

Abstract
Vascular endothelial cells are a critical component of the hematopoietic microenvironment that regulates blood cell production. Recent studies suggest the existence of functional cross-talk between hematologic malignancies and vascular endothelium. Here, we show that human acute myeloid leukemia (AML) localizes to the vasculature in both patients and in a xenograft model. A significant number of vascular tissue-associated AML cells (V-AML) integrate into vasculature in vivo and can fuse with endothelial cells. V-AML cells acquire several endothelial cell-like characteristics, including the up-regulation of CD105, a receptor associated with activated endothelium. Remarkably, endothelial-integrated V-AML shows an almost 4-fold reduction in proliferative activity compared to non-vascular associated AML. Primary AML cells can be induced to down regulate the expression of their hematopoietic markers in vitro and differentiate into phenotypically and functionally-defined endothelial-like cells. After transplantation, these leukemia-derived endothelial cells are capable of giving rise to AML. Taken together, these novel functional interactions between AML cells and normal endothelium along with the reversible endothelial cell potential of AML suggest that vascular endothelium may serve as a previously unrecognized reservoir for acute myeloid leukemia.
Keywords: Acute Myeloid Leukemia, endothelium, xenograft, cell fusion, vascular niche
Introduction
Despite recent advances in detecting the presence of minimal residual leukemia, it remains challenging to distinguish those patients who are cured from those destined to develop recurrent disease (1, 2). Most studies of the potential mechanisms responsible for AML relapse have focused on identifying mutations and other leukemia cell intrinsic factors that confer drug resistance. Although it is well established that the tumor microenvironment is an important determinate of solid tumor initiation, progression and resistance to therapy (3-5), relatively little is known about the role of the microenvironment in regulating the survival of acute leukemia.
Our previous studies of normal hematopoietic stem cells (HSCs) provided us with evidence that the functional relationship between hematopoiesis and vascular endothelium originally identified during early development persists into adult life (6, 7). In both mice and humans, the adult HSC generates both blood and blood vessels (8-12). Furthermore, recent reports show that vascular endothelial cells within the bone marrow microenvironment are important for regulating self-renewal and differentiation of HSCs and lineage committed progenitors (13-17). Interactions between the vascular niche and hematopoietic malignancies may therefore play an important role in the pathophysiology of leukemia. Supportive of this possibility, increased microvascular density is found in the bone marrow of patients with hematologic malignancies (18, 19) and may correlate with more aggressive disease (20-23). Functionally significant, cytokine-mediated cross-talk between endothelial cells and AML has been demonstrated in tissue culture (24-27). Thus, developing an in depth understanding of interactions between the vascular microenvironment and malignant hematopoiesis is a promising area of investigation (28).
In this study we directly tested the possibility that AML functionally interacts with the vascular endothelium in vivo. Our results demonstrate that incorporation into the endothelial cell layer by attachment or cell fusion induces a relatively quiescent phenotype in AML cells. Moreover, AML cells that adopt endothelial cell characteristics have the potential to give rise to leukemia upon transplantation. This previously unrecognized potential of AML to both closely associate with and integrate into vascular endothelium has important implications for disease relapse. Targeting the interactions between AML and endothelial cells may be therapeutically useful.
Materials and Methods
Primary AML samples
Cells were obtained from AML patients undergoing leukapheresis or bone marrow aspiration after informed consent and in accordance with Oregon Health & Science University and University of Florida institutional review board policies. Cells were processed within 24 hrs of collection, cryopreserved in DMSO and stored in liquid nitrogen.
Xenotransplantation
NOD/LtSz-scid/IL2Rγ cnull (NSG) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in a breeding colony under specific pathogen-free conditions in the animal care facilities at both institutions. 8-12 week old mice were irradiated with 200-250 cGy from a cesium irradiator immediately prior to transplant. Human AML cells (1-3×106 cells per mouse) or AML-derived ECFCs (1-3×105 cells per mouse) were injected intravenously. Mice were allowed to drink freely for 6 days from water bottles containing 0.8mg/ml Bromodeoxy uridine (BrdU; Sigma), freshly prepared every other day.
ECFC culture
The ECFC protocol developed by Yoder and colleagues was employed (29). AML cells were suspended in EGM-2 medium and plated in culture dishes coated with type 1 rat tail collagen (BD Biosciences, Bedford, MA) at a density of 1 × 106 to 1 × 107 cells per well. 24-hours after plating, non-adherent cells were removed and remaining adherent cells fed with fresh EGM-2 medium. Medium was changed every day for the first 7 days and then every other day until ECFCs were generated.
Immunostaining
Deparaffinized patient liver biopsy sections were stained with hCD33 (1:100, Novocastra) or hCD31 (1:100, Dako JC70A) followed by goat anti-mouse biotin. Signal was detected by staining with horse radish peroxidase Elite Standard ABC reagent (Vector laboratories) and a DAB staining kit (Vector Laboratories). For samples that were also stained with hCD45, the slides were incubated in PBS at 60°C for 30 minutes, blocked and then incubated with anti-hCD45 (BD), followed by goat anti-mouse biotin and detection with a Standard Alkaline Phosphatase ABC kit in conjunction with Vector Blue staining reagent (Vector Laboratories). Livers from xenografted mice were harvested, fixed in 4% PFA, dehydrated in 30% sucrose, cryopreserved in optimal cutting temperature (OCT) compound (Sakura Finetek). Fixed femurs from xenografted mice were decalcified in 14% EDTA prior to cryopreservation. Sorted cells and allowed to settle overnight onto slides in 2% PFA. Cells and tissue cryosections (5-7 microns) were incubated with blocking buffer (5% goat serum, 1% albumin from bovine serum, 0.05% Tween 20 in PBS) for up to 1 hour, incubated overnight at 4°C with rat anti-mCD31 (1:50, MEC13.3, BD), and then stained with goat anti-rat Cyanine (Cy) 3 (1:200-1:400, Millipore Chemicon). Slides were then incubated with mouse anti-hCD45 (1:100; clone HI30, BD) for 1 hour at room temperature followed by goat anti-mouse conjugated to Alexa488 for 1 hour (1:200-1:400, Invitrogen). Alternatively, cryosectioned livers were incubated in mouse anti-CD105 and isolectin GS-IB4 conjugated to Alexafluor 647 (1:25, Invitrogen) overnight at 4°C, followed by goat anti-mouse Cy3. Sections were then stained with hHLAABC-biotin (1:100, eBioscience) followed by streptavidin DyLight488 (1:100, Vector Labs). For BrdU labeling studies, cryosections were unmasked in 2N HCl at 37° for 10 minutes, blocked with IMAGE-iT (Invitrogen), incubated overnight with rat anti-BrdU (1:100, AbD Serotec) and isolectin GS-IB4 conjugated to Alexafluor 647, followed by goat anti-rat Cy3. All immunofluorescently labeled cells were counterstained with 4,6 diamino-2-phenylindole (DAPI) and mounted in FluoromountG (Southern Biotech). For immunofluorescence staining of AML-derived ECFCs, mouse antibodies directed against hCD14 (1:50; clone 2Q1233, Abcam), hCD45 (1:100; clone HI30, BD), and hCD105 (1:50, clone 4G11, Abcam) were used. Following primary antibody incubation, cells were washed and exposed to donkey anti-mouse- fluorescein isothiocyanate (FITC) secondary antibody (1:500, Invitrogen) for 1-hour at room temperature in the dark. Cells were then washed and incubated with normal buffered formalin (Sigma) for 10 min followed by a final rinse in Tris buffered saline. For colorimetric detection of AML engraftment in mouse bone marrow, bones were decalcified, embedded in paraffin and sectioned, and an ARK kit (Dako) was employed following the manufacturer's instructions. Mouse anti hCD45 (1:50, Dako) was applied for 20 minutes at 95° C in Target Retrieval Solution (Dako), and positive signal was detected using DAB+ (Dako). Samples were counterstained with hematoxylin QS (Vector Laboratories).
Microscopy
Samples were examined and photographed with a Zeiss Axiophot 200 microscope with an LD 40X 0.60 NA Achroplan objective or an EC Plan 100X 1.3 NA Neofluor oil objective and a true color or a monochromatic AxioCam camera and standard epifluorescence filters for FITC,Cy3, Cy5 and DAPI (Zeiss). Images were digitally combined using Axio Vision software (Zeiss). Alternatively, samples were examined using a fluorescent Leica DM 2500 microscope (Leica Microsystems), 10×/0.40, 20×/0.70, 40×/0.75 or 63×/1.20 (water-corrected) objective and color camera (Optronics) in conjunction with MagnaFire 2.1C software (Optronics). For Z stack analysis, deconvolution microscopy was performed on a high resolution wide field Core DV system (Applied Precision) comprised of an Olympus IX71 inverted microscope with a proprietary XYZ stage, a solid state module for fluorescence and a Nikon Coolsnap ES2 HQ. Z- stacks (0.2 or 0.5 micron) were acquired in a 1024×1024 format with a 60× 1.42 NA Plan Apo N objective in FITC, TRITC and DAPI. The images were deconvolved with the appropriate optical transfer function using an iterative algorithm of 10 iterations. Deconvolved images were analyzed with Softworx (Applied Precision) or Imaris (Bitplane) software. Z stacks were also obtained with Olympus FluoView FV1000 confocal microscope at 0.5 micron intervals using Olympus FV10-ASW 2.1c software with a 60× 1.42 NA Plan Apo N objective. Images were analyzed using Olympus FluoView software.
Flow cytometry and fluorescence activated cell sorting (FACS)
Peripheral blood and bone marrow cells from xenografts were processed as previously described (10). V-AML was isolated from bones that were crushed with a mortar and pestle and collagenased as described by Hooper et al (30). Non-parenchymal cells from the liver of chimeric mice were generated essentially as described (31), following a single collagenase digestion. Antibodies were purchased from BD Biosciences unless otherwise indicated. For human cell engraftment, mCD45.1-PE-Cy7 (eBioscience), hCD13-PE, hCD33-PE, hCD45-APC, hCD3-FITC were used and cells were analyzed with a BD FACSCalibur or a BD LSRII flow cytometer. Following staining with mCD45.1-FITC, hCD33-PE, hCD13-PE, mCD31-APC, V-AML cells were then isolated by FACS sorting using a BD InFlux with a 150 micron nozzle. ECFCs were stained with hCD14-APC, hCD146-PE, hCD105-PE (Invitrogen) and hUEA-1-FITC (Sigma) and hCD45-PE, hCD115-PE, hCD144-PE, hVEGF-R2-PE (R&D Systems) and Dil-ac-LDL (BD Biosciences) uptake by incubating cells with 10 μg/ml Dilac-LDL in EGM-2 media for 4 hours at 37°C prior to analysis. Analysis was performed using a BD FACSCanto II flow cytometer. For all flow cytometry, dead cells were excluded using propidium iodide and scatter gates and doublets were excluded using the pulse width parameter.
Assessment of FLT3-ITD status in single cells
Nested PCR using the external FLT3 primers 5’TCTGCAGAACTGCCTATTCCT3’ and 5’AACTGTGCCTCCCATTTTTGTG3’ and the internal FLT3 primers (32) 5’CTTTCAGCATTTTGACGGCAAC3’ and 5’GCAATTTAGGTATGAA AGCCAGC3’ was performed on DNA isolated from single cells and control DNA isolated from bulk populations of AML and mobilized peripheral blood cells from normal donors using a Qiagen DNeasy Kit according to the manufacturer's instructions. Single cells were sorted into 96-well PCR plates containing 5μL DNA lysis buffer per well. Cell lysis was performed at 50°C for 30 minutes followed by 99°C for 5 minutes. Single cell DNA was amplified using 0.2mM external primers at 96°C for 5 minutes, followed by 10 cycles of 96°C for 2 minutes, 55°C for 1 minute, 72°C for 1 minute, followed by an additional 25 cycles of 96°C for 1 minute, 55°C for 1 minute, 72°C for 1 minute, and a final 72°C elongation step for 3 minutes. Control DNAs (5ng) were subject to the external PCR in a 25μL volume, diluted 1:40 in nuclease-free water, and 5μL was subsequently used for template in the internal nested PCR reaction using internal primers at a concentration of 0.2mM under the following conditions: 95°C for 3 minutes, followed by 35 cycles at 96°C for 30 seconds, 56°C for 30 seconds, and 72°C for 1 minute. Amplicons were subject to electrophoresis on a 2% agarose DNA gel and photographed using a Bio-Rad Gel-Doc U.V. system.
RT-PCR
10-cell aliquots of primary AML cells or HLMVEC were sorted into 96-well PCR plates containing 5μL of 5x RT-PCR buffer and 20U rRNasin (Promega) per well, covered and stored at -80°C. RT-PCR was performed on sorted cells using the Qiagen One Step RT-PCR kit was performed according to manufacturer's instructions. Semi-nested multiplex PCR for human gene expression was performed in 25ul reactions containing 0.2mM dNTPs, 0.6μM of each primer (Table S1) and 1μL RT-PCR enzyme mix (Qiagen) by incubating at 50°C for 30 minutes and then 95°C for 15 minutes. The first PCR reaction was then performed with 10 cycles of 96°C for 2 minutes, 55°C for 1 minute and 72°C for 1 minute, followed by 30 cycles of 96°C for 1 minute, 55°C for 1 minute, 72°C for 1 minute, and a final elongation step of 72°C for 5 minutes. The amplified PCR products were diluted 1:40 in nuclease-free water, and 5μL was used for template in a second, semi-nested PCR reaction containing 1× PCR Buffer (Promega), 1mM MgCl2, 0.2mM of each dNTP (Invitrogen 10297-018), 1.5U GoTaq-Flexi DNA polymerase (Promega), and 0.2mM of each primer. The following PCR conditions were used: 95°C for 3 minutes, followed by 25 cycles (for CD105) or 30 cycles (for CD45) of 96°C for 30 seconds, 56°C for 30 seconds, and 72°C for 1 minute, and a final 72°C elongation step for 5 minutes. Amplicons were subject to electrophoresis on a 2% agarose DNA gel and photographed on a Bio-Rad Gel-Doc U.V. system. For CD45 amplification, CD45-Fwd 5’-CAGGCATGGTTTCCACATTC-3’ and CD45-R1 5’-CTACAAA TATTGGTTCGCTGC-3’ were used in the first PCR reactions and CD45-Fwd and CD45-R2 5’-GCTTCAGGGAGCTTTTCTGGGGC-3’ were used in the second reaction.
Fluorescence in situ hybridization (FISH)
Analysis of AML-specific mutations was performed using CEP 9 SpectrumGreen and LSI MLL 11q23 DNA probe sets (Abbott Molecular) essentially following the manufacturer's protocol. Sorted V-AML cells were hybridized with mouse IDetect pancentromeric-FITC probe and a human IDetect pancentromeric IDYE 556 probe following the manufacturer's protocol (ID Labs Biotechnology). Slides were mounted in Vectashield with DAPI (Vector labs).
Statistical analysis
Statistical significance was determined using an unpaired, two-tailed Student's t-test. P values less than 0.05 were considered significant.
Results
AML localizes to vascular endothelium in patients and xenografted mice
To dissect the functional relationship between AML and endothelium in vivo, primary human AML cells (Table 1) were transplanted into an immunodeficient NOD/SCID IL2Rγcnull (NSG) mouse model (Figure 1A,B). Typically, the frequency of AML cells was highest in the bone marrow, but the collapsed and distorted architecture of the marrow venous sinusoids precluded definitive localization of individual AML cells relative to the vascular endothelium (Figure 1C). However, infiltrates of AML cells were also found in other tissues. The liver, a common site for extramedullary hematopoiesis in myeloproliferative disorders and myeloid leukemia (33-35), consistently displayed relatively high levels of AML involvement and provided us with an opportunity to unambiguously study the relationship between AML cells and venous endothelium (Figure 1D). Using species-specific antibodies, we identified a marked accumulation of AML cells near mouse endothelium (Figure 1E). This leukemic infiltrate was particularly prominent around the portal veins, and herein, we will refer to these vessel-associated AML cells as V-AML.
Table 1.
Patient characteristics.
| Patient ID | Age | Gender | Disease Status | FAB | WBC | Karyotype | FLT3-ITD | NPM-1 mutation |
|---|---|---|---|---|---|---|---|---|
| 1 | 22 | M | de novo | M2 | 253 | normal | positive | negative |
| 2 | 52 | M | de novo | M2 | 293 | normal | positive | positive |
| 3 | 67 | F | de novo | M1 | 182 | del5q | positive | negative |
| 4 | 65 | M | relapsed | NS | 217 | t(13;18) | positive | negative |
| 5 | 35 | M | relapsed | M4 | 133 | inv 2; trisomy 8 | positive | negative |
| 6 | 38 | M | de novo | NS | 82.8 | normal | negative | positive |
| 7 | 72 | M | de novo | M5 | 175 | monosomy 7 | negative | negative |
| 8 | 49 | F | de novo | M4 | 90 | NA | NA | NA |
| 9 | 50 | F | refractory | NS | 125 | tetrasomy 8 | NA | |
| 10 | 61 | M | relapsed | M4 | 31.7 | complex | negative | NA |
| 11 | 70 | F | relapsed | M1 | 64.4 | normal | positive | NA |
| 12 | 72 | M | refractory | M2 | 1.1 | normal | positive | NA |
| 13 | 48 | F | de novo | M2 | 9.8 | complex | negative | negative |
| 14 | 84 | M | de novo | M5 | 0.8 | complex | negative | negative |
| 15 | 32 | F | de novo | M4 | 2.6 | normal | negative | positive |
| 16 | 75 | M | relapsed | M1 | 19.6 | trisomy 13 | positive | NA |
Leukapheresis samples from patients 1-7 were used to generate the in vivo xenografts in Figures 1-4 and Supplementary Figures 1-3. Patients 8-10 were the source of autopsy liver samples shown in Figure 1. AML bone marrow samples from patients 10-16 were used in Figures 5-6 and Supplementary Figure 4. WBC: white blood cell count ×103 per μL. NA: not available.
Figure 1. AML localizes to vascular endothelium in vivo.

(A) Transplantation schema. (B) Engraftment analysis of primary AML in NSG mice. Representative flow cytometry data showing the frequency of AML cells from one donor in the peripheral blood (PB) and bone marrow (BM) of xenografted animals. Each diamond in the scatter plot on the right represents an individual mouse. (C) AML cells in the bone marrow of an NSG recipient femur. Tissue sections were stained with antibodies to mouse CD31 (red), human CD45 (green). Arrowheads indicate sinusoids, which are compressed in regions with high levels of human cell engraftment. Nuclei are stained with DAPI (blue). (D) Infiltrates of primary human AML cells (outlined with dashed lines) are found immediately adjacent to portal veins (PV) in the livers of NSG recipient mice (H&E stain). (E) Human CD45+ AML cells (hCD45; green) localize next to mouse CD31+ portal vein endothelial cells (mCD31+, red). Nuclei are blue (DAPI). (F-H) Perivascular accumulation of AML cells around the portal vessels in human liver. (F) H&E stained section. (G) Extensive infiltration of CD33+ cells (brown) in a portal triad (BD: bile duct; A: artery) is shown. (H) CD45+ AML cells (blue) surround a CD31+ (brown) portal vessel (PV). Scale bars for all images are 20 microns.
To ensure that this finding of AML localization to portal vessels was not unique to our NSG xenograft model system, we evaluated liver tissue obtained from a cohort of 30 AML patients at autopsy. Seven patients (23%) showed a periportal infiltrate of AML. The pattern of leukemic infiltration in the human liver tissue (Figure 1 F-H) was indistinguishable from the AML infiltration in the liver of our NSG mouse model (Figure 1 D-E). In this cohort, one patient with newly diagnosed AML died before induction therapy could begin, a second had primary induction therapy failure and died within 5 weeks, and the third patient had a long history of refractory AML. Therefore, perivascular liver involvement can be detected throughout the course of active disease in patients with AML. Clinically significant liver dysfunction attributable to AML is infrequent (36, 37) and none of the patients in our study demonstrated this. However, subclinical hepatic involvement is quite common and usually unrecognized. Specifically, in an autopsy series of 585 AML patients (38) the frequency of perivascular liver involvement by AML at autopsy ranged from 28% to 71%. Taken together, our results show that a perivascular infiltration of the liver (V-AML) is a common finding in primary AML xenografts and in patients with AML.
AML binds to ECs and can integrate into vascular endothelium in vivo
High resolution imaging of the livers of xenografted mice revealed a subset of V-AML cells that was tightly associated with mCD31+ vascular endothelial cells in the portal vessels. Z-stack analysis confirmed co-localization of these human and mouse cell surface markers on the luminal side of the membrane of individual V-AML cells (Figure 2 A-B). These mCD31+hCD45+ V-AML cells comprised up to 2% of the total portal endothelial cells (Figure 2C). Importantly, when total bone marrow from primary recipients was harvested and transferred into secondary recipients, mCD31+hCD45+ V-AML cells were again readily detected in the liver, demonstrating that their capacity to generate V-AML is a conserved property of serially transplantable leukemic bone marrow (Figure 2C).
Figure 2. Subpopulations of AML cells tightly adhere to vascular endothelium in vivo.
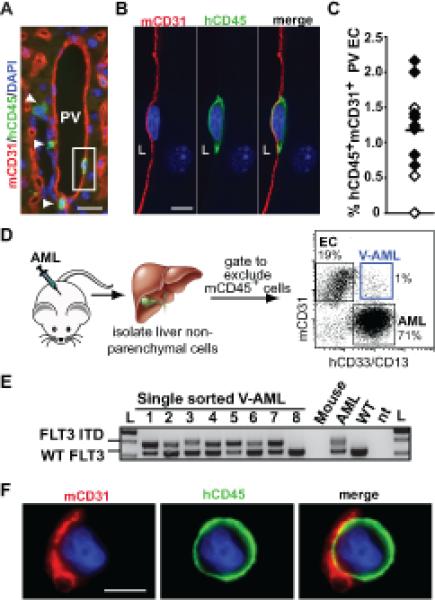
(A) An example of a single V-AML cell (in box; hCD45+, green) with a region of cell membrane that is so adherent to mouse CD31 (mCD31+, red) labeled portal vein (PV) endothelium that the mouse and human markers overlap. Arrowheads indicate V-AML cells adjacent to the endothelium but not tightly associated with mCD31+ portal ECs. (B) Z-stack analysis of the boxed cell shown in panel A. L indicates the PV lumen. A representative Z plane is shown. (C) Quantitation of AML integration into PV endothelium in individual primary (black diamonds) and secondary (white diamonds) NSG recipient mice (n=11). A minimum of 140 nucleated endothelial cells from non-adjacent tissue sections were scored for each mouse. The bar indicates the mean. (D) Strategy for isolating V-AML from the livers of AML recipient mice. Representative FACS sorting gates are shown for mouse endothelial cells (EC), AML and V-AML. (E) Genomic PCR analysis of FLT3 in thrice sorted, single V-AML cells. The FLT3-ITD was detected in 96% of V-AML cells analyzed, confirming their leukemic origin. An example of a rare single cell containing only wild type FLT3 is shown (Lane 8). FLT3-ITD PCR data from pooled mouse cells, bulk input AML cells, and normal peripheral human peripheral blood cells (WT) are shown. nt: no template control. (F) IF analysis of sorted V-AML revealed the presence of mouse CD31+ membranes on AML cells. Scale bars: (A): 20 microns; (B): 10 microns; (E): 5 microns.
Using FACS (Figure 2D, and Supplementary Figure 1A), mCD31+ V-AML was isolated from the livers of every AML xenografted mouse tested (n=28). Non-specific binding of mCD31 was specifically excluded by assaying primary AML samples prior to transplant (Supplementary Figure 1B). Genomic analysis of single cells isolated from FLT3-ITD+ AML recipient mice (n=6) revealed that 96% mCD31+V-AML cells harbored the FLT3-ITD (Fig. 2E), indicative of their leukemic origin. Consistent with the in situ liver studies (Figure 2A-B), evaluation of sorted cell populations by immunofluorescence and confocal imaging demonstrated that nearly all individual sorted cells co-expressing mouse and human markers displayed a uniform hCD45 staining pattern with discrete patches of mCD31+ membrane (Figure 2F and Supplementary Figure 1C,D). A bone marrow population of mCD31+ V-AML cells with identical cell surface marker expression was also found (Supplementary Figure 2). The distinct pattern of hCD45 and mCD31 co-expression in mCD31+ V-AML cells is indicative of a very tight physical association and membrane coupling between AML cells and mouse endothelial cells in the bone marrow and liver.
A small subset of V-AML cells displayed a uniform, circumferential co-expression pattern of both hCD45 and mCD31 in both liver tissue sections (Figure 3A, Supplementary Figure 3A) and in sorted mCD31+V-AML cells (Figure 3B). Some of these cells were binucleated (Figure 3B), consistent with a cell fusion event. AML/mouse EC hybrid cells in which the expression of mouse and human markers did not overlap were also observed (Supplementary Figure 3B). To assess cell fusion at the chromosomal level, we performed FISH using species-specific pan-centromeric probes. This approach revealed a subpopulation of sorted V-AML cells that contained both mouse and human centromeres (Figure 3C) at the same frequency (~1% of total CD31+ V-AML) fused/hybrid mouse EC and AML cells were identified using cell surface marker expression (Supplementary Figure 1D).
Figure 3. A subset of V-AML cells fuses with endothelium.
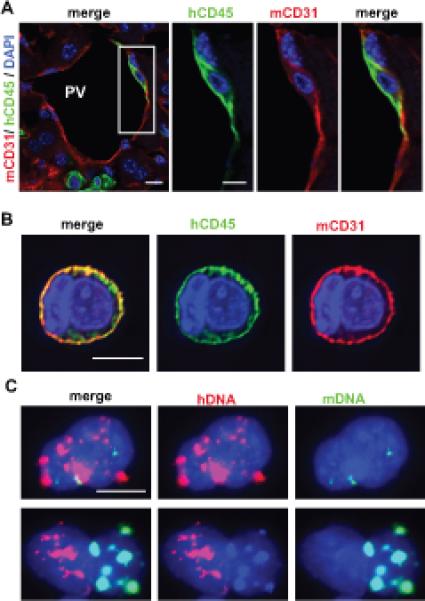
(A) Confocal, single Z-stack image of a mouse portal vessel (PV). A V-AML cell (CD45+, green) that also exhibits circumferential mCD31 staining (red) is shown within the boxed region. Panels on the right show single color and merged channels at a higher magnification. (B) Z-stack imaging of a single FACS sorted V-AML cell is shown. Circumferential, overlapping hCD45 and mCD31 expression is observed in the cell membrane, consistent with AML and EC syncytium formation. These syncytia typically represent ~1% of the total sorted mCD31+ V-AML population. (C) Interphase FISH analysis of sorted V-AML cells showing the presence of human (red) and mouse (green) centromeres. Scale bars: 10 microns.
V-AML expresses CD105 and adopts a quiescent phenotype in vivo
We and others have reported that the progeny of normal hematopoietic stem cells, including common myeloid progenitors, can integrate into existing functional vascular endothelium and express endothelial cell markers (8-12, 39, 40). These findings, in addition to integration of V-AML into the vasculature shown above (Figures 2A, 3A), raised the possibility that V-AML develops some endothelial characteristics. To investigate this further, we determined whether the tight association of AML cells with endothelium is correlated with their expression of endothelial cell related genes. It is now recognized that many cell surface markers originally thought to be endothelial cell-specific are also expressed in normal and malignant hematopoietic cells (41, 42). Consequently, we first screened primary AML samples to identify those lacking the expression of individual endothelial associated markers and then tested these samples further in vivo. Human endoglin (CD105) was the only one of 8 endothelial markers that was not expressed in some of our patient samples (Supplementary Table 1). Consistent with our hypothesis, sorted V-AML cells derived from CD105-negative primary AML samples had readily detectable CD105 mRNA expression by RT-PCR (Figure 4A). In tissue sections, some AML cells closely associated with the endothelium in vivo also expressed CD105 (Figure 4B). These results demonstrate that the close association of AML with endothelium can induce the expression of CD105, a membrane receptor associated with activated endothelium and tumor angiogenesis (43).
Figure 4. V-AML cells up regulate CD105 expression and become quiescent.
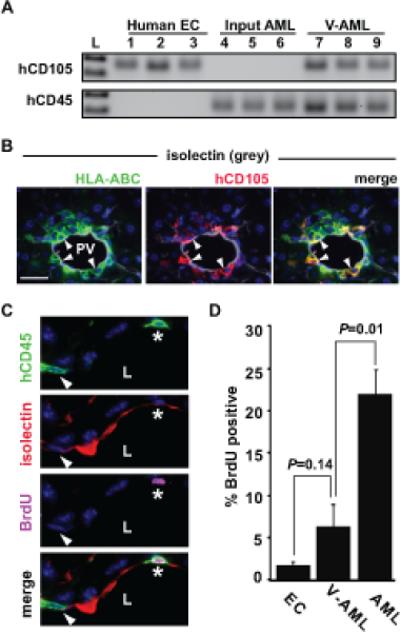
(A) Input primary AML cells express hCD45 but not hCD105 by RT-PCR. By contrast, V-AML cells sorted from the liver of an NSG recipient show an induction of CD105 expression. Each lane shows ten sorted cells. (B) hCD105 expression in a subpopulation of V-AML that are tightly associated with the endothelium. Arrowheads indicate hCD105+ (red), isolectin+ (grey) HLA-ABC+ (green) V-AML cells. DNA is labeled with DAPI (blue). (C) Example of BrdU uptake in PV-adherent V-AML cells. In the top panel, two hCD45+ (green) V-AML cells (*, arrowhead) that appear to share membrane with mouse endothelial cells are shown. The second panel shows mouse endothelial isolectin GS-IB4 expression (red). In the third panel, BrdU (pink) is detected in one of the two V-AML cells (*). Bottom panel is a merged image. L indicates the PV lumen. (D) Quantitation of BrdU uptake in mouse endothelial cells, AML and V-AML tightly associated to mouse CD31+ PV endothelium. VAML cells adherent to or incorporated into the endothelial layer of the PV proliferate significantly less than AML cells present throughout the rest of the liver. The mean ± SEM is shown for pooled samples. A minimum of 600 PV ECs, 75 PV integrated V-AML cells and 2500 AML cells were scored in non-adjacent sections from each liver. Scale bars: 20 microns
To investigate whether the integration of AML cells into vascular endothelium reduced their proliferative activity, AML engrafted NSG mice were maintained on drinking water containing BrdU (Figure 4C). After 6 days of continuous labeling, a mean of 22% of non-vascular tissue associated AML cells were BrdU positive. By contrast, only 6% of the mCD31+ V-AML cells tightly associated with the portal vein endothelium were BrdU labeled (P=0.01). Interestingly, the low frequency of proliferating mCD31+ V-AML cells was similar to the frequency observed in adjacent normal mouse CD31+ endothelial cells. Together, our results demonstrate that a subpopulation of AML cells adopt certain endothelial characteristics, including integration into existing endothelium, CD105 expression and decreased proliferation.
AML can give rise to endothelial colony forming cells in vitro
AML cells were cultured using conditions that specifically support the growth of human endothelial progenitors (29), also known as endothelial colony forming cells (ECFC, Figure 5A). Similar to normal endothelial cells and ECFCs, AML-derived ECFCs readily formed vascular tubes in the presence of Matrigel (Figure 5B), demonstrated the uptake of acetylated LDL, and expressed endothelial cell markers, including CD105, CD146, UEA-1 and VEGFR2 (Figure 5C and Supplementary Figure 4). ECFCs derived from AML cells also down-regulated the expression the pan-hematopoietic marker CD45 as well as the myeloid lineage markers CD14 and CD115. Consistent with their endothelial phenotype, AML-derived ECFC did not grow in standard myeloid colony forming media (44). To exclude a significant contribution from pre-existing normal endothelial progenitors, patient-specific AML markers were utilized. FISH analysis of single cells derived from these colonies revealed the presence of either trisomy 9 or the MLL rearrangement, the corresponding molecular marker for the individual patient's AML (Figure 5D). Thus, under what is considered the most stringent conditions for assaying endothelial progenitors (29), primary human AML can generate cells with an endothelial-like phenotype at the clonal level. We will subsequently refer to these in vitro-generated, leukemia-derived endothelial colony forming cells as L-ECFC.
Figure 5. AML has endothelial cell potential in vitro.
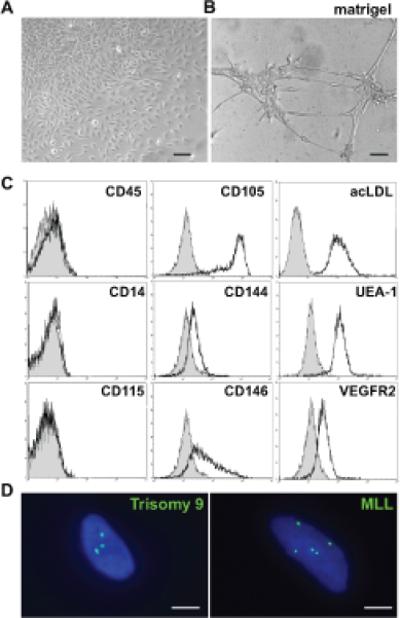
(A) Leukemia-derived endothelial colony forming cells (L-ECFC) from the bone marrow of AML patients forms vascular tubules (B) when cultured in Matrigel. (C) Endothelial cell marker expression is upregulated in cultured L-ECFC by flow cytometry whereas hematopoietic cell marker expression is down regulated. Isotype controls are shown in grey. (D) FISH analysis of single L-ECFC cells shows the presence of the AML-specific mutations (green) including trisomy 9 and the MLL re-arrangement. Nuclei are stained blue (DAPI). Scale bars: (A,B) 100 microns; (D) 10 microns.
AML-derived endothelium can adopt a leukemic phenotype
The differentiation of AML into endothelial-like cells raises the intriguing possibility that vascular endothelium may represent a reservoir for occult residual leukemia. To begin to test this hypothesis, mCD31+ V-AML cells isolated from the livers of chimeric mice (Figure 2D) were transplanted into secondary NSG recipients (3-10x103 cells/mouse) and non-vascular associated AML cells isolated from the same livers were also transplanted into NSG mice as a control (3x104 cells/mouse). Neither population gave rise to detectable AML (data not shown). This result was not surprising as the frequency of leukemia initiating cells is quite low (45). By contrast, when L-ECFCs, which could be generated in large numbers from the bone marrow of AML patients, were transplanted into NSG recipients, the mouse bone marrow showed clusters of human CD45 expressing cells (Figure 6A-D) that co-expressed the myeloid lineage marker CD33. The presence of molecular markers identified in the original, pre-transplant leukemia confirmed the leukemic origin of the LECFC-derived cells (Figure 6E,F). These findings demonstrate that human AML-derived endothelial cells can retain leukemia initiating potential in vivo despite initially down regulating their hematopoietic cell surface markers and adopting an endothelial cell-like phenotype.
Figure 6. AML-derived endothelium can adopt a leukemic phenotype.
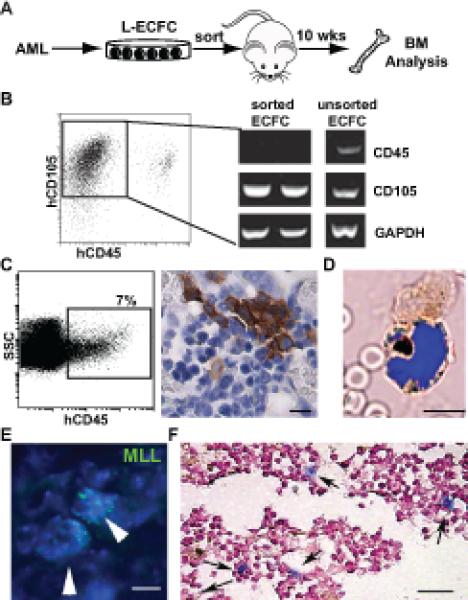
(A) Experimental strategy to assess in vivo leukemic potential of AML-derived ECFCs. (B) Flow cytometry gating used for the purification of CD105+CD45neg L-ECFC prior to transplant. RT-PCR analysis of sorted CD105+CD45neg cells confirmed the absence of CD45 expression. (C) Analysis of bone marrow from CD105+CD45neg L-ECFC recipient NSG mice. A significant population of human CD45+ cells is detected in mouse bone marrow by flow cytometry (left panel); and in tissue sections by immunohistochemistry (DAB brown). (D) Localization of hCD33+ expressing cells (brown) in the bone marrow of an L-ECFC engrafted NSG mouse. A representative merged brightfield and fluorescent image (DAPI, blue) is shown. (E) Multiple copies of human MLL gene (green, arrowheads) confirm the leukemic origin of the human donor cells in bone marrow from L-ECFC engrafted mice. All nuclei are stained with DAPI. (F) Detection of cytoplasmic NPM1 (blue, arrows) following transplant of L-ECFC derived from an NPM1 mutant donor patient. Scale bars: (C): 10 microns; (D-E): 5 microns; (F): 50 microns.
Discussion
In this study of the functional and developmental relationship between AML and endothelium, we identified a subset of AML that localizes to endothelium (V-AML) in vivo. Moreover, some V-AML cells physically integrate into the vasculature and become quiescent. Importantly, we demonstrate that AML cells with endothelial cell characteristics have the potential to give rise to leukemia upon transplantation. Our results provide evidence that functional relationships between the vasculature and hematopoiesis may contribute to the pathophysiology of AML.
Several reports have shown that hematopoietic cells are capable of contributing to blood vessel formation, during embryonic development and in adults (8, 10, 11, 40, 43, 46). In this study, we found that AML can adopt a spectrum of endothelial cell properties and that some AML cells integrate into the vasculature in xenografts, in part by a cell fusion mechanism. Moreover, standard endothelial progenitor cell culture conditions are capable of inducing primary AML cells to down regulate their hematopoietic phenotype and express a CD45neg, CD105+, acLDL+, VEGFR2+, UEA1+ endothelial phenotype (L-ECFC). When transplanted into NSG recipients, these L-ECFC can convert back to their leukemic phenotype. Our findings of endothelial cell potential in AML are consistent with an intriguing study by Hoffman and colleagues which evaluated the liver endothelium of patients with polycythemia vera (35). They demonstrated the presence of the JAK2V617F mutation in isolated portal vein endothelial cells and concluded that this clonal myeloproliferative disorder, which often progresses to AML, involves primitive cells with the potential to differentiate into both hematopoietic and endothelial lineages. Together, these findings highlight that both normal and malignant myeloid cells have endothelial potential by at least 2 different mechanisms.
Normal hematopoietic stem cells are found in close proximity to vascular endothelium (13) and vascular endothelial cells are increasingly recognized as an important component of the normal and malignant hematopoietic microenvironments (47, 48). Our study revealed that perivascular infiltration of the liver is common in primary AML xenografts and in patients with AML (Figure 1). These findings are similar to the reported high frequency of subclinical, liver involvement seen at autopsy (38) and the perivascular distribution of AML observed in patients with leukemic involvement of the skin (49). Interestingly, we observed three classes of V-AML cells: AML cells adjacent to the endothelium; AML cells touching/adherent to the endothelium and AML cells that are integrated into the endothelium. In both the liver and bone marrow of transplanted mice, V-AML cells were so tightly attached to host endothelial cells, that extensive collagenase treatment did not disrupt the association between their cell membranes (Figure 2, Supplementary Figures 1-3). The presence of these tightly adherent V-AML cells is consistent with the results of our previous studies of human AML cell lines and endothelium in vitro (50).
Previous studies suggested that proximity and adherence of AML cells to endothelium have dramatically different effects on AML cell behavior in vitro. Whereas the overall proliferation of most primary AMLs is increased when cultured in the presence of ECs (25), we recently demonstrated that EC-adherent AML cells become quiescent in vitro relative to non-adherent AML cells (50). Moreover, EC-adherent AML is less sensitive to cytarabine than non-adherent ECs. In our present in vivo study, V-AML cells attached to or integrated into the endothelial cell layer became quiescent relative to non-vascular associated AML. Whether the small subpopulation of endothelial-AML cells produced by cell fusion is capable of proliferation in vivo remains to be determined. Future in vivo experiments will also ascertain whether the decreased proliferation observed in some V-AML cells confers resistance to cell cycle targeted therapies. In terms of clinical translation, we have recently shown that targeting ECs with vascular disrupting agents (51) and anti-angiogenic therapy (52) cause regression of AML in experimental models. We have translated these findings into an ongoing phase I clinical trial of an anti-vascular agent, combretastatin, in patients with relapsed and refractory AML (ClinicalTrials.gov NCT0108656).
In conclusion, our results reveal alternative developmental fates of AML cells and support the hypothesis that leukemic cells can directly contribute to vascular endothelium (51). Specifically, we show that AML can either differentiate into endothelial-like cells or fuse with established endothelium. Both of these fates may endow AML cells with pro-survival attributes. Our results strongly suggest that endothelium may serve as a previously unrecognized site of residual disease and lends further support to the concept of disrupting AML-endothelial cell interactions as a targeted therapeutic approach for AML.
Supplementary Material
Acknowledgements
We thank Pamela Canaday from the OHSU flow cytometry core for FACS. Funding was provided by the NIH to W.H.F. (R01 HL069133), C.R.C. (K08 DK067359), and E.W.S. (R01 HL70738). The Leukemia & Lymphoma Society also supported C.R.C. with a Translational Research Program grant (6264-08) and a Scholar in Clinical Research award (2400-13).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Supplementary information is available at Leukemia's website.
References
- 1.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011 Feb 10;29(5):487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 2.Buccisano F, Maurillo L, Del Principe MI, Del Poeta G, Sconocchia G, Lo-Coco F, et al. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood. 2012 Jan 12;119(2):332–341. doi: 10.1182/blood-2011-08-363291. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012 Mar 20;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011 May 6;8(5):482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dvorak HF, Weaver VM, Tlsty TD, Bergers G. Tumor microenvironment and progression. J Surg Oncol. 2011 May 1;103(6):468–474. doi: 10.1002/jso.21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey AS, Fleming WH. Converging roads: evidence for an adult hemangioblast. Exp Hematol. 2003 Nov;31(11):987–993. doi: 10.1016/j.exphem.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Cogle CR, Scott EW. The hemangioblast: cradle to clinic. Exp Hematol. 2004 Oct;32(10):885–890. doi: 10.1016/j.exphem.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 8.Grant MB, May WS, Caballero S, Brown GA, Guthrie SM, Mames RN, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002 Jun;8(6):607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 9.Cogle CR, Wainman DA, Jorgensen ML, Guthrie SM, Mames RN, Scott EW. Adult human hematopoietic cells provide functional hemangioblast activity. Blood. 2004 Jan 1;103(1):133–135. doi: 10.1182/blood-2003-06-2101. [DOI] [PubMed] [Google Scholar]
- 10.Bailey AS, Jiang S, Afentoulis M, Baumann CI, Schroeder DA, Olson SB, et al. Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood. 2004 Jan 1;103(1):13–19. doi: 10.1182/blood-2003-05-1684. [DOI] [PubMed] [Google Scholar]
- 11.Bailey AS, Willenbring H, Jiang S, Anderson DA, Schroeder DA, Wong MH, et al. Myeloid lineage progenitors give rise to vascular endothelium. Proc Natl Acad Sci U S A. 2006 Aug 29;103(35):13156–13161. doi: 10.1073/pnas.0604203103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madlambayan GJ, Butler JM, Hosaka K, Jorgensen M, Fu D, Guthrie SM, et al. Bone marrow stem and progenitor cell contribution to neovasculogenesis is dependent on model system with SDF-1 as a permissive trigger. Blood. 2009 Nov 5;114(19):4310–4319. doi: 10.1182/blood-2009-03-211342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005 Jul 1;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 14.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006 Dec;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010 Aug 12;466(7308):829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010 Mar 5;6(3):251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012 Jan 26;481(7382):457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000 Jan 1;95(1):309–313. [PubMed] [Google Scholar]
- 19.Ribatti D. Is angiogenesis essential for the progression of hematological malignancies or is it an epiphenomenon? Leukemia. 2009 Mar;23(3):433–434. doi: 10.1038/leu.2008.381. [DOI] [PubMed] [Google Scholar]
- 20.Fragioudaki M, Tsirakis G, Pappa CA, Aristeidou I, Tsioutis C, Alegakis A, et al. Serum BAFF levels are related to angiogenesis and prognosis in patients with multiple myeloma. Leuk Res. 2012 Aug;36(8):1004–1008. doi: 10.1016/j.leukres.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Korkolopoulou P, Thymara I, Kavantzas N, Vassilakopoulos TP, Angelopoulou MK, Kokoris SI, et al. Angiogenesis in Hodgkin's lymphoma: a morphometric approach in 286 patients with prognostic implications. Leukemia. 2005 Jun;19(6):894–900. doi: 10.1038/sj.leu.2403690. [DOI] [PubMed] [Google Scholar]
- 22.Lundberg LG, Hellstrom-Lindberg E, Kanter-Lewensohn L, Lerner R, Palmblad J. Angiogenesis in relation to clinical stage, apoptosis and prognostic score in myelodysplastic syndromes. Leuk Res. 2006 Mar;30(3):247–253. doi: 10.1016/j.leukres.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Rajkumar SV, Greipp PR. Angiogenesis in multiple myeloma. Br J Haematol. 2001 Jun;113(3):565. doi: 10.1046/j.1365-2141.2001.02690.x. [DOI] [PubMed] [Google Scholar]
- 24.Hatfield K, Oyan AM, Ersvaer E, Kalland KH, Lassalle P, Gjertsen BT, et al. Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br J Haematol. 2009 Jan;144(1):53–68. doi: 10.1111/j.1365-2141.2008.07411.x. [DOI] [PubMed] [Google Scholar]
- 25.Hatfield K, Ryningen A, Corbascio M, Bruserud O. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int J Cancer. 2006 Nov 15;119(10):2313–2321. doi: 10.1002/ijc.22180. [DOI] [PubMed] [Google Scholar]
- 26.Hatfield KJ, Evensen L, Reikvam H, Lorens JB, Bruserud O. Soluble mediators released by acute myeloid leukemia cells increase capillary-like networks. Eur J Haematol. 2012 Dec;89(6):478–490. doi: 10.1111/ejh.12016. [DOI] [PubMed] [Google Scholar]
- 27.Liesveld JL, Rosell KE, Lu C, Bechelli J, Phillips G, Lancet JE, et al. Acute myelogenous leukemia--microenvironment interactions: role of endothelial cells and proteasome inhibition. Hematology. 2005 Dec;10(6):483–494. doi: 10.1080/10245330500233452. [DOI] [PubMed] [Google Scholar]
- 28.Trujillo A, McGee C, Cogle CR. Angiogenesis in acute myeloid leukemia and opportunities for novel therapies. J Oncol. 2012;2012:128608. doi: 10.1155/2012/128608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Critser PJ, Yoder MC. Endothelial colony-forming cell role in neoangiogenesis and tissue repair. Curr Opin Organ Transplant. 2010 Feb;15(1):68–72. doi: 10.1097/MOT.0b013e32833454b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009 Mar 6;4(3):263–274. doi: 10.1016/j.stem.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996 Mar;12(3):266–273. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 32.Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001 Jan 1;97(1):89–94. doi: 10.1182/blood.v97.1.89. [DOI] [PubMed] [Google Scholar]
- 33.Fritz J, Vogel W, Bares R, Horger M. Radiologic spectrum of extramedullary relapse of myelogenous leukemia in adults. AJR Am J Roentgenol. 2007 Jul;189(1):209–218. doi: 10.2214/AJR.06.1500. [DOI] [PubMed] [Google Scholar]
- 34.Navarro M, Crespo C, Perez L, Martinez C, Galant J, Gonzalez I. Massive intrahepatic extramedullary hematopoiesis in myelofibrosis. Abdom Imaging. 2000 Mar-Apr;25(2):184–186. doi: 10.1007/s002619910041. [DOI] [PubMed] [Google Scholar]
- 35.Sozer S, Fiel MI, Schiano T, Xu M, Mascarenhas J, Hoffman R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. 2009 May 21;113(21):5246–5249. doi: 10.1182/blood-2008-11-191544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathews E, Laurie T, O'Riordan K, Nabhan C. Liver involvement with acute myeloid leukemia. Case Rep Gastroenterol. 2008;2(1):121–124. doi: 10.1159/000120756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisen A, Dovrish Z, Hadari R, Lew S, Amital H. Jaundice and acute liver failure as the first manifestation of acute myeloid leukemia. Isr Med Assoc J. 2008 Oct;10(10):733–735. [PubMed] [Google Scholar]
- 38.Barcos M, Lane W, Gomez GA, Han T, Freeman A, Preisler H, et al. An autopsy study of 1206 acute and chronic leukemias (1958 to 1982). Cancer. 1987 Aug 15;60(4):827–837. doi: 10.1002/1097-0142(19870815)60:4<827::aid-cncr2820600419>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 39.Larrivee B, Niessen K, Pollet I, Corbel SY, Long M, Rossi FM, et al. Minimal contribution of marrow-derived endothelial precursors to tumor vasculature. J Immunol. 2005 Sep 1;175(5):2890–2899. doi: 10.4049/jimmunol.175.5.2890. [DOI] [PubMed] [Google Scholar]
- 40.Jiang S, Walker L, Afentoulis M, Anderson DA, Jauron-Mills L, Corless CL, et al. Transplanted human bone marrow contributes to vascular endothelium. Proc Natl Acad Sci U S A. 2004 Nov 30;101(48):16891–16896. doi: 10.1073/pnas.0404398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kukk E, Wartiovaara U, Gunji Y, Kaukonen J, Buhring HJ, Rappold I, et al. Analysis of Tie receptor tyrosine kinase in haemopoietic progenitor and leukaemia cells. Br J Haematol. 1997 Jul;98(1):195–203. doi: 10.1046/j.1365-2141.1997.1732989.x. [DOI] [PubMed] [Google Scholar]
- 42.Watarai M, Miwa H, Shikami M, Sugamura K, Wakabayashi M, Satoh A, et al. Expression of endothelial cell-associated molecules in AML cells. Leukemia. 2002 Jan;16(1):112–119. doi: 10.1038/sj.leu.2402326. [DOI] [PubMed] [Google Scholar]
- 43.Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM, et al. Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011 Jun;31(6):2283–2290. [PubMed] [Google Scholar]
- 44.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007 Mar 1;109(5):1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest. 2011 Jan 4;121(1):384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park C, Ma YD, Choi K. Evidence for the hemangioblast. Exp Hematol. 2005 Sep;33(9):965–970. doi: 10.1016/j.exphem.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Colmone A, Sipkins DA. Beyond angiogenesis: the role of endothelium in the bone marrow vascular niche. Transl Res. 2008 Jan;151(1):1–9. doi: 10.1016/j.trsl.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 48.Doan PL, Chute JP. The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia. 2012 Jan;26(1):54–62. doi: 10.1038/leu.2011.236. [DOI] [PubMed] [Google Scholar]
- 49.Cho-Vega JH, Medeiros LJ, Prieto VG, Vega F. Leukemia cutis. Am J Clin Pathol. 2008 Jan;129(1):130–142. doi: 10.1309/WYACYWF6NGM3WBRT. [DOI] [PubMed] [Google Scholar]
- 50.Pezeshkian B, Donnelly C, Tamburo K, Geddes T, Madlambayan GJ. Leukemia Mediated Endothelial Cell Activation Modulates Leukemia Cell Susceptibility to Chemotherapy through a Positive Feedback Loop Mechanism. PLoS One. 2013;8(4):e60823. doi: 10.1371/journal.pone.0060823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madlambayan GJ, Meacham AM, Hosaka K, Mir S, Jorgensen M, Scott EW, et al. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood. 2010 Sep 2;116(9):1539–1547. doi: 10.1182/blood-2009-06-230474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyner JW, Yang WF, Bankhead A, Fan G, Fletcher LB, Bryant J, et al. Kinase Pathway Dependence in Primary Human Leukemias Determined by Rapid Inhibitor Screening. Cancer Res. 2012 Oct 18; doi: 10.1158/0008-5472.CAN-12-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.