

Introduction
Danon disease is an X-linked dominant skeletal and cardiac muscle disorder with multisystem clinical manifestations. It was first described in boys presenting with cardiomyopathy, skeletal myopathy, and varying degrees of intellectual disability.1 As histological findings of glycogen buildup in muscle tissue similar to those seen in Pompe disease were noted, the condition was originally considered to be a lysosomal storage disease and was termed glycogen storage disease type IIb. In 2000, Nishino et al. identified the genetic defects in the lysosome-associated membrane protein 2 (LAMP2) gene, encoding the LAMP2 protein.2 Most Danon disease mutations lead to an absence of LAMP2 protein expression,2 a situation more problematic in males who are hemizygous for LAMP2. For reasons not yet fully understood, reduction in LAMP2 disrupts intracytoplasmic trafficking and leads to accumulation of autophagic material and often glycogen in skeletal muscle and cardiac muscle cells (Figure 1).2 Major clinical features include skeletal and cardiac myopathy, cardiac conduction abnormalities, mild intellectual difficulties, and retinal disease. Males are typically affected earlier and more severely than females. The disease is unfamiliar to many practitioners and the majority of published data stem from case reports with a brief clinical review published in 2002.4 Our aim was to perform a systematic review of Danon disease, provide a comprehensive clinical and molecular update, and propose diagnostic and management guidelines for clinicians and researchers working with Danon disease patients.
Figure 1. Histological Images From Skeletal Muscle Biopsy and Endomyocardial Biopsy3.
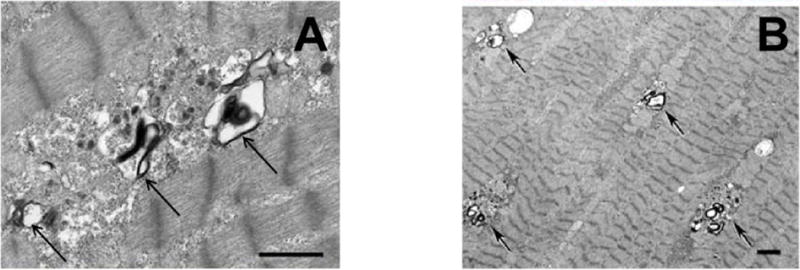
Electron microscopy shows intracytoplasmic vacuoles (arrows) containing autophagic material and glycogen in both a) skeletal muscle (Bar 1 μm) and b) endomyocardial tissue biopsy (Bar 1 μm).
Reprinted from Taylor et al3 with permission of the publisher. Copyright © 2007, Nature Publishing Group. Authorization for this adaptation has been obtained both from the owner of the copyright in the original work and from the owner of copyright in the translation or adaptation.
Materials and Methods
A literature review was performed using PubMed to identify articles and case reports in the English literature between 1981 and August 2013 on the clinical description, molecular mechanism, genetics, and treatment of Danon disease. Combinations of medical subject heading terms including ‘Danon disease’, ‘LAMP2’, ‘Antopol disease’, ‘Lysosomal Glycogen Storage Disease Without Acid Maltase Deficiency’, and ‘Glycogen Storage Disease IIb’ were used. Identified articles and case reports were reviewed and the related reference lists were also searched to include additional studies.
Information on molecular mechanisms, genetic mutations, and treatment approaches for Danon disease were extracted from the literature; endpoint data including age of symptom onset (cardiomyopathy or skeletal muscle weakness), age of heart transplantation, and age of death were retrieved. Mutation data were obtained from our database and the publically available Human Gene Mutation Database (http://www.hgmd.org/). De-identified clinical data from our own Danon disease registry were reviewed under a protocol approved by the Colorado Multiple Institutional Review Board.
Epidemiology
The prevalence of Danon disease is unknown and as cases have been described around the world it can likely affect any ethnic group.5 The observed prevalence may be rising due to increased detection from wider availability of LAMP2 testing included in clinical genetic cardiomyopathy gene testing panels. One study identified Danon disease in two of 50 (4%) pediatric patients with hypertrophic cardiomyopathy.6 Arad et al. found Danon disease in four of 24 patients (17%) among a subgroup with both thickened left ventricular walls and pre-excitation on electrocardiogram.7 In another selected population, three Danon disease patients (33%) were present in a subgroup of nine male patients with both vacuolar myopathy on muscle biopsy and hypertrophic cardiomyopathy.8
Molecular Mechanism
The LAMP2 gene codes for three major LAMP2 protein isoforms generated by alternative splicing. The LAMP2 protein is a type 1 membrane protein predominantly located in the lysosomal compartment and its structure consists of a large luminal domain that is heavily glycosylated, a transmembrane region, and a short carboxy-terminal cytoplasmic tail.9 The short LAMP-2A cytoplasmic tail is thought to serve as a receptor for uptake of certain proteins into the lysosome for degradation, a process termed chaperone-mediated autophagy.9, 10 Interestingly, about 2–3% of LAMP2 is present in the plasma membrane, and this percentage increases during malignancy and scleroderma.9
The LAMP2 protein isoforms, LAMP-2A, LAMP-2B, and LAMP-2C, differ only at the carboxy-terminal lysosomal transmembrane domain and at the short cytosolic tail.11 While LAMP-2A is more ubiquitously expressed, there are indications that LAMP-2B is expressed at a higher level in the heart, skeletal muscle and brain.12 Due to differences in the cytosolic tail and expression patterns, it is possible that each LAMP2 isoform has a unique biological role.
Different roles for the LAMP2 isoforms have been proposed in the autophagy process. Several studies have established the role of LAMP-2A in chaperone-mediated autophagy.10, 13 More recently, the LAMP-2C protein has been implicated in novel types of autophagy, termed “RNautophagy” and “DNautophagy”, which are involved in the uptake and degradation of RNA and DNA, respectively, mainly in the brain.14, 15
The vast majority of LAMP2 mutations affect all three isoforms and to-date isoform-specific mutations have only been reported for the LAMP-2B isoform (c.1097–1098 delAA, c. −1137–1140 del TATA/ins GCTGGTCCCAAT, c.1150G>C, c.1201 A>G, c.1204 A>T).2, 12, 16–18 Given that all known mutations affect at least the LAMP-2B isoform, LAMP-2B deficiency seems to be a necessary and central feature to the pathogenesis of Danon disease. Further supporting this notion is the observation that the tissues most affected in Danon disease (myocardium, skeletal muscle, and brain) manifest higher LAMP-2B isoform expression.
LAMP2 Mutations
At the time of this review, 68 LAMP2 mutations are reported in the English literature (62) and unpublished data from our registry (6) (Supplemental Table 1 and Figure 2). While the inheritance pattern is X-linked dominant, de novo mutations have also been reported.6, 7 The exon-skipping mutation c.928G>A (skips exon 7) is the most frequent mutation reported in the LAMP2 gene,19 although mutations may be present in every exon. Most mutations are nonsense or frameshift mutations predicted to truncate the LAMP2 protein, resulting in absence of the transmembrane and cytoplasmic domains and likely disabling its function as a lysosomal membrane protein.2 Splicing, large deletion, large duplication, insertion/deletion, and missense mutations that cause LAMP2 deficiency of all isoforms have also been described. Splicing mutations are most prevalent in exon 6 with none being reported in introns 3 or 4. Five mutations restricted tothe LAMP-2B isoform have been reported to cause Danon disease.2, 16, 18
Figure 2. LAMP2 Mutations in Danon Disease.
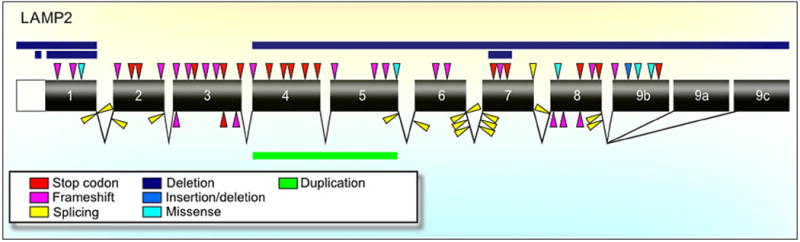
This visual display (not to scale) includes locations of previously reported and novel LAMP2 mutations causing Danon disease. Relative positions of mutations are reflected via arrows and bars (deletion and duplications indicated by bars only). Refer to Supplemental Table 1 for specific mutation information.
Based on mutation and clinical data (published and unpublished from our registry), we assessed genotype-phenotype correlations (Figure 3). Mutation types (nonsense, frameshift, etc.) were used as categories of genotype, while age of symptom onset (onset of cardiomyopathy and/or skeletal myopathy) was used as a measure of phenotypic severity. Of the 68 LAMP2 mutations, 35 mutations had reported data on age of symptom onset providing data on 73 cases. Nonsense, frameshift, and large deletion/duplication mutations showed the earliest age of onset with mean and standard deviation of ages of symptom onset for males of 13.5±4.9 years, 12.1±8.4 years, and 12.3±7.5 years, respectively, and for females of 29.9±10.0 years and 21.6±11.9 years (no case report for females with large deletion/duplication), respectively. Splicing mutations showed a trend of presenting later with ages of symptom onset for males and females of 15.4±8.1 years (p=0.31) and 37.3±12.3 years (p=0.059), respectively. Missense mutations showed the latest age of onset compared to all other mutations at 47.6±19.1 years for males (p=0.015; n=5 males). Interestingly, four of these five male patients were from a single family with a missense mutation restricted to the LAMP-2B isoform (c.1150 G>C),18 with an average age of onset of 54.2±13.8 years; a single female carrier in this family, formerly healthy, died suddenly at age 28 due to cardiomyopathy18, 20 and had irregular LAMP2 protein distribution on her muscle biopsy but no clear reduction in total LAMP2 protein by Western blotting. The fifth male patient carried a Trp321Arg missense mutation common to all isoforms21 and leading to classic findings at age 21. These cases suggest that missense mutations, particularly those restricted to a single isoform (e.g. LAMP-2B) produce a muted phenotype compared to pan-isoform protein-truncating mutations.
Figure 3. Genotype-Phenotype Correlation.
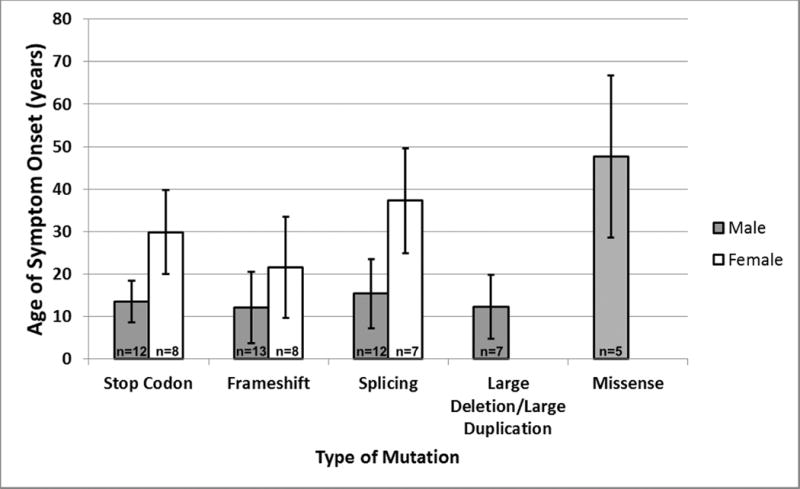
Age of first-symptom-onset by gender and mutation type is depicted. Mutation types were used as categories of genotype, while age of symptom onset (usually cardiomyopathy but also included skeletal myopathy) was used as a measure of phenotype severity. The utilization of age of death as a measure of phenotype severity (not shown) displayed similar trends, although not enough cases were present in several mutation categories to assess statistical significance.
n = number of Danon disease patients
Clinical and Diagnostic Manifestations
Danon disease presents classically with the clinical triad of cardiomyopathy, skeletal myopathy, and intellectual disability1 in boys. Other less prevalent symptoms may also be present, including retinal disease,22, 23 hepatic disease1, 8, 24, 25 and pulmonary disease.24 Birth and perinatal histories of Danon disease patients are usually unremarkable. The earliest reported onset of symptoms was at age 4 months in a male patient who presented with hypotonia and cardiac failure.19 Further diagnostic examination revealed severe obstructive hypertrophic cardiomyopathy on echocardiography and marked vacuolar myopathy on muscle biopsy.19 Females generally present later in childhood or early adulthood and have a more protracted course. The gender differences lend themselves to later presentations in females where the average ages of first symptom, cardiac transplantation, and death occur 10–15 years later in females (27.9, 33.7, and 34.6 years in females and 12.1, 17.9, and 19.0 years in males, respectively).16 Clinical features of males and females are described below separately due to the different ages of presentation and clinical courses by gender (see also Supplemental Table 2).
Clinical and Diagnostic Manifestations in Males
Due to haploinsufficiency of the X-linked LAMP2 gene, males with LAMP2 mutations are more severally affected than females and symptom onset is noted at an earlier age (13.3±8.0 years for males and 28.9±14.2 years for females; p=0.0008; (LAMP2 missense mutations excluded from analysis)). Data extrapolated from the two largest case series on Danon disease show 100% of affected males having cardiomyopathy, 80–90% having skeletal muscle weakness, and 70–100% of affected males reporting some form of cognitive impairment.16, 25 Furthermore, symptomatic respiratory disease and gastrointestinal disease were reported in 13/26 (50%) and 20/26 (77%) affected males, respectively.16
Cardiomyopathy
Danon cardiomyopathy is progressive and typically manifests a hypertrophic phenotype, with preserved ejection fraction and normal cavity dimensions early in the course of disease,26 and later progression to dilated features in 11–12% of males.16, 25 Hypertrophy can be extreme, and one report noted the largest heart by weight ever reported in medical literature from a 14-year old male with a ventricular septal thickness of 65 mm and a weight of 1,425 grams at autopsy.26 Concentric left-ventricular hypertrophy may evolve into dilated cardiomyopathy during follow-up evaluations. The extent and severity of cardiomyopathy is the major prognostic factor and cardiac transplantation may be inevitable for most males in the second and third decades.16 Postmortem examination of affected myocardium displays significant fibrosis and necrosis.27 Both atrial and ventricular arrhythmias are seen. Sudden cardiac death, presumably from ventricular arrhythmia, is the ultimate cause of death in many, noted in 2/7 (29%) patients in one high-profile case series.26
Cardiac Electrical Abnormalities
Electrical conduction abnormalities are also common, presenting in 86–100% of affected males.16, 25 Pre-excitation with a Wolf-Parkinson-White (WPW) syndrome pattern is the most common electrocardiogram finding present in 69% of cases.16 Since the prevalence of WPW patterns in Danon disease is approximately 45 and five times greater in Danon disease than in idiopathic and familial hypertrophic cardiomyopathy, respectively, the presence of WPW pattern in a young male with hypertrophic cardiomyopathy strongly suggests Danon disease.28 Arrhythmias and cardiac ablation procedures are common in 53% and 41%, respectively.16 The mechanism of pre-excitation anomalies is not known but could be due to myocardial hypertrophy29 or directly resulting from abnormal autophagy.25 Supporting the latter hypothesis are mutations in PRKAG2 which result in impaired cellular autophagy and pre-excitation findings.30 In a PRKAG2 mouse model, Arad et al. found that the annulus fibrosus, which electrically insulates the ventricles from the atria, was disrupted by glycogen-filled myocytes, suggesting that microscopic atrioventricular connections provide the anatomic substrate for ventricular preexcitation.31
Myopathy
Skeletal myopathy manifests as progressive proximal muscle weakness of the shoulders, neck, and legs1 in 80–90% of males.16, 25 Weakness is seldom debilitating and patients usually retain the ability to walk as adults. A study utilizing a hand-held dynamometer to measure muscle strength showed that compared to healthy males, Danon disease males showed a significantly lower overall generalized strength compared to controls with an average 60±5% decrease.32 Elevated serum creatine kinase (CK) levels with an average level of 944±327 U/L25 may be present. The relative lack of severe and progressive skeletal myopathy may contribute to the favorable rehabilitation outcomes following cardiac transplantation noted by our group (Taylor, unpublished data, 2013). Skeletal muscle biopsy (Figure 1a) shows intracytoplasmic vacuoles containing autophagic material and glycogen1, 25 with absent expression of LAMP2 protein in males.
Neurological Manifestations
Learning disability and cognitive deficits were reported in 70–100% of affected males, although the majority wasdescribed as mild16, 25 and affected males are able to learn to read, hold jobs, enter relationships, and usually live independently. In spite of the high prevalence of cognitive problems, a careful characterization of neurocognitive problems has not been published. One psychiatric case report on an affected 19-year male described significant psychosis, suicidal ideations, and attention-deficit hyperactivity disorder.33 Another 38-year-old male patient transplanted 15 years previously developed depression, cognitive decline with dementing features, and severe paranoia necessitating psychiatric hospitalization (un-published data). It is unclear currently if either psychiatric episode was related to Danon disease. Charcot-Marie-Tooth features, including pes cavus, distal lower limb atrophy with mild axonal neuropathy and sensory loss were also described in a 24-year-old male patient with Danon disease, suggestive of a neuropathic disease instead of a primary muscular disorder.34 Other mental symptoms in males include speech and language delay,27, 35 attention deficit,33 behavioral problems,25, 33 and dysmetria.36
Retinal involvement is common and visual problems are present in 69% of males16 who show a higher degree of ophthalmic manifestations than females,23 including a near-complete and diffuse loss of pigment in the retinal pigment epithelium.22 Thiadens et al. reported central scotoma, serious color vision disturbances, and a cone-rod pattern of amplitude reduction on electroretinogram in male patients.20 Cone rod dystrophy was also reported with a late onset but severe dystrophy (loss of photoreceptors and retinal pigment epithelium cells).20
Clinical and Diagnostic Manifestations in Females
Girls and women with LAMP2 mutations are generally less severely affected. In the two largest case series, 6–47% reported cognitive disabilities, 61–100% had evidence of cardiomyopathy, and 33–50% demonstrated skeletal muscle weakness.16, 25
Cardiomyopathy
In contrast to the predominantly hypertrophic cardiomyopathy phenotype in males, affected females show an approximately equal prevalence of dilated cardiomyopathy (28%) and hypertrophic cardiomyopathy (33%); eventually 18% received cardiac transplantation.16 Explanted hearts show severe interstitial fibrosis, hypertrophic cardiomyocytes with vacuolization, and myofibrillar disarray.37
Cardiac Electrical Abnormalities
Electrical conduction abnormalities are present in 80–100% of affected women,16, 25 although only 27% display WPW pattern on electrocardiogram.16 A subset of women is severely affected as demonstrated by one Italian family where four of six affected women died suddenly between ages 37 to 54 years. Wolff-Parkinson-White pattern and atrioventricular block were present in two of the women, although the latter arrhythmogenic condition (AV block) may have been exacerbated by the administration of beta-blocker or non-dihydropyridine calcium-channel blocker medication. Approximately a third of females received cardiac ablation procedures, 31% underwent defibrillator implantation.16 It is suggested that cardiac magnetic resonance (cMRI) might be beneficial to detect fibrosis implying a high arrhythmogenic and sudden-death risk.37
Myopathy
Muscle weakness is usually mild to asymptomatic in females. A quantitative comparison of overall generalized skeletal muscle strength showed that affected females were only 30±5% weaker than healthy females.32 Furthermore, CK elevations are present in just over half of females5 with a mean reported value of only 106±104 U/L.16
Neurological Manifestations
Reported learning and cognitive problems are less frequent in female, while complaints of unspecified neuropathy and muscle cramping are higher in females.16 Visual problems were also reported in up to 64% of affected females in one series16 and affected females may have peripheral pigmentary retinopathy.22 In contrast to the diffuse and near-complete loss of retinal pigment in male patients, carrier females demonstrate a peppered and granular retinal pigment epithelium appearance, which is proposed to be due to lionization.22
Diagnosis and Differential Diagnosis
The differential diagnoses for Danon disease are presented in Table 1. Major clinical features that suggest and ultimately confirm Danon disease include an X-linked dominant inheritance pattern, hypertrophic cardiomyopathy in young males, muscle weakness, and some degree of cognitive difficulties. Supporting diagnostic studies include normal acid maltase levels on muscle biopsy1 (or increasingly by blood-spot analysis), immunohistochemistry showing LAMP2 protein deficiency,2 autophagic vacuole accumulation by electron microscopy, and genetic mutation analysis of LAMP2 gene.27 The non-invasive nature of DNA-based methods and the inclusion of LAMP2 gene testing in hypertrophic cardiomyopathy genetic testing panels likely favor genetic testing as the most common route to identifying Danon disease. Males show elevated serum creatine kinase levels around two to three times the normal value.1, 6–8, 25, 35, 36, 40, 41 Liver function tests display elevated levels of aspartate transaminase, alanine aminotransferase, and lactate dehydrogenase.1, 6, 25, 35, 40 Hepatic synthetic function is usually normal, although hepatomegaly was reported in 5/14 (35%) males in one study.25
Table 1.
Differentiating Danon Disease from other Vacuolar Myopathies.
| Condition | OMIM Number | Inheritance Pattern | Cardiomyopathy | Skeletal Myopathy | Intellectual Disability | Deposition of complement C5b-9 on muscle fibers | Molecular Defect |
|---|---|---|---|---|---|---|---|
| Danon disease | 300257 | X-linked dominant | X | X | X | Mutation in LAMP2 gene | |
| Pompe disease | 232300 | Autosomal recessive | X | X | Mutation in GAA gene (reduced/absent acid maltase) | ||
| X-linked myopathy with excessive autophagy (XMEA) | 310440 | X-linked recessive | X | X | Reduced/absent VMA21 protein | ||
| X-linked congenital autophagic vacuolar myopathy | *38 | X-linked recessive | X | X | Genetic locus/causative gene unknown | ||
| Infantile autophagic vacuolar myopathy (AVM) | 609500 | X-linked recessive | X | X | X | Genetic locus/causative gene unknown | |
| Autophagic vacuolar myopathy with late-onset and multiorgan involvement | *39 | X-linked | X | X | X | Genetic locus/causative gene unknown | |
| “Glycogen storage disease of the heart, lethal congenital” | 261740 | Autosomal recessive | X | Mutation in PRKAG2 gene | |||
| Chloroquine-induced myopathy | ** | Drug-induced | X | X | Chloroquineinhibits lysosomal enzymes |
Differential diagnoses and associated clinical signs in patients presenting with vacuolar myopathy are displayed. An X-linked dominant inheritance pattern, cardiomyopathy, skeletal myopathy, intellectual disability, LAMP2 protein deficiency, confirmed LAMP2 gene mutation, and normal acid maltase levels are clinical signs used to diagnose Danon disease. Note that not every Danon disease patient presents with all listed clinical symptoms, and LAMP2 protein deficiency may not always be present. However, the presence of a LAMP2 gene mutation confirms Danon disease.
= OMIM number currently not present (but reference included)
= no OMIM numbers for non-genetic disorders
Treatment Guidelines
Diagnostic and management guidelines for Danon disease have not been published. In Table 2, we propose therapeutic approaches for each specific clinical manifestation. Clinicians may also refer to the 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy with the caveat that Danon disease may present earlier and progress more rapidly than other forms of hypertrophic cardiomyopathy, especially in males.42
Table 2.
Suggested Treatment Guidelines for Danon Disease Manifestations.
| Manifestations | Diagnostic Testing* | Recommended Treatment |
|---|---|---|
| Cardiomyopathy |
|
|
| Skeletal myopathy |
|
|
| Intellectual Disability |
|
|
| Ocular Disease |
|
|
| Genetic |
|
|
This table is intended as a basic guide, and is by no means complete or definitive. Suggested frequencies of evaluations are provided for reference and should be adjusted on the basis of individual patient symptoms and disease progression.
ECG = electrocardiogram; BNP = brain natriuretic peptide; LVEF = left ventricular ejection fraction; ICD = implantable cardioverter-defibrillator
Newly diagnosed patients may be best served by a team approach that includes a primary-care physician in conjunction with several specialties including cardiology, genetics, neurology, ophthalmology, rehabilitation medicine, and physical therapy. Regular cardiology evaluations are critical given the nature of the expected cardiac progression; frequently, input from both Advanced Heart Failure and Transplant and Clinical Cardiac Electrophysiology sub-specialties will be beneficial. Newly diagnosed patients should typically be studied by electrocardiogram and echocardiography, along with measurements of serum natriuretic peptide (BNP) levels and consideration for 24-hour Holter or event monitoring. cMRI may be sensitive for fibrotic changes that may predict higher risks of arrhythmogenic events.37 Our experience is that progression of moderate hypertrophic disease can be quite rapid in some males and therefore evaluations every three to six months, including consideration of transplant evaluation, may be appropriate in patients with evidence of significant cardiac involvement. Girls and women with progressive cardiac disease should be treated similarly.
Implantable cardioverter-defibrillator (ICD) therapy should be strongly considered in patients with symptomatic arrhythmias, moderate to severe hypertrophy, substantial fibrosis burden on cMRI imaging, and/or family history of premature sudden death.37 Cardiac ablation has also achieved some success in eliminating arrhythmias in Danon disease patients;7, 43, 44 however, anecdotally several patients in our registry have required multiple ablation procedures possibly reflecting diffuse and progressive fibrosis that is challenging to eliminate by ablation therapy.
Other symptoms in Danon disease, including skeletal myopathy, intellectual disability, and eye disease are considered mild and not life-threatening. Physicians should aim to prevent progressive loss of muscle strength and flexibility in affected patients through standard physical therapy and light exercise. Assessment of muscle strength, particularly the proximal muscles of the shoulder, neck, and legs, should be performed during scheduled physical examination visits.
Intellectual difficulties should be anticipated and identified for early intervention. From registry data, most boys have areas of academic weakness in mathematics and reading. We suggest that comprehensive neuropsychological exams may be useful to assess other neurocognitive problems such as attention-deficit hyperactivity and autism-spectrum disorders. Enrollment in a rehabilitation center for educational, psychological, and social support is suggested. Eye-related problems can present in the form of choriocapillary ocular atrophy,6, 36 diminished retinal pigmentation,22, 23 lens changes, myopia, and abnormal visual fields,22 maculopathy,34 and cone rod dystrophy.20, 45 A baseline exam with a retinal specialist seems prudent with prospective ophthalmologic examinations based on initial findings.
Genetic counseling should also be offered to affected families so that they are knowledgeable about the inheritance pattern and reproductive risks. With improved survival from cardiac transplantation, the expectation is that men may be more capable of fathering children. Thus, both men and women of sufficient health and reproductive potential should be advised of the inheritance risks for LAMP2 mutations to be transmitted to future offspring. As has been suggested previously, this may be ideally performed in centers with expertise in cardiomyopathy genetic counseling.46
Conclusion
In summary, Danon disease is a rare cardiac and skeletal muscle disorder caused by LAMP2 mutations and presenting with systemic symptoms of cardiomyopathy, skeletal myopathy, and intellectual disability. Symptom severity tends to be much greater in affected males and Danon disease should be strongly suspected in young males presenting with pre-excitation and moderate to severe cardiac hypertrophy. Family history analysis may reveal affected females and both symptomatic males and females in affected families should receive diagnostic genetic testing and case-specific therapy, particularly cardiac treatment. The literature supports that truncating mutations tend to cause more detrimental phenotypes, and that the LAMP2B isoform may play a crucial role in Danon disease pathogenesis. In addition, along with previously published LAMP2 mutations in Danon disease, we report novel LAMP2 mutations from patients in our registry. Finally, the overall major goals of our proposed management guidelines include slowing progression to heart failure, eliminating arrhythmias, slowing muscle loss and flexibility, and preventing loss of cognition. Future research efforts should investigate the role of the LAMP2 gene in Danon disease by identifying biochemical pathways involving LAMP2 protein and describing the molecular function of each LAMP2 isoform.
Supplementary Material
Acknowledgments
Sources of Funding
NIH 1R01HL109209-01A1, NIH/NCATS UL1 TR001082, MDA 67944, Department of Medicine at University of Colorado Anschutz Medical Campus
Footnotes
Journal Subject Codes:
Etiology: [109] Clinical genetics, Heart failure: [11] Other heart failure, Hypertension: [15] Hypertrophy, Hypertension: [16] Myocardial cardiomyopathy disease
Disclosures
None.
References
- 1.Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31:51–57. doi: 10.1212/wnl.31.1.51. [DOI] [PubMed] [Google Scholar]
- 2.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Primary lamp-2 deficiency causes x-linked vacuolar cardiomyopathy and myopathy (danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 3.Taylor MR, Ku L, Slavov D, Cavanaugh J, Boucek M, Zhu X, Graw S, Carniel E, Barnes C, Quan D, Prall R, Lovell MA, Mierau G, Ruegg P, Mandava N, Bristow MR, Towbin JA, Mestroni L, Registry FC. Danon disease presenting with dilated cardiomyopathy and a complex phenotype. J Hum Genet. 2007;52:830–835. doi: 10.1007/s10038-007-0184-8. [DOI] [PubMed] [Google Scholar]
- 4.Froissart R, Maire I. Danon disease. Orphanet Encyclopedia. 2004 https://www.orpha.net/data/patho/GB/uk-Danon.pdf.
- 5.Nishino I. Autophagic vacuolar myopathy. Semin Pediatr Neurol. 2006;13:90–95. doi: 10.1016/j.spen.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Charron P, Villard E, Sébillon P, Laforêt P, Maisonobe T, Duboscq-Bidot L, Romero N, Drouin-Garraud V, Frébourg T, Richard P, Eymard B, Komajda M. Danon’s disease as a cause of hypertrophic cardiomyopathy: A systematic survey. Heart. 2004;90:842–846. doi: 10.1136/hrt.2003.029504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arad M, Maron BJ, Gorham JM, Johnson WH, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 8.Fanin M, Nascimbeni AC, Fulizio L, Spinazzi M, Melacini P, Angelini C. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in danon disease. Am J Pathol. 2006;168:1309–1320. doi: 10.2353/ajpath.2006.050646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuda M. Biogenesis of the lysosomal membrane. Subcell Biochem. 1994;22:199–230. doi: 10.1007/978-1-4615-2401-4_7. [DOI] [PubMed] [Google Scholar]
- 10.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 11.Majer F, Pelak O, Kalina T, Vlaskova H, Dvorakova L, Honzik T, Palecek T, Kuchynka P, Masek M, Zeman J, Elleder M, Sikora J. Mosaic tissue distribution of the tandem duplication of lamp2 exons 4 and 5 demonstrates the limits of danon disease cellular and molecular diagnostics. J Inherit Metab Dis. 2014;37:117–124. doi: 10.1007/s10545-013-9617-z. [DOI] [PubMed] [Google Scholar]
- 12.Konecki DS, Foetisch K, Zimmer KP, Schlotter M, Lichter-Konecki U. An alternatively spliced form of the human lysosome-associated membrane protein-2 gene is expressed in a tissue-specific manner. Biochem Biophys Res Commun. 1995;215:757–767. doi: 10.1006/bbrc.1995.2528. [DOI] [PubMed] [Google Scholar]
- 13.Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000;113(Pt 24):4441–4450. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- 14.Fujiwara Y, Furuta A, Kikuchi H, Aizawa S, Hatanaka Y, Konya C, Uchida K, Yoshimura A, Tamai Y, Wada K, Kabuta T. Discovery of a novel type of autophagy targeting rna. Autophagy. 2013;9:403–409. doi: 10.4161/auto.23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujiwara Y, Kikuchi H, Aizawa S, Furuta A, Hatanaka Y, Konya C, Uchida K, Wada K, Kabuta T. Direct uptake and degradation of dna by lysosomes. Autophagy. 2013;9 doi: 10.4161/auto.24880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucek D, Jirikowic J, Taylor M. Natural history of danon disease. Genet Med. 2011;13:563–568. doi: 10.1097/GIM.0b013e31820ad795. [DOI] [PubMed] [Google Scholar]
- 17.Hong D, Shi Z, Wang Z, Yuan Y. Danon disease caused by two novel mutations of the lamp2 gene: Implications for two ends of the clinical spectrum. Clin Neuropathol. 2012;31:224–231. doi: 10.5414/NP300465. [DOI] [PubMed] [Google Scholar]
- 18.van der Kooi AJ, van Langen IM, Aronica E, van Doorn PA, Wokke JH, Brusse E, Langerhorst CT, Bergin P, Dekker LR, dit Deprez RH, de Visser M. Extension of the clinical spectrum of danon disease. Neurology. 2008;70:1358–1359. doi: 10.1212/01.wnl.0000309219.61785.b3. [DOI] [PubMed] [Google Scholar]
- 19.Bertini E, Donati MA, Broda P, Cassandrini D, Petrini S, Dionisi-Vici C, Ballerini L, Boldrini R, D’Amico A, Pasquini E, Minetti C, Santorelli FM, Bruno C. Phenotypic heterogeneity in two unrelated danon patients associated with the same lamp-2 gene mutation. Neuropediatrics. 2005;36:309–313. doi: 10.1055/s-2005-872844. [DOI] [PubMed] [Google Scholar]
- 20.Thiadens AA, Slingerland NW, Florijn RJ, Visser GH, Riemslag FC, Klaver CC. Cone-rod dystrophy can be a manifestation of danon disease. Graefes Arch Clin Exp Ophthalmol. 2012;250:769–774. doi: 10.1007/s00417-011-1857-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musumeci O, Rodolico C, Nishino I, Di Guardo G, Migliorato A, Aguennouz M, Mazzeo A, Messina C, Vita G, Toscano A. Asymptomatic hyperckemia in a case of danon disease due to a missense mutation in lamp-2 gene. Neuromuscul Disord. 2005;15:409–411. doi: 10.1016/j.nmd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Prall FR, Drack A, Taylor M, Ku L, Olson JL, Gregory D, Mestroni L, Mandava N. Ophthalmic manifestations of danon disease. Ophthalmology. 2006;113:1010–1013. doi: 10.1016/j.ophtha.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 23.Schorderet DF, Cottet S, Lobrinus JA, Borruat FX, Balmer A, Munier FL. Retinopathy in danon disease. Arch Ophthalmol. 2007;125:231–236. doi: 10.1001/archopht.125.2.231. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in lamp-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 25.Sugie K, Yamamoto A, Murayama K, Oh SJ, Takahashi M, Mora M, Riggs JE, Colomer J, Iturriaga C, Meloni A, Lamperti C, Saitoh S, Byrne E, DiMauro S, Nonaka I, Hirano M, Nishino I. Clinicopathological features of genetically confirmed danon disease. Neurology. 2002;58:1773–1778. doi: 10.1212/wnl.58.12.1773. [DOI] [PubMed] [Google Scholar]
- 26.Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, Wright GB, Almquist AK, Baffa JM, Saul JP, Ho CY, Seidman J, Seidman CE. Clinical outcome and phenotypic expression in lamp2 cardiomyopathy. JAMA. 2009;301:1253–1259. doi: 10.1001/jama.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balmer C, Ballhausen D, Bosshard NU, Steinmann B, Boltshauser E, Bauersfeld U, Superti-Furga A. Familial x-linked cardiomyopathy (danon disease): Diagnostic confirmation by mutation analysis of the lamp2gene. Eur J Pediatr. 2005;164:509–514. doi: 10.1007/s00431-005-1678-z. [DOI] [PubMed] [Google Scholar]
- 28.MARRIOTT HJ. Electrocardiographic abnormalities, conduction disorders and arrhythmias in primary myocardial disease. Prog Cardiovasc Dis. 1964;7:99–114. doi: 10.1016/s0033-0620(64)80013-x. [DOI] [PubMed] [Google Scholar]
- 29.Riggs JE, Schochet SS, Gutmann L, Shanske S, Neal WA, DiMauro S. Lysosomal glycogen storage disease without acid maltase deficiency. Neurology. 1983;33:873–877. doi: 10.1212/wnl.33.7.873. [DOI] [PubMed] [Google Scholar]
- 30.Oliveira SM, Ehtisham J, Redwood CS, Ostman-Smith I, Blair EM, Watkins H. Mutation analysis of amp-activated protein kinase subunits in inherited cardiomyopathies: Implications for kinase function and disease pathogenesis. J Mol Cell Cardiol. 2003;35:1251–1255. doi: 10.1016/s0022-2828(03)00237-2. [DOI] [PubMed] [Google Scholar]
- 31.Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG. Transgenic mice overexpressing mutant prkag2 define the cause of wolff-parkinson-white syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107:2850–2856. doi: 10.1161/01.CIR.0000075270.13497.2B. [DOI] [PubMed] [Google Scholar]
- 32.Stevens-Lapsley JE, Kramer LR, Balter JE, Jirikowic J, Boucek D, Taylor M. Functional performance and muscle strength phenotypes in men and women with danon disease. Muscle Nerve. 2010;42:908–914. doi: 10.1002/mus.21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatz DE, Sharma A, Germer KE, Rolfsmeyer EA, Bowen JM. Psychosis in a patient with danon cardiomyopathy. Gen Hosp Psychiatry. 2010;32:328–329. doi: 10.1016/j.genhosppsych.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Laforêt P, Charron P, Maisonobe T, Romero NB, Villard E, Sebillon P, Drouin-Garraud V, Dubourg O, Fardeau M, Komajda M, Eymard B. Charcot-marie-tooth features and maculopathy in a patient with danon disease. Neurology. 2004;63:1535. doi: 10.1212/01.wnl.0000141858.80738.aa. [DOI] [PubMed] [Google Scholar]
- 35.Dworzak F, Casazza F, Mora M, De Maria R, Gronda E, Baroldi G, Rimoldi M, Morandi L, Cornelio F. Lysosomal glycogen storage with normal acid maltase: A familial study with successful heart transplant. Neuromuscul Disord. 1994;4:243–247. doi: 10.1016/0960-8966(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 36.Lacoste-Collin L, Garcia V, Uro-Coste E, Arné-Bes MC, Durand D, Levade T, Delisle MB. Danon’s disease (x-linked vacuolar cardiomyopathy and myopathy): A case with a novel lamp-2 gene mutation. Neuromuscul Disord. 2002;12:882–885. doi: 10.1016/s0960-8966(02)00179-7. [DOI] [PubMed] [Google Scholar]
- 37.Miani D, Taylor M, Mestroni L, D’Aurizio F, Finato N, Fanin M, Brigido S, Proclemer A. Sudden death associated with danon disease in women. Am J Cardiol. 2012;109:406–411. doi: 10.1016/j.amjcard.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 38.Yan C, Tanaka M, Sugie K, Nobutoki T, Woo M, Murase N, Higuchi Y, Noguchi S, Nonaka I, Hayashi YK, Nishino I. A new congenital form of x-linked autophagic vacuolar myopathy. Neurology. 2005;65:1132–1134. doi: 10.1212/01.wnl.0000178979.19887.f5. [DOI] [PubMed] [Google Scholar]
- 39.Kaneda D, Sugie K, Yamamoto A, Matsumoto H, Kato T, Nonaka I, Nishino I. A novel form of autophagic vacuolar myopathy with late-onset and multiorgan involvement. Neurology. 2003;61:128–131. doi: 10.1212/01.wnl.0000069605.00498.bd. [DOI] [PubMed] [Google Scholar]
- 40.Byrne E, Dennett X, Crotty B, Trounce I, Sands JM, Hawkins R, Hammond J, Anderson S, Haan EA, Pollard A. Dominantly inherited cardioskeletal myopathy with lysosomal glycogen storage and normal acid maltase levels. Brain. 1986;109(Pt 3):523–536. doi: 10.1093/brain/109.3.523. [DOI] [PubMed] [Google Scholar]
- 41.Katsumi Y, Tokonami F, Matsui M, Aii H, Nonaka I. [a case of glycogen storage disease with normal acid maltase accompanied with the abnormal platelet function] Rinsho Shinkeigaku. 1994;34:827–831. [PubMed] [Google Scholar]
- 42.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW, Guidelines ACoCFAHATFoP, Surgery AAfT, Echocardiography ASo, Cardiology ASoN, America HFSo, Society HR, Interventions SfCAa, Surgeons SoT 2011 accf/aha guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: Executive summary: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation. 2011;124:2761–2796. doi: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- 43.Piotrowska-Kownacka D, Kownacki L, Kuch M, Walczak E, Kosieradzka A, Fidzianska A, Krolicki L. Cardiovascular magnetic resonance findings in a case of danon disease. J Cardiovasc Magn Reson. 2009;11:12. doi: 10.1186/1532-429X-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z, Funke BH, Cripe LH, Vick GW, Mancini-Dinardo D, Peña LS, Kanter RJ, Wong B, Westerfield BH, Varela JJ, Fan Y, Towbin JA, Vatta M. Lamp2 microdeletions in patients with danon disease. Circ Cardiovasc Genet. 2010;3:129–137. doi: 10.1161/CIRCGENETICS.109.901785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brodie S. Cone-rod dystrophy in danon disease. Graefes Arch Clin Exp Ophthalmol. 2012;250:633. doi: 10.1007/s00417-012-1936-5. [DOI] [PubMed] [Google Scholar]
- 46.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA, America HFSo. Genetic evaluation of cardiomyopathy–a heart failure society of america practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.