk AMLs, and 1 adverse-risk AML; in contrast, the 6 specimens with polyclonal growth were cytogenetically classified as favorable-risk (n=3) or intermediate-risk (n=3, including 2 cases with normal karyotype and NPM1 mutation but without FLT3/ITD). The complete remission rate was similar among patients whose specimens yielded polyclonal CFU-GMs derived from CD34+/CD33− cells relative to those with clonal progenitor growth (4/6 [67%] vs. 7/10 [70%]). However, the overall survival of the 6 patients whose specimens yielded polyclonal CFU-GMs derived from CD34+/CD33− or unsorted cells was significantly longer than that for the 10 patients whose specimens yielded a clonal progenitor growth pattern after long-term culture of CD34+/CD33− cells (P=0.045; Figure 2A). Likewise, among the 11 patients who achieved a complete remission, the relapse-free survival of the 4 patients whose specimens yielded polyclonal CFU-GMs derived from CD34+/CD33− or unsorted cells was significantly longer than that for the 7 patients whose specimens yielded a clonal progenitor growth pattern after long-term culture of CD34+/CD33− cells (P=0.03; Figure 2B).
Figure 2. Clinical Significance of AML Progenitor Cell Heterogeneity.
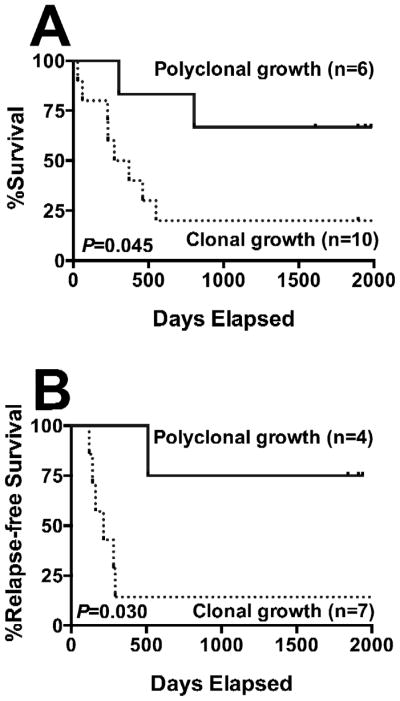
(A) Overall survival and (B) relapse-free survival of adult patients with newly diagnosed non-APL AML undergoing standard induction chemotherapy, stratified based on polyclonal vs. clonal growth of CFU-GMs derived from FACS-isolated CD34+/CD33− cells after long-term (≥4 weeks) in vitro co-culture.
Hierarchical maturation-linked mutation acquisition in hematopoietic progenitor cells
The data presented thus far suggested that mutations necessary for clonal expansion of either normal or preleukemic progenitors that fail to achieve clonal dominance may only be acquired in some patients in more mature progenitor cells, and that these progenitors may have more limited proliferative potential. Our analyses further suggested that this pattern of mutation acquisition could be found in at least a subset of leukemias with CBF translocations. To study mutation acquisition in CBF leukemias in more detail, we investigated 57 specimens from 44 pediatric and 13 adult patients with newly diagnosed CBF AML (t(8;21) [n=22] or inv(16)/t(16;16) [n=35]); in 41 patients, at least 1 additional mutation was clinically recognized as follows: KIT (n=29), FLT3/ITD (n=10), FLT3/ALM (n=1), and NRAS (n=1).
We first analyzed CD34+/CD33− and CD34+/CD33+ cells from CBF AML specimens with known 2nd mutations. Thirty-six of 41 tested specimens (88%) had paired molecular data available for analysis: in CD34+/CD33− cells of 25 specimens, both mutations were detected, while either the CBF translocation only or neither mutation were found in 9 specimens and 2 specimens, respectively. In contrast, within CD34+/CD33+ progenitors, both CBF and complementing alterations were found in all but 1 specimen, in which only the CBF aberration was detected (Table 3; data on individual specimens are summarized in Supplemental Table 2). We also subjected uncultured CD34+/CD33− and CD34+/CD33+ cells to CFC assays and obtained informative data from 20 specimens. As summarized in Table 4, we found heterogeneity among the CFU-GMs derived from CD34+/CD33− cells across these specimens with regard to mutation patterns. In 11 specimens, CD34+/CD33− cells yielded CFU-GMs that carried only CBF translocations but not complementing mutations. In contrast, in 7 specimens, CFU-GMs were derived from CD34+/CD33− cells that carried both mutations. In the remaining 2 specimens, only presumably normal CFU-GMs were derived from CD34+/CD33− cells whereas corresponding CFU-GMs derived from CD34+/CD33+ cells carried either both mutations or the CBF translocations alone; the latter suggested origination from a preleukemic cell that had not yet acquired the requisite complementing mutation necessary for frank leukemic transformation. Importantly, in no instance did we identify the complementing mutation but not the CBF aberration.
TABLE 3.
Disease characteristics and detection of AML-associated somatic mutations in CD34+/CD33− and CD34+/CD33+ progenitors from patients with CBF leukemias and complementing second mutation
| CD34+/CD33− Cells | CD34+/CD33+ Cells* | |||||
|---|---|---|---|---|---|---|
| Mutations | Samples | Both Mutations | CBF+ Only | No Mutation | Both Mutations | CBF+ Only |
| t(8;21) + FLT3/ITD | N=6 | 5 | 1 | 0 | 6 | 0 |
| t(8;21) + KIT | N=9 | 5 | 4 | 0 | 8 | 1 |
| inv(16) + FLT3/ITD | N=3 | 2 | 1 | 0 | 3 | 0 |
| inv(16) + FLT3/ALM | N=1 | 0 | 1 | 0 | 1 | 0 |
| inv(16) + KIT | N=16 | 13 | 2 | 1 | 16 | 0 |
| inv(16) + NRAS | N=1 | 0 | 0 | 1 | 1 | 0 |
Note: no case was found that lacked both mutations in CD34+/CD33+ cells.
TABLE 4.
Growth and molecular analysis of CFU-GMs derived from uncultured CD34+/CD33− and CD34+/CD33+ progenitor cells from patients with CBF leukemias and complementing second mutation
| Sample | Mutation | CD34+/CD33− Cells | CD34+/CD33+ Cells |
|---|---|---|---|
| 1. RO50397 | t(8;21) + KIT | No mutation | CBF+ only as well as both mutations |
| 2. 792590 | t(8;21) + KIT | No mutation | CBF+ only as well as both mutations |
| 3. 71494 | t(8;21) + FLT3/ITD | CBF+ only | --- |
| 4. 600685 | t(8;21) + FLT3/ITD | CBF+ only | --- |
| 5. 715096 | t(8;21) + FLT3/ITD | CBF+ only | --- |
| 6. RO50077 | t(8;21) + FLT3/ITD | CBF+ only | CBF+ only as well as both mutations |
| 7. 719733 | t(8;21) + KIT | CBF+ only | --- |
| 8. 765326 | inv(16) + KIT | CBF+ only | --- |
| 9. 766940 | inv(16) + KIT | CBF+ only | |
| 10. 66830 | inv(16) + KIT | CBF+ only | --- |
| 11. 764821 | inv(16) + KIT | CBF+ only | CBF+ only |
| 12. 775184 | inv(16) + KIT | CBF+ only | CBF+ only as well as both mutations |
| 13. 26450 | inv(16) + NRAS | CBF+ only | CBF+ only |
| 14. 66980 | t(8;21) + KIT | CBF+ only as well as both mutations | CBF+ only as well as both mutations |
| 15. 715381 | t(8;21) + KIT | CBF+ only as well as both mutations | --- |
| 16. 750501 | t(8;21) + KIT | CBF+ only as well as both mutations | |
| 17. 58340 | t(8;21) + FLT3/ITD | CBF+ only as well as both mutations | --- |
| 18. 71863 | inv(16) + FLT3/ITD | CBF+ only as well as both mutations | No mutation |
| 19. 706269 | inv(16) + KIT | CBF+ only as well as both mutations | CBF+ only as well as both mutations |
| 20. 774353 | t(8;21) + KIT | Both mutations | CBF+ only as well as both mutations |
These initial studies therefore provide the first evidence of sequential acquisition of complementing mutations in development of clonal dominance and frank leukemia, with CBF translocations being the earlier mutation. Further assessment of the stage-specific acquisition of mutations in 16 patients with CBF leukemias and complementing second mutation was therefore carried out using the same experimental strategy as depicted in Figure 1A. Informative data are summarized in Table 5. After short-term culture of CD34+/CD33− progenitor cells, 5 out of 6 specimens that gave rise to CFU-GMs harbored the CBF mutation, but only 3 of these 5 specimens also carried the second mutation. However, after 4–8 weeks of culture, of 5 specimens that gave rise to CFU-GMs, 2 harbored the CBF mutation and none harbored the second mutation, suggesting the least mature cells in these leukemias were predominantly normal or preleukemic. Consistent with this notion, CFU-GMs derived from the more mature CD34+/CD33+ progenitor cells harbored both mutations in 4/5 cases after short-term and 2/4 after long-term culture, and in all 3 cases where the least mature cells lacked CBF and known complementing mutations, more mature precursors derived following long-term culture of CD34+/CD33+ progenitors or unsorted cells harbored either the CBF translocation alone or both mutations. Overall, these studies indicated heterogeneity among progenitor cells in CBF leukemias, with the least mature cells being predominantly or completely preleukemic or normal, and expansion of frankly leukemic cells as a result of the acquisition of additional mutations present in more mature precursors, consistent with hierarchical mutation acquisition in this AML subset.
TABLE 5.
Growth and molecular analysis of CFU-GMs derived from short-term (2 weeks) or long-term (4–8 weeks) culture of primary hematopoietic cells from patients with CBF leukemias and complementing second mutation
| Sample | Mutation | CD34+/CD33− Cells | CD34+/CD33+ Cells |
|---|---|---|---|
| Short-term culture (2 weeks) | |||
| 1. 798451 (BM) | inv(16) + KIT | No mutation | ---* |
| 2. 781469 (BM) | t(8;21) + KIT | --- | CBF+ only |
| 3. 26450 (PB) | inv(16) + NRAS | CBF+ only | ---* |
| 4. 001-001-012 (PB) | inv(16) + FLT3/ITD | CBF+ only | CBF+ only as well as both mutations |
| 5. 791406 (BM) | t(8;21) + FLT3/ITD | Both mutations | Both mutations |
| 6. 792590 (BM) | t(8;21) + KIT | Both mutations | Both mutations |
| 7. 774353 (BM) | t(8;21) + KIT | Both mutations | Both mutations |
| Long-term culture (4–8 weeks) | |||
| 1. RO50397 (BM) | t(8;21) + KIT | No mutation | No mutation** |
| 2. 766411 (BM) | inv(16) + KIT | --- | CBF+ only |
| 3. 774353 (BM) | t(8;21) + KIT | No mutation | Both mutations |
| 4. 792590 (BM) | t(8;21) + KIT | No mutation | CBF+ only as well as both mutations |
| 5. 26450 (PB) | inv(16) + NRAS | CBF+ only | --- |
| 6. 775184 (BM) | inv(16) + KIT | CBF+ only | --- |
CBF+ only CFC-GMs were isolated from short-term cultured unsorted cells.
CBF+ only CFC-GMs were isolated from long-term cultured unsorted cells.
Abbreviations: “---“ denotes no CFU-GM growth; BM, bone marrow; PB, peripheral blood.
DISCUSSION
Because of the technical limitations of studying very rare cells, the hurdle of identifying the molecular characteristics of the stem cells driving leukemic growth has remained.15, 16 As a result, the nature of the stem/progenitor cells involved in human leukemogenesis remains controversial,2–4, 6, 7 and the temporal sequence of mutation acquisition at the stem/progenitor cell level is unclear. As evidenced by our data, the combined use of E4ORF1+-transduced ECs, SR1, and hypoxia offers a novel tool to decipher early and late mutational events during leukemogenesis and to examine clonal evolution in individual leukemias on a functional level in progenitor cells, thus providing a significant advancement for the study of AML transformation. To our knowledge, the investigations presented herein are the first to assess acquisition of somatic mutations during human leukemogenesis based on functional characterization of the cells in which these lesions occur. Overall, our studies identify significant diversity among the progenitor cells across individual AML patients, with a subset of leukemias that is characterized by apparently normal cells in the least mature precursors and expansion of leukemic cells and mutation acquisition in more mature precursors with limited differential potential. Our findings indicate that this progenitor cell diversity has important clinical implications, as the treatment outcome of the patients with these leukemias was very favorable. Thus, our data suggest that the nature of progenitor cells involved in the leukemogenic process is directly related to prognosis and therapeutic response in general and may be pivotal for the success of treatments that target differentiation antigens such as CD33.
Unlike with the use of G6PD isoenzymes, as utilized in the initial clonal analyses of human AML,8 the use of the DNA methylation status of a highly polymorphic CAG tandem repeat sequence in the HUMARA gene allows clonal studies in most women,44 permitting a generalizable assessment of clonality. Our data are consistent with the classic G6PD-based observations, supporting further the notion of progenitor cell heterogeneity in human AML and indicating that complementing mutations may be acquired by the malignant clone (or, at a minimum, the clone expands) only within more mature, committed myeloid progenitors in some leukemias; in these, highly immature precursors thus show a predominantly polyclonal growth, as evidenced by a polyclonal growth pattern of CFU-GMs that are derived after long-term culture of CD34+/CD33− cells in our assay. In other leukemias, as evidenced by a clonal growth pattern of CFU-GMs derived after long-term culture of CD34+/CD33− cells in our assay, mutations that lead to clonal disease expansion are acquired among highly immature precursors. However, our findings extend previous results by providing first evidence of an intricate relationship between the nature of involved AML progenitor cell and clinical characteristics. Specifically, the investigations presented herein are the first to indicate that the leukemias in which clonal expansion of leukemic cells and mutation acquisition only occurs in more mature precursors intrinsically have a better prognosis than those in which AML expansion occurs in the least mature precursors. This favorable leukemia subset may thus resemble APL, a highly curable leukemia for which small studies have indicated predominant involvement of committed CD33+ myeloid progenitors.45 Further support for the notion that mutations occurring in committed myeloid progenitors may result in less resistant disease and better response to conventional chemotherapy than those occurring in pluripotent stem/progenitor cells comes from correlative studies that found that leukemias in which somatic abnormalities (such as FLT3/ITD or loss of chromosome 7) could already be detected in CD34+/CD33− progenitors or derived CFCs were more likely resistant to chemotherapy and had a higher relapse risk and worse outcome than leukemias in which these lesions were not detectable at this progenitor maturation stage.39, 40 Possibly unlike known somatic mutations, however, which may have been acquired as a secondary event in the leukemogenic process and mark a disease subclone, X chromosome inactivation patterns allow for the determination of the maturation stage of the earliest clonally involved cell. And finally, this notion is supported by recent data from murine AML models indicating that the cell of origin influences the biology of the resulting leukemia and, importantly, responsiveness to chemotherapeutics.46
Of note, this favorable subset of non-APL leukemias was heterogeneous with regards to known cytogenetic and molecular abnormalities, including some but not all cases of normal karyotype AML with NPM1 mutation and CBF leukemias studied; the latter is consistent with observations by Fialkow et al. who found 3 of the 4 CBF leukemia among those with clonal dominance limited to granulocytes and monocytes.8 These observations immediately suggested heterogeneity in the maturation-linked acquisition of somatic mutations in this cytogenetically well-defined subtype of AML. Using CBF leukemias with complementing RTK or NRAS mutations as a paradigm, our comparison of CFU-GMs derived from uncultured as well as cultured cells indeed points towards significant heterogeneity of the transformation process in this AML subset. Specifically, while isolated CD34+/CD33− cells from some specimens yielded CFU-GMs with both mutations either upon direct analysis or after short-term culture, others produced colonies harboring only the CBF mutation and/or lacked both mutations. Moreover, after long-term culture, isolated CD34+/CD33− cells yielded CFU-GMs carrying the CBF translocation alone or neither the CBF nor complementing mutation. In contrast, isolated CD34+/CD33+ cells yielded colonies harboring both mutations after short- and long-term culture in many cases. Thus, although either the CBF translocation and/or the complementing mutation may arise in CD34+/CD33− cells as suggested by our short-term culture assays, frankly leukemic cells carrying both mutations may primarily expand in more mature precursors. Together, these findings indicate a predominance of preleukemic or presumably normal cells in the least mature precursors after prolonged culture in many cases. Such highly immature progenitor cells carrying the initial somatic clonal aberration alone may be long-lived and could underlie the phenomenon of clonal hematopoiesis in remission and the persistence of small populations of cells harboring CBF translocations in seemingly cured patients with CBF AML.47–49 The absence of the complementing mutation within such highly immature hematopoietic progenitors with great self-renewal potential may also explain why such mutations (e.g. FLT3/ITD) are absent upon disease recurrence in some but not all patients.
Therapeutically, the concept of AML progenitor cell heterogeneity may be essential for the design and success of stem cell-directed therapies as the cellular origin of the cells involved in the transformation process likely defines the display of antigens that could serve as drug targets. In fact, early recognition that some AMLs may predominantly or entirely involve committed CD33+ myeloid progenitors led to efforts targeting underlying malignant stem cells with antibodies recognizing CD33, as exemplified by the development of the immunoconjugate, GO.7 Recent randomized trials have demonstrated that GO improves survival in some patients with AML, in particular those with CBF translocations.50, 51 Our data indicating that, in many CBF AMLs, at least the complementing mutation may expand only within a more mature, committed CD33+ myeloid progenitor may now allow to reconcile the clinical efficacy of GO in this disease subset with origin of the progenitor cells involved in the malignant transformation process. Specifically, successful depletion of CD33+ cells would eradicate the fully transformed, proliferative clone and leave the CD33− preleukemic progenitor cells behind, i.e. re-set the clock to a pre-malignant stage. For a cure, such preleukemic cells may need to be controlled or eliminated by other means, e.g. other chemotherapeutic agents or immunological mechanisms.
Together, our findings indicate that clonal involvement of the least mature precursors may bode for poor prognosis. Diversity in the progenitor cells involved in leukemic transformation may offer an explanation for the differences in chemotherapy responsiveness and outcome of individual patients with AML, even within cytogenetically well-defined disease subsets. Further, in studies of the hierarchical, maturation-linked mutation acquisition in human CBF AML, we found the CBF translocation present in highly immature precursors, possibly resulting in a preleukemic state that then evolved into frank AML with acquisition of the complementing mutation or expansion in more mature precursors. In other cases, expansion of both mutations may occur only at the level of a more mature precursor. Ultimately, knowledge of this progenitor cell heterogeneity may provide the rationale for a stem cell-based AML classification that would facilitate prognostication and utilization of cell surface antigen-targeted therapies.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by grants from the National Cancer Institute/National Institutes of Health (P30-CA015704-35S6 [to R.B.W.], U10-CA098453 [NIH COG Chair Grant, Children’s Hospital of Philadelphia], and U10-CA32102 and U10-CA38926 [to SWOG]), Alex’s Lemonade Stand Foundation (to R.B.W.), the Hope Foundation (to R.B.W.), the Ronald McDonald House Charities of Southern California and Couples Against Leukemia (to R.B.W.), the Leukemia & Lymphoma Society (Specialized Center for Research [SCOR] grant #7008-08 to I.D.B.), a St. Baldrick’s Foundation Career Development Award (to J.A.P.), and a CureSearch Research Fellowship Award (to J.A.P.).
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
Presented in part at the 52nd and 55th Annual Meetings of the American Society of Hematology (December 4–7, 2010, Orlando, FL; and December 7–10, 2013, New Orleans, LA)
Supplementary information is available at Leukemia’s website.
References
- 1.Welch JS, Link DC. Genomics of AML: clinical applications of next-generation sequencing. Hematology Am Soc Hematol Educ Program. 2011;2011:30–35. doi: 10.1182/asheducation-2011.1.30. [DOI] [PubMed] [Google Scholar]
- 2.Passegué E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11842–11849. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stubbs MC, Armstrong SA. Therapeutic implications of leukemia stem cell development. Clin Cancer Res. 2007;13(12):3439–3442. doi: 10.1158/1078-0432.CCR-06-3090. [DOI] [PubMed] [Google Scholar]
- 4.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 5.Lane SW, Gilliland DG. Leukemia stem cells. Semin Cancer Biol. 2010;20(2):71–76. doi: 10.1016/j.semcancer.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Majeti R. Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene. 2011;30(9):1009–1019. doi: 10.1038/onc.2010.511. [DOI] [PubMed] [Google Scholar]
- 7.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fialkow PJ, Singer JW, Raskind WH, Adamson JW, Jacobson RJ, Bernstein ID, et al. Clonal development, stem-cell differentiation, and clinical remissions in acute nonlymphocytic leukemia. N Engl J Med. 1987;317(8):468–473. doi: 10.1056/NEJM198708203170802. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein ID, Singer JW, Andrews RG, Keating A, Powell JS, Bjornson BH, et al. Treatment of acute myeloid leukemia cells in vitro with a monoclonal antibody recognizing a myeloid differentiation antigen allows normal progenitor cells to be expressed. J Clin Invest. 1987;79(4):1153–1159. doi: 10.1172/JCI112932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernstein ID, Singer JW, Smith FO, Andrews RG, Flowers DA, Petersens J, et al. Differences in the frequency of normal and clonal precursors of colony-forming cells in chronic myelogenous leukemia and acute myelogenous leukemia. Blood. 1992;79(7):1811–1816. [PubMed] [Google Scholar]
- 11.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest. 2011;121(1):384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vargaftig J, Taussig DC, Griessinger E, Anjos-Afonso F, Lister TA, Cavenagh J, et al. Frequency of leukemic initiating cells does not depend on the xenotransplantation model used. Leukemia. 2012;26(4):858–860. doi: 10.1038/leu.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112(3):568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 18.Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115(10):1976–1984. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 20.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 21.Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U S A. 2011;108(12):5009–5014. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–152. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica. 2012;97(7):966–974. doi: 10.3324/haematol.2011.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ailles LE, Gerhard B, Kawagoe H, Hogge DE. Growth characteristics of acute myelogenous leukemia progenitors that initiate malignant hematopoiesis in nonobese diabetic/severe combined immunodeficient mice. Blood. 1999;94(5):1761–1772. [PubMed] [Google Scholar]
- 25.Rombouts WJC, Blokland I, Löwenberg B, Ploemacher RE. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia. 2000;14(4):675–683. doi: 10.1038/sj.leu.2401731. [DOI] [PubMed] [Google Scholar]
- 26.Rombouts WJC, Martens ACM, Ploemacher RE. Identification of variables determining the engraftment potential of human acute myeloid leukemia in the immunodeficient NOD/SCID human chimera model. Leukemia. 2000;14(5):889–897. doi: 10.1038/sj.leu.2401777. [DOI] [PubMed] [Google Scholar]
- 27.Lumkul R, Gorin NC, Malehorn MT, Hoehn GT, Zheng R, Baldwin B, et al. Human AML cells in NOD/SCID mice: engraftment potential and gene expression. Leukemia. 2002;16(9):1818–1826. doi: 10.1038/sj.leu.2402632. [DOI] [PubMed] [Google Scholar]
- 28.Monaco G, Konopleva M, Munsell M, Leysath C, Wang RY, Jackson CE, et al. Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells. 2004;22(2):188–201. doi: 10.1634/stemcells.22-2-188. [DOI] [PubMed] [Google Scholar]
- 29.Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107(3):1166–1173. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez PV, Perry RL, Sarry JE, Perl AE, Murphy K, Swider CR, et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia. 2009;23(11):2109–2117. doi: 10.1038/leu.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrews RG, Torok-Storb B, Bernstein ID. Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood. 1983;62(1):124–132. [PubMed] [Google Scholar]
- 33.Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. 1984;8(4):521–534. doi: 10.1016/0145-2126(84)90001-8. [DOI] [PubMed] [Google Scholar]
- 34.Andrews RG, Takahashi M, Segal GM, Powell JS, Bernstein ID, Singer JW. The L4F3 antigen is expressed by unipotent and multipotent colony-forming cells but not by their precursors. Blood. 1986;68(5):1030–1035. [PubMed] [Google Scholar]
- 35.Andrews RG, Singer JW, Bernstein ID. Precursors of colony-forming cells in humans can be distinguished from colony-forming cells by expression of the CD33 and CD34 antigens and light scatter properties. J Exp Med. 1989;169(5):1721–1731. doi: 10.1084/jem.169.5.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernstein ID, Andrews RG, Berenson R, Bensinger W, Singer JW, Buckner CD. Isolation of human hematopoietic stem cells. In: Champlin RE, Gale RP, editors. New Strategies in Bone Marrow Transplantation. Wiley-Liss; New York: 1991. pp. 201–207. [Google Scholar]
- 37.Robertson MJ, Soiffer RJ, Freedman AS, Rabinowe SL, Anderson KC, Ervin TJ, et al. Human bone marrow depleted of CD33-positive cells mediates delayed but durable reconstitution of hematopoiesis: clinical trial of MY9 monoclonal antibody-purged autografts for the treatment of acute myeloid leukemia. Blood. 1992;79(9):2229–2236. [PubMed] [Google Scholar]
- 38.Bock TA. Assay systems for hematopoietic stem and progenitor cells. Stem Cells. 1997;15(Suppl 1):185–195. doi: 10.1002/stem.5530150824. [DOI] [PubMed] [Google Scholar]
- 39.Johnston DL, Meshinchi S, Opheim KE, Pallavicini MG, Feusner J, Woods WG, et al. Progenitor cell involvement is predictive of response to induction chemotherapy in paediatric acute myeloid leukaemia. Br J Haematol. 2003;123(3):431–435. doi: 10.1046/j.1365-2141.2003.04633.x. [DOI] [PubMed] [Google Scholar]
- 40.Pollard JA, Alonzo TA, Gerbing RB, Woods WG, Lange BJ, Sweetser DA, et al. FLT3 internal tandem duplication in CD34+/CD33− precursors predicts poor outcome in acute myeloid leukemia. Blood. 2006;108(8):2764–2769. doi: 10.1182/blood-2006-04-012260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uchida T, Ohashi H, Aoki E, Nakahara Y, Hotta T, Murate T, et al. Clonality analysis by methylation-specific PCR for the human androgen-receptor gene (HUMARA-MSP) Leukemia. 2000;14(1):207–212. doi: 10.1038/sj.leu.2401631. [DOI] [PubMed] [Google Scholar]
- 42.Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329(5997):1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6(3):251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen GL, Prchal JT. X-linked clonality testing: interpretation and limitations. Blood. 2007;110(5):1411–1419. doi: 10.1182/blood-2006-09-018655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grimwade D, Enver T. Acute promyelocytic leukemia: where does it stem from? Leukemia. 2004;18(3):375–384. doi: 10.1038/sj.leu.2403234. [DOI] [PubMed] [Google Scholar]
- 46.Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27(4):852–860. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyamoto T, Nagafuji K, Akashi K, Harada M, Kyo T, Akashi T, et al. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long-term remission patients with t(8;21) acute myelogenous leukemia. Blood. 1996;87(11):4789–4796. [PubMed] [Google Scholar]
- 48.Muto A, Mori S, Matsushita H, Awaya N, Ueno H, Takayama N, et al. Serial quantification of minimal residual disease of t(8;21) acute myelogenous leukaemia with RT-competitive PCR assay. Br J Haematol. 1996;95(1):85–94. doi: 10.1046/j.1365-2141.1996.d01-1877.x. [DOI] [PubMed] [Google Scholar]
- 49.Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000;97(13):7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–377. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 51.Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508–1516. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.