

Abstract
Deamidation of asparagine residues, a post-translational modification observed in proteins, is a common degradation pathway in monoclonal antibodies (mAbs). The kinetics of deamidation is influenced by primary sequence as well as secondary and tertiary folding. Analytical hydrophobic interaction chromatography (HIC) is used to evaluate hydrophobicity of candidate mAbs and uncover post-translational modifications. Using HIC, we discovered atypical heterogeneity in a highly hydrophobic molecule (mAb-1). Characterization of the different HIC fractions using LC/MS/MS revealed a stable succinimide intermediate species localized to an asparagine-glycine motif in the heavy chain binding region. The succinimide intermediate was stable in vitro at pH 7 and below and increased on storage at 25°C and 40°C. Biacore evaluation showed a decrease in binding affinity of the succinimide intermediate compared with the native asparagine molecule. In vivo studies of mAb-1 recovered from a pharmacokinetic study in cynomolgus monkeys revealed an unstable succinimide species and rapid conversion to aspartic/iso-aspartic acid. Mutation from asparagine to aspartic acid led to little loss in affinity. This study illustrates the importance of evaluating modifications of therapeutic mAbs both in vitro and in serum, the intended environment of the molecule. Potential mechanisms that stabilize the succinimide intermediate in vitro are discussed.
Keywords: deamidation, succinimide, IgG1, monoclonal antibody, aspartate, iso-aspartate
Introduction
Recombinant monoclonal antibodies (mAbs) are an important class of therapeutics with over 30 molecules approved and sales of ~$50 billion in 2010.1-4 The exquisite specificity of mAbs for antigen has resulted in therapies for a variety of indications in oncology, chronic inflammation, cardiovascular disease, transplantation and infectious diseases.1,5 Therapeutic proteins manufactured for clinical use are subject to a variety of chemical and physical degradative pathways during cell culture, purification, formulation and storage. Characterization of these post-translational modifications (PTMs) is important to understand how they affect the stability and potency of the molecules.
Deamidation of asparagine to aspartic (Asp) or iso-aspartic (iso-Asp) acid, a common degradation pathway observed in proteins, has been identified and characterized in both constant and variable regions of mAbs.6-9 Deamidation is minimal at pH 5–6; however, at pH 6 and above (Fig. 1) nucleophilic attack of a α-amino group (from the peptide bond) on the side chain carbonyl group of the asparagine residue results in formation of a metastable cyclic succinimide intermediate.10 The succinimide intermediate is then hydrolyzed to either Asp (α peptide linkage) or iso-Asp (β peptide linkage) end products. Hydrolysis of the β peptide linkage is generally favored over the α peptide linkage due to asymmetry of the succinimide intermediate, resulting in a 3:1 ratio of iso-Asp:Asp acid residues.5,6,8 At basic pH of 10, the rate of deamidation through the succinimide species is very rapid. The cyclic intermediate (succinimide) is not formed when the pH is less than 5; instead, the side chain is directly hydrolyzed to Asp, and the isomeric species is not formed.11,12 While pH is important for succinimide formation, other factors such as temperature, primary structure, higher order structure, composition of buffer and ionic strength are reported to play significant roles.13-20 Small hydrophilic residues following an asparagine residue in the primary sequence, for example, will promote deamidation, with Asn:Gly motif being the most labile followed by Asn:Ser and Asn:Thr motifs.21-26

Figure 1. Deamidation at neutral or basic pH > 6 proceeds through the cyclic imide succinimide followed by hydrolysis to Asp/iso-Asp with a resulting mass change of +1Da.
Therapeutic proteins that circulate in blood for an extended time, at an elevated temperature and in a crowded environment will undergo deamidation. Thus, studying the deamidation process in vivo is important because loss of binding and potency resulting from deamidation in the CDR region have been reported.25,27 In addition, a number of studies have raised concern about the immunogenicity of proteins containing the hydrolyzed iso-Asp species.28,29 The succinimide intermediate can also undergo racemization resulting in D-Asp residues being incorporated into the molecule.14,30 Thus, pharmacokinetic (PK) information on the therapeutic antibody and an estimate of the kinetics of deamidation for different asparagine residues on the molecule are important for assessing the exposure and criticality of the PTM.
Deamidation can be monitored using a variety of techniques because the process involves changes in charge, hydrophobicity and mass. Thus, monitoring is done using capillary isoelectric focusing and ion exchange chromatography, which assess charge; reverse phase chromatography and hydrophobic interaction chromatography (HIC), which assess hydrophobicity; or mass spectrometry, which assesses mass.9-11 Evaluating deamidation in vivo generally involves using antigen cross-linked to sepharose beads to recover and purify the therapeutic mAb from other serum proteins. Recovery from animal serum can also be accomplished using antibodies that specifically recognize the Fc portion of human antibodies. The purified antibody is enzymatically digested and peptides analyzed by LC/MS/MS.27
HIC is widely used in the biotechnology industry for purification of antibodies. This mode of chromatography takes advantage of differences in relative hydrophobicity of molecules as its mode of separation.31,32 Analytical HIC is used for understanding stability in different formulations because highly hydrophobic proteins have a higher likelihood to self-associate and form aggregates. In addition, there are many examples of the use of HIC for analytical purposes to resolve PTMs such as N-terminal glutamine cyclization, oxidation of tryptophan and methionine residues, and deamidation.33-35 Here, we report use of HIC to discover a stable succinimide intermediate species that easily separated on the basis of hydrophobicity. The levels of the succinimide intermediate increased on storage at 25°C and 40°C at pH 7 and below, and, using Biacore, we found that the binding affinity of purified mAb-1 with one or two succinimide species is decreased compared with the native asparagine residue or a mutant molecule that contained Asp at this site. While stable succinimide species have been reported for other mAbs,35,36 we show that in vivo the succinimide intermediate is unstable and rapidly hydrolyzed to Asp/iso-Asp. These studies emphasize the relevance of evaluating PTMs both in standard formulation buffers and in circulation.37 We describe a model to help explain how the succinimide intermediate is stabilized in solution.
Results
Analytical hydrophobic interaction chromatography
HIC is a widely used analytical method to separate proteins on the basis of the hydrophobicity of the molecule. During HIC analysis, the molecule being analyzed is denatured in a salt buffer exposing hydrophobic patches that interact with the stationary phase. The stationary phase used for this analysis is a butyl functional group cross-linked to a 2.5 micron non-porous particle. As the gradient decreases the concentration of salt in the mobile phase, molecules are eluted, with molecules having lower relative hydrophobicity eluting first. An IgG1 molecule (mAb-1) was found to be very hydrophobic and also displayed atypical heterogeneity on the HIC column (Fig. 2).

Figure 2. Analysis of mAb-1 by HIC showing atypical heterogeneity (peaks 1–5).
To understand the source of the heterogeneity, we fractionated the 5 major peaks labeled in Figure 2 (peaks 1–5) using HIC. Each fraction was exchanged from the high salt buffer to 10 mM phosphate, 10 mM citrate pH 6 and reanalyzed by HIC to confirm purity. Upon reanalysis, we discovered that injection of any of the fractionated peaks 1, 2 and 3 resulted in redistribution to a mixture of peaks 1, 2 and 3 where peak 3 was the predominant species (75%), peak 2 comprised 20% and peak 1 was ~5% (data not shown). Digestion of mAb-1 with Lys-C into the Fab and Fc fragments localized the heterogeneity to the Fab portion of the molecule. We speculated that these were different Fab conformers formed during desalting.
Characterization of heterogeneity by LC/MS/MS
Intact and reduced LC/MS analysis was performed on peaks 3 and 4. Analysis at the intact level indicated that the MW of the predominant species in fractionated peak 3 matched the theoretical MW of the glycosylated (G0) mAb-1 and the only observed heterogeneity was the expected heterogeneity due to glycosylation. The predominant species observed in fractionated peak 4, however, had a MW that was lower than the theoretical MW by ~15 Da (data not shown).
Analysis at the reduced level indicated that the MW of the light chain (LC) in both fractionated peaks 3 and 4 matched the theoretical MW of the LC (data not shown). The predominant species of the deconvoluted LC/MS spectra of the heavy chain (HC) in fractionated peak 3 matched the theoretical MW of the glycosylated HC (50,885 Da) and the only observed heterogeneity was due to glycosylation (Fig. 3A). The predominant species observed in the deconvoluted LC/MS spectra of the HC in fractionated peak 4 matched the theoretical MW; however, an additional peak with a MW that was ~17–18 Da less than the theoretical MW was also observed (Fig. 3B). The ratio of the two observed species was ~1:1 suggesting that the source of the heterogeneity existed on one of the two HC subunits prior to reduction.

Figure 3. Reverse phase ESI-Q-TOF MS analysis of (A) reduced HIC fractionated peak 3 and (B) reduced HIC fractionated peak 4 showing the MW of the deconvoluted mass spectra of the HC. The theoretical MW of the reduced HC of mAb-1 (G0) is 50,885 Da.
To isolate the 17–18 Da heterogeneity observed on the HC, we performed tryptic digests on fractionated peaks 3 and 4 and analyzed by LC/MS. Digestion was performed under basic conditions (pH 8), the optimal pH for trypsin activity. The molecular feature extraction tool of the Agilent MassHunter Bioconfirm software was used to extract and tabulate all the peptides observed in the LC/MS of the tryptic digest for both fractionated peaks 3 and 4. We could not identify any peptides in fractionated peak 4 that had a lower MW of 17–18 Da than the expected tryptic peptides from an in silico digest. In fractionated peak 4, however, we observed a significant increase in deamidation (increase in MW of 1 Da) of a 32 amino acid peptide containing the CDR3 region of the HC (HC99–130). The increase in deamidation was not observed in fractionated peak 3, which suggests that it was not an artifact of the digestion procedure.
Deamidation of asparagine proceeds through the cyclic imide succinimide followed by hydrolysis to Asp/iso-Asp (Fig. 1) with a mass change of +1Da. Previous reports have shown stabilization of a succinimide intermediate with a mass decrease of 17 Da.27,36 Hydrolysis of the succinimide intermediate to either Asp/iso-Asp proceeds much more rapidly under basic conditions. Digestion at pH 8 drives any succinimide-containing peptides to the hydrolyzed Asp/iso-Asp residues. The succinimide intermediate can be stabilized under acidic digest conditions (pH-5) and peptides containing the intermediate have been positively identified by mass spectrometry.36,38
Tryptic digests of HIC fractionated peaks 3 and 4 were therefore performed under acidic conditions (pH 5) and the corresponding peptides analyzed by LC/MS. A search of the peptides under these modified conditions identified a peptide in peak 4 with a -17 Da modification in the 32 amino acid HC tryptic peptide (HC99–130) localized to the CDR3 region (Fig. 4A). The aforementioned peak containing the modification had an approximate ratio of 1:1 of relative area percent of the extracted ion chromatograms (EICs) in comparison to the unmodified peptide. This result confirmed the observation made in the reduced LC/MS (described above) and provided further evidence that the source of heterogeneity in this fractionated peak existed on one of the two HC subunits prior to reduction and digestion. In peak 3 (Fig. 4B), the predominant unmodified tryptic peptide HC99–130 is observed with a small amount of modified peptide, most likely due to inconsistency in HIC fractionation.

Figure 4. Reverse phase LC/MS extracted ion chromatograms of acidic tryptic digests of (A) HIC fractionated peak 4 and (B) peak 3. The EIC shows the HC99–130 tryptic peptide (MW = 3583.5 Da) and the corresponding tryptic peptide containing a 17 Da modification (MW = 3566.5 Da).
The MS/MS spectra of the modified peptide (Fig. 5A) was analyzed and confirmed to be comparable to the MS/MS spectra of the unmodified HC99–130 peptide (Fig. 5B), but with changes associated with a loss of 17 Da in a segment of the peptide. Analysis of the MS/MS spectra isolated the modification to the Asn105. Modification at the Asn105 was inferred from the 17 Da decrease in the Y series ions (Y26 to the end of the Y series in the modified peptide). Cleavage at Y25 between the Asn105 and the following Gly residue was observed in the unmodified peptide, and was not observed in the modified peptide. This difference was likely due to stabilization from the cyclic imide.

Figure 5. MS/MS spectra obtained from the CID fragmentation of (A) HC99–130 tryptic peptide with a 17 Da mass shift and (B) unmodified HC99–130 tryptic peptide from HIC fractionated peak 4.
MS analysis of HIC fractionated peak 4 revealed that the major sources of heterogeneity were deamidation of Asn105 in the CDR3 region of the HC and stabilization of the succinimide intermediate on one HC arm, which occurred at levels of 10–20% depending on purification conditions. Analysis of peak 5 revealed modification on both HC arms (data not shown). Additional heterogeneity found on the trailing end of peak 3 revealed Asp and iso- Asp modifications (data not shown). We also confirmed the site of the modification by analyzing point mutations of Asn105 to either Gly, His, Gln, Glu, Lys or Arg. In all 6 mutations, we did not observe the heterogeneity by HIC analysis corresponding to the succinimide modification. HIC analysis of a point mutation of Asn105 to Asp105 showed similar heterogeneity however, which suggested stabilization of the cyclic intermediate from dehydrolysis of Asp as well (data not shown).
Binding affinity by surface plasmon resonance analysis
Two primary concerns associated with the succinimide intermediate are loss of binding to antigen and decreased stability as a result of the modification. To provide sufficient material for evaluation, a preparative scale Butyl HP Sepharose column (GE Healthcare Biosciences) was used to provide pure fractions of the unmodified species and sufficient material of the one and two HC succinimide105 modifications (Fig. 6).
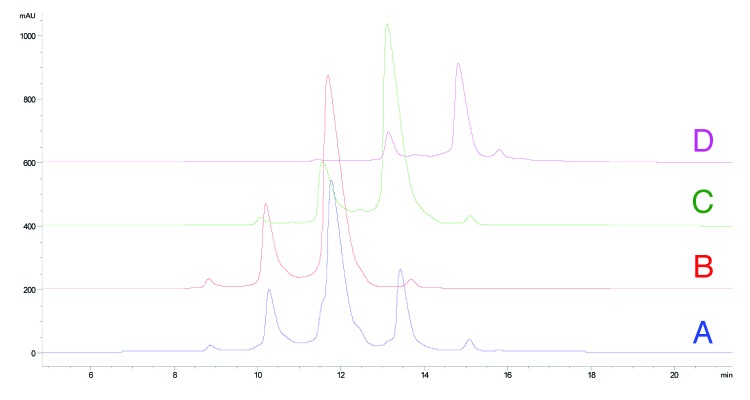
Figure 6. HIC fractionation of mAb-1 (A) unfractionated mAb-1, (B) fractionated unmodified (HC Asn105) mAb-1 (peak 3), (C) fractionated mAb-1 containing one HC succinimide105 species (peak 4), (D) fractionated mAb-1 containing two HC succinimide105 species (peak 5).
Previous studies have found a 50% decrease in binding affinity for a Fab containing succinimide modification in the CDR2 of the HC of an IgG136 and a 10% decrease in binding affinity for a succinimide modification in the CDR1 of one and two LC arms of an IgG2.35,36 The kinetics of binding for mAb-1 fractions to antigen was determined by surface plasmon resonance (SPR) on a Biacore T-200 instrument (GE Healthcare). As shown in Table 1, we observed a significant decrease in the binding affinity of mAb-1 containing one or two succinimide105 modifications on the HC compared with the HIC fraction (peak 3) containing no modifications (HC Asn105). Remarkably, evaluation of binding affinity of mAb-1 containing a point mutation of HC Asn105 to Asp105 showed comparable affinity with Asn105.
Table 1. Binding affinity (KD (M) of mAb-1 with no modification (HC Asn105); one HC succinimide105 modification; two HC succinimide105 modifications and HC Asn105 to Asp105 point mutation.
| ka (M−1s−1) | kd (s−1) | KD, pM | |
|---|---|---|---|
| (2) HC unmodified-Asn105 (peak3) | 5.6E+06 | 4.5E-06 | 0.8 |
| (1) HC succinimide105 (peak4) | 5.6E+06 | 2.9E-05 | 5.1 |
| (2) HC succinimide105 (peak5) | 6.0E+06 | 6.6E-05 | 10.9 |
| Asn105 to Asp105 mutation | 4.8E+06 | 1.1E-05 | 2.3 |
In vitro stability of the succinimide intermediate
To determine the rate of formation of succinimide and Asp/iso-Asp, the fractionated unmodified HC Asn105 (peak 3) was placed on stability at varying pH of 4, 5, 6, 7 and 8 and at temperatures of 5°C, 25°C and 40°C for up to 3 mo. The samples were at 1 mg/ml in 10 mM phosphate, 10 mM citrate. At various time points, the samples were analyzed by HIC to determine change in various deamidation species. The percent succinimide reported by HIC was calculated as the percentage of succinimide at HC 105 over total Asn105, Asu105 and Asp/iso-Asp105.
At pH 6, the targeted pH of the formulation, we found a rapid increase in the relative percent of succinimide at the more extreme conditions of 40°C and 25°C (Fig. 7A). At 40°C, we saw an increase in total succinimide from under 2% at t = 0 to greater than 35% after 3 weeks, and at 25°C, we saw an increase to ~30% after 3 mo. Even at the expected storage conditions of 5°C, we observed an increase from ~2% to ~4% in 3 mo. Evaluation at pH 5 showed that the increase in the relative percent succinimide at the more extreme conditions of 40°C and 25°C was substantially reduced (Fig. 7B). At pH 5 at 40°C, total succinimide increased from under 2% at t = 0 to ~10% after 3 weeks and at 25°C an increase to ~10% after 3 mo. At the expected storage conditions of 5°C, there was a slight increase in succinimide; however, the relative area percent of the succinimide still remained below 2% after 3 mo. Across the entire pH range evaluated and at 25°C for 3 weeks, mAb-1 is most stable to deamidation at pH 5 (Fig. 8). Further analysis also indicated that the HC succinimide intermediate remained stable against hydrolysis at pH 6 and 7 and no significant increase in hydrolysis to Asp/iso-Asp was observed until the pH was raised to pH 8.
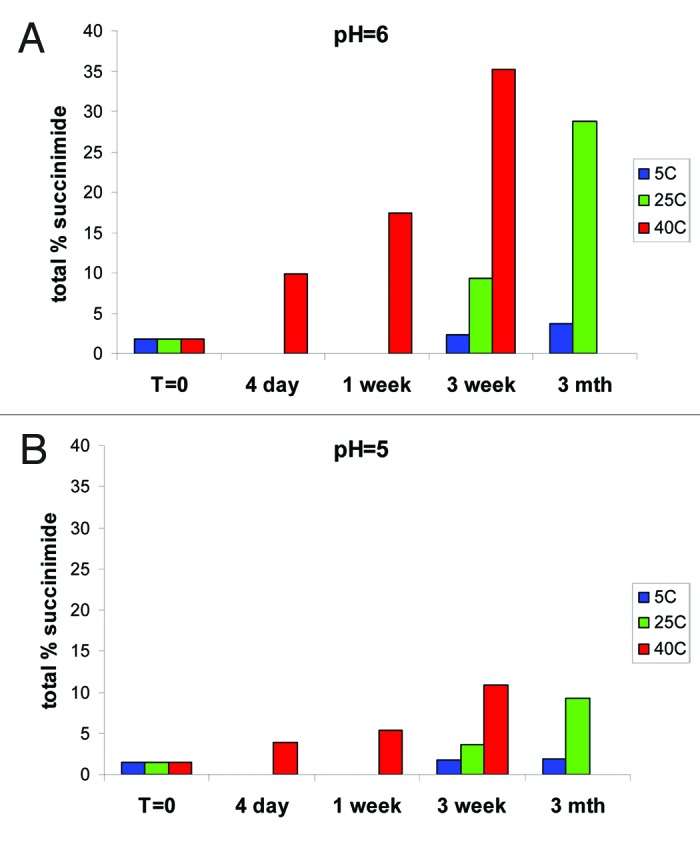
Figure 7. In vitro stability of peak 3 at (A) pH 6 showing rapid formation of succinimide at elevated temperatures and (B) pH 5 with slower kinetics of succinimide formation.
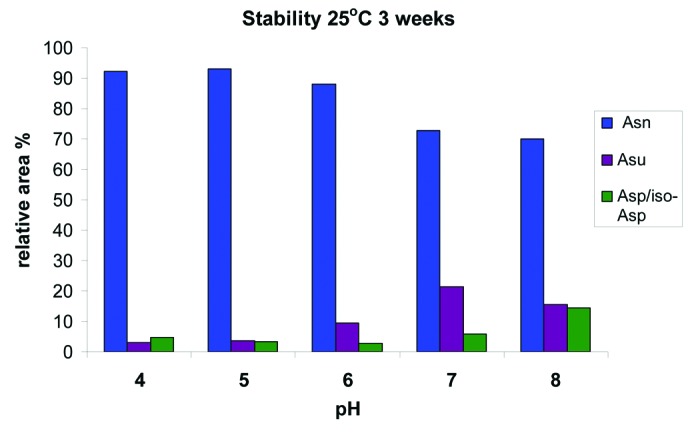
Figure 8. In vitro stability of HIC fractionated peak 3 at pH of 4, 5, 6, 7 and 8 demonstrating the relative increase of the succinimide (Asu) and Asp/iso-Asp species after 3 weeks at 25°C.
Recovery of mAb from cynomolgus monkey serum
During early development of a candidate therapeutic, a high-dose tolerability study is typically performed in cynomolgus monkeys, which assures adequate material over several days to enable recovery and characterization. In this study, cynomolgus monkeys were dosed at 60 mg/kg with mAb-1 starting at day 0 and then dosed every 7 d to day 21 (4 doses in total). Blood samples were collected immediately following dosing at 0.1 h and then at varying time points to 50 d. Samples for analysis were taken at 0.1 h, 7 d, 14 d, 21 d, 28 d and 50 d. mAb-1 was purified from the serum using immobilized goat anti-human IgG (Fcɤ). The captured antibodies were digested with trypsin under acidic conditions (pH 5) and analyzed by LC/MS on an Agilent ESI-Q-TOF. The specific peptides (HC99–130) representing unmodified HC Asn105, HC Asu105 and HC Asp/iso-Asp105 were extracted from the data using Agilent Bioconfirm MFE software. Relative percentages of each were calculated from the EICs (Fig. 9). The level of Asp/iso-Asp is a sum of two peaks with the same MW that eluted at different retention times; the earlier eluting species is iso-Asp and the later Asp based on the observed ratios of 3:1 iso-Asp:Asp.

Figure 9. Representative reverse phase LC/MS extracted ion chromatogram of acidic tryptic digests of mAb-1 recovered from serum showing the EIC’s of HC99–130 tryptic peptide (MW = 3583.5 Da) and the corresponding tryptic peptide containing the succinimide (Asu) modification (MW = 3566.5 Da) and the Asp and iso-Asp modification (MW 3584.5 Da).
The results at the different time-points analyzed were plotted and compared with the in vitro data obtained from samples at pH 7 and stored at 40°C (Fig. 10). Deamidation of Asn105 to one of the 3 end products proceeded much more rapidly in vivo. The most striking result was loss of stabilization of the succinimide intermediate observed in vivo. At day 7, for example, the in vivo studies showed that the Asp/iso-Asp deamidation species were the predominant species and levels of that peptide were at 65% relative area percent with no succinimide intermediate observed. In contrast, in the in vitro analysis the Asp/iso-Asp species were at less than 10% at the same time point and the succinimide species increased to approximately 30%. We observed stabilization of the relative area percent of the different species in the in vivo study from day 7 to day 28 due to re-dosing every 7 d out to day 21. By day 50, however, no residual HC Asn105 was found and only the Asp and iso-Asp deamidation species was observed.
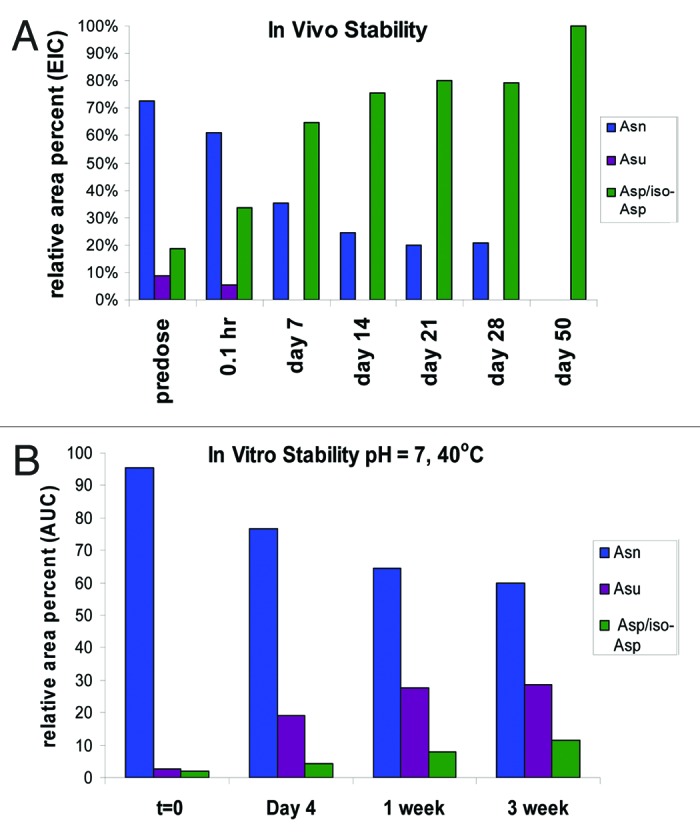
Figure 10. (A) In vivo stability of mAb-1 recovered from serum (B) In vitro stability of HIC fractionated peak 3 at pH 7 and temperature of 40°C for 3 weeks. Studies in vivo show increase in total deamidation over time and loss of stability of the succinimide (Asu) intermediate.
Hydrodynamic size analysis by light scattering and molecular modeling
By SPR, a loss in binding upon cyclization of HC Asn105 to succinimide was seen, but binding was restored upon hydrolysis to Asp, which suggests that the succinimide intermediate introduces a different fold in the binding region. This is readily apparent from the increase in hydrophobicity observed upon cyclization compared with Asn105 or Asp/iso-Asp105. Additional evidence is also available from size exclusion chromatography (SEC) results in which a species with a smaller apparent hydrodynamic size than the intact monomer of mAb-1, based on later elution from SEC, was observed. Analysis of HIC fractionated peak 3 (unmodified mAb-1) and peak 4 (one HC succinimide105) by SEC demonstrated that the later eluting species was a result of the cyclization of the HC Asn105 (data not shown). Static light scattering experiments verified that the later eluting species by SEC had the same MW as the monomer species (Fig. 11A). Further, quasi elastic light scattering (QELS) experiments demonstrated that the later eluting shoulder species (Fig. 11C) had a smaller hydrodynamic size (4.2 nM) than the mAb-1 monomer (Fig. 11B), which had a hydrodynamic size (5.3 nM) comparable to previously reported values for other IgG molecules.39-43

Figure 11. (A) SEC-Static Light scattering data of mAb1 showing similar MW of monomer and a shoulder species observed in mAb-1. SEC QELS data of mAb1 showing: (B) native monomer species with expected hydrodynamic radius of 5.3 nM, and (C) later eluting shoulder species with one HC succinimide 105 with a smaller hydrodynamic radius of 4.2 nM.
We modeled the Fab region of mAb-1 using YASARA (Yasara Biosciences) to better understand the local environment that helped stabilize the succinimide species and led to the lower apparent hydrodynamic radius observed by QELS. The homology model was built on a user-defined limited library of Fabs and mAbs downloaded from the Protein Data Bank. The model showed that the susceptible asparagine residue, HC Asn105, is located on a solvent-exposed loop containing several hydrophobic residues in its vicinity (Fig. 12A). In this model, we speculate that, following nucleophilic attack by the backbone nitrogen of HC Gly106 to the gamma carbonyl of HC Asn105, the resulting cyclic imide ring is likely to sit parallel to the plane of an adjacent HC Trp104 side chain (Fig. 12B). We postulate that π interaction between the succinimide moiety and tryptophan side chain could help stabilize the cyclic imide and provide some degree of solvent shielding. Further, upon cyclization of the Asn105 to succinimide, changes to the tertiary structure of the CDR3 region results in a more compact structure (Fig. 12C and D), which supports the change in hydrodynamic size observed by QELS.

Figure 12. (A) Hydrophobic rich region in CDR3. There are four hydrophobic residues with high solvent accessibility shown in the figure: Phe103, Trp104, Tyr107 and Tyr108. (B) Expanded view of Asn(succinimide)105 and Trp104. The two π bond rich systems have close to a planar relationship as is typical for π stacking interactions. (C) Tertiary alignment of molecular models of Fabs. The energy minimized model of the CDR3 containing an asparagine at position 105 is shown in magenta and the energy minimized region showing the cyclic imide (succinimide105) is shown in teal. Changes to both structures are observed in both the backbone and side chain orientations within CDR3. (D) Comparison of the backbones of the two energy minimized regions of the native structure and the stable cyclic imide.
Discussion
In the study reported here, we used HIC to screen for molecules with acceptable hydrophobicity and discovered a therapeutic mAb-1 that contained a stable succinimide species. Typically, succinimide, formed either by deamidation of asparagine or de-hydrolysis of Asp, is a short-lived intermediate and is rapidly hydrolyzed to Asp/iso-Asp. In contrast to the expected behavior, the succinimide intermediate we found was stable in vitro at pH 7 and below and increased on storage at 25 ° and 40°C. Homology modeling suggested that the succinimide intermediate is stabilized via π interaction with the side chain of an adjacent tryptophan residue.
Using SPR analysis, we found a moderate decrease in binding affinity of mAb-1, which contained one succinimide105 modification on the HC, compared with a fraction containing no modifications (HC Asn105). With two succinimide105 modifications, the affinity is decreased by a log factor. These findings are corroborated by other studies that report a 50% decrease in binding affinity for a Fab containing succinimide modification in the CDR2 of the HC of an IgG136 and a 10% decrease in binding affinity for a succinimide modification in the CDR1 of one and two LC arms of an IgG2.35,36 It is likely that the succinimide intermediate introduces a different fold in the binding region of the mAb-1 because we observe a significant increase in hydrophobicity by HIC and different migration by SEC. In addition, SEC-QELS identified a decrease in the apparent hydrodynamic size of the succinimide-containing molecule and modeling studies corroborate a significant change in HC Fab following cyclization of HC Asn105 to succinimide. Further studies are warranted to better understand how various residues in HC CDR3 contribute to succinimide stabilization and an altered fold in this binding region. In studying single point mutation of Asn105 to Asp105, we found comparable binding affinity, which suggests that, when deamidation proceeds to completion, binding affinity to antigen is not altered. This is important because the succinimide intermediate was not stabilized in the highly crowded, reducing environment of blood and deamidation rapidly proceeded to Asp/iso-Asp end products. This study illustrates the importance of evaluating post-translational modifications of therapeutic mAbs in vitro, as well as in serum, the intended environment of the molecule.
Materials and Methods
Materials
Ammonium sulfate, tris base, sodium acetate trihydrate and sodium phosphate (Na2HPO4–7H20) were purchased from JT Baker. Dithiothreitol (DTT), N-ethylmaleimide (NEM), tris(2-carboxyethyl) phosphine-HCl (TCEP), guanidine HCL and acetonitrile were purchased from Sigma-Aldrich. Formic acid and trifluoroacetic acid (TFA) were purchased from EMD Chemicals. Modified trypsin, was purchased from Promega. The mAbs used in this study were produced at AbbVie Bioresearch Center.
Fractionation and analysis using hydrophobic interaction chromatography
Samples for fractionation or analysis were first diluted to 2 mg/mL in 15 mM histidine, pH 5.5. Samples were further diluted 2X in 2M ammonium sulfate, 20 mM sodium phosphate, pH 6.0 to a final concentration of 1 mg/mL containing the same amount of chaotrope (1 M ammonium sulfate) as starting gradient. A 25 to 100 µg injection was separated on a Tosoh Biosciences TSKgel Butyl NPR, 4.6 x 100 mm with a 2% B / min gradient using 1M ammonium sulfate, 20 mM sodium phosphate, pH 6.0 (MPA) and 20 mM sodium phosphate, pH 6.0 (MPB).
Reduced LC/MS analysis
Samples were diluted to 1 mg/mL and reduced with the addition of 100 mM DTT followed by incubation at 37°C for 30 min. The reaction was quenched by lowering pH with formic acid. Samples were analyzed on an Agilent ESI-QTOF model 6510 mass spectrometer coupled to an Agilent 1200 capillary HPLC system (Agilent Technologies). The samples were desalted for introduction into the mass spectrometer using a polymeric reverse phase protein microtrap from Michrom Bioresources. The samples were loaded under aqueous conditions and washed for 5 min. using 0.02% TFA, 0.08% formic acid in water as MPA to desalt the sample prior to gradient elution into the mass spectrometer using 0.02% TFA, 0.08% formic acid in acetonitrile as MPB. The mass spectrometer was run in positive ion mode with a capillary voltage of 4750 V, drying gas temperature = 350°C, fragmentor of 350 V and skimmer of 100 V.
Tryptic digestion (basic digestion)
To 100 µl of a 0.125 mg/mL mAb in 6M guanidine HCl, 100 mM Tris, pH 8 was added 0.5 μl of 1M dithiothreitol (DTT) for a final DTT concentration of 5 mM. The sample was incubated at 37°C for 30 min. to denature and reduce mAb-1. Then 2.5 μl of 500 mM iodoacetic acid (IAA) was added for a final IAA concentration of 12.5 mM and the sample further incubated at 37°C for 30 min. to alkylate. The reduced and alkylated sample was desalted using Zeba desalt spin cartridges (Thermo Scientific) and exchanged into 10 mM Tris pH 8.0. Trypsin digestion was done at enzyme:protein ratio of 1:20 at 37°C for 6 h. The digestion was quenched with the addition of formic acid to lower pH.
Tryptic digestion (acidic conditions)
To 100 µl of a 0.5 mg/mL mAb in 4M guanidine HCl, 50 mM acetate, pH 5 was added 2 ul of 500 mM tris (2-carboxyethyl) phosphine-HCl (TCEP) for a final TCEP concentration of 10 mM. The sample was incubated at 37°C for 30 min to denature and reduce. Then 10 ul of 250 mM N-ethylmaleimide (NEM) was added for a final NEM concentration of 25 mM and incubated at 37°C for 30 min. The reduced and alkylated sample was desalted using Zeba desalt spin cartridges and exchanged into 50 mM acetate pH 5.0. Trypsin digestion was done at an enzyme:protein ratio of 1:10 at 37°C for 2.5 h. An additional 2.5 µl of 1 µg/µl trypsin was added and the sample was incubated at 37°C for an additional 2.5 h. The sample was quenched with the addition of formic acid to lower pH.
Peptide mapping mass spectrometry
Samples were analyzed on an Agilent ESI-QTOF model 6510 coupled to an Agilent 1200 capillary HPLC system (Agilent Technologies). Peptides were resolved for introduction into the mass spectrometer with an Agilent Zorbax-C18, 1.0 x 150 mm, 3.5 µm reverse phase C18 column using a binary gradient with MPA - 0.02% TFA, 0.08% formic acid in water and MPB - 0.02% TFA, 0.08% formic acid in acetonitrile. The mass spectrometer was run in positive ion mode with a capillary voltage of 4750 V, drying gas temperature = 325°C, fragmentor of 185 V and skimmer of 70 V.
Recovery of mAb-1 from cynomolgus monkey serum
Goat anti-human IgG (Fcγ) (min x BvMsHs Sr Prot, GE Healthcare Biosciences) was immobilized on CNBr activated Sepharose 4 Fast Flow resin (GE Healthcare Biosciences). The immobilized antibody was used to capture the mAb-1 from cynomolgus monkey serum. Following capture the immobilized antibody was washed with 1X PBS and then eluted by denaturing with 4M guanidine HCl. The denatured protein was immediately reduced and alkylated for tryptic digest under acidic conditions for analysis by LC/MS.
Surface Plasmon Resonance analysis
The kinetics of antigen binding to mAb-1 and its HIC fractions were determined by SPR (Biacore T200 instrument, GE Healthcare Life Sciences). Briefly, mAb-1 and its HIC fractions were captured by goat anti-human IgG antibodies specific to the Fc region of human IgG covalently linked to the carboxymethyl dextran matrix of the CM5 biosensor chip via free amine groups using an amine coupling kit from GE Healthcare Life Sciences according to the manufacturer’s protocol. Approximately 5000 RU of goat anti-human IgG Fc were immobilized on the chip surface on flow cells 2, 3 and 4. A modified CM surface with similarly conjugated goat IgG antibodies in flow cell-1 was used as a reference surface.
mAb-1 and its HIC fractions were diluted in running buffer (HBS-EP+ buffer containing 0.1 mg/ml BSA) to 1 μg/mL and injected over the goat anti-human IgG Fc surface on flow cells 2, 3 and 4 to achieve capture levels of ~100–120 RU. Recombinant human antigen was injected at various concentrations (0, 0.39, 0.78, 1.56, 3.125, 6.25, and 12.5 nM) at a flow rate of 50 µL/min for 5 min over captured mAbs and the reference surface to ascertain association rates. The dissociation phase consisted of 15 min and 60 min of continuous flow of the running buffer (50 μL/min). The chip surface was regenerated with 30 sec injection of 10 mM glycine (pH 1.5) at a flow rate of 50 µL/min. Evaluation of each interaction was performed in duplicate.
Affinity (KD, M), association rate (ka, M−1s−1) and dissociation rate constants (kd, s−1) were calculated by 1:1 Langmuir binding with mass transport incorporated using Biaevaluation software, based on the values extracted from the data using global fit analysis (allowing identical values for each parameter in the data set, except for Rmax that was set local to account for variation in mAbs capture level).
Static and Quasi elastic electric light scattering to determine molecular weight and hydrodynamic radius
Samples for analysis were first diluted to 1 mg/mL in Milli-Q water. A 50 µg injection was separated on a G3000SWxl, 7.8 mm ID, 30 cm length, 5 µm (Tosoh Biosciences) isocratically with 0.1 M sodium sulfate, 0.1 M sodium phosphate, 1 mM azide, pH 6.8 mobile phase. Static light scattering (SLS) detection was achieved using a Dawn HELEOS 18 angle detector (Wyatt Technology). Concentration detection for SLS was achieved using an Optilab REX refractive index detector (Wyatt Technology). Quasi elastic light scattering was collected in line through the 12th angle of the Dawn HELEOS detector on a Dynapro Nanostar detector (Wyatt Technology).
Acknowledgments
The authors thank Robert Hickman and Yumiko Kobayashi for assistance with semi-preparative scale purification; Michael Siedler and Vineet Kumar for assistance with stability studies; Alexander Ivanov for assistance with SPR analysis; Edit Tarcsa for providing serum samples and Chung-Ming Hsieh and Suju Zhong for providing point mutations. Finally, we acknowledge Gary Welch and George Avgerinos for their support of these studies.
Glossary
Abbreviations:
- CDR
complementarity-determining regions
- HIC
hydrophobic interaction chromatography
- RPLC
reverse phase liquid chromatography
- cIEF
capillary isoelectric focusing
- PTM
post-translational modification
- PK
pharmacokinetic
- MPA and MPB
mobile phase A and B
- LC/MS/MS
liquid chromatography/mass spectrometry/mass spectrometry
- mAb
monoclonal antibody
- SPR
surface plasmon resonance
- SEC
size exclusion chromatography
- MW
molecular weight
- HC
heavy chain
- LC
light chain
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Reichert JM. Which are the antibodies to watch in 2012? MAbs. 2012;4:1–3. doi: 10.4161/mabs.4.1.18719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3:76–99. doi: 10.4161/mabs.3.1.13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reichert JM. Antibodies to watch in 2010. MAbs. 2010;2:84–100. doi: 10.4161/mabs.2.1.10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhimolea E, Reichert JM. World Bispecific Antibody Summit, September 27-28, 2011, Boston, MA. MAbs. 2012;4:4–13. doi: 10.4161/mabs.4.1.18821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Correia IR. Stability of IgG isotypes in serum. MAbs. 2010;2:221–32. doi: 10.4161/mabs.2.3.11788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson NE, Robinson AB. Molecular clocks. Proc Natl Acad Sci U S A. 2001;98:944–9. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson NE, Robinson AB. Deamidation of human proteins. Proc Natl Acad Sci U S A. 2001;98:12409–13. doi: 10.1073/pnas.221463198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson NE. Protein deamidation. Proc Natl Acad Sci U S A. 2002;99:5283–8. doi: 10.1073/pnas.082102799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sreedhara A, Cordoba A, Zhu Q, Kwong J, Liu J. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm Res. 2012;29:187–97. doi: 10.1007/s11095-011-0534-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Yip H, Katta V. Identification of isomerization and racemization of aspartate in the Asp-Asp motifs of a therapeutic protein. Anal Biochem. 2011;410:234–43. doi: 10.1016/j.ab.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 11.Cournoyer JJ, Pittman JL, Ivleva VB, Fallows E, Waskell L, Costello CE, et al. Deamidation: Differentiation of aspartyl from isoaspartyl products in peptides by electron capture dissociation. Protein Sci. 2005;14:452–63. doi: 10.1110/ps.041062905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cournoyer J, O’Connor P. Analysis of Deamidation in Proteins Comprehensive Analytical Chemistry. 2008 52:375-410 [Google Scholar]
- 13.Stephenson RC, Clarke S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J Biol Chem. 1989;264:6164–70. [PubMed] [Google Scholar]
- 14.Radkiewicz JL, Zipse H, Clarke S, Houk KN. Neighboring side chain effects on asparaginyl and aspartyl degradation: an ab initio study of the relationship between peptide conformation and backbone NH acidity. J Am Chem Soc. 2001;123:3499–506. doi: 10.1021/ja0026814. [DOI] [PubMed] [Google Scholar]
- 15.Oliyai C, Patel JP, Carr L, Borchardt RT. Solid state chemical instability of an asparaginyl residue in a model hexapeptide. J Pharm Sci Technol. 1994;48:167–23. [PubMed] [Google Scholar]
- 16.Oliyai C, Patel JP, Carr L, Borchardt RT. Chemical pathways of peptide degradation. VII. Solid state chemical instability of an aspartyl residue in a model hexapeptide. Pharm Res. 1994;11:901–8. doi: 10.1023/A:1018998312503. [DOI] [PubMed] [Google Scholar]
- 17.Oliyai C, Borchardt RT. Chemical pathways of peptide degradation. VI. Effect of the primary sequence on the pathways of degradation of aspartyl residues in model hexapeptides. Pharm Res. 1994;11:751–8. doi: 10.1023/A:1018944800691. [DOI] [PubMed] [Google Scholar]
- 18.Oliyai C, Borchardt RT. Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm Res. 1993;10:95–102. doi: 10.1023/A:1018981231468. [DOI] [PubMed] [Google Scholar]
- 19.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem. 1987;262:785–94. [PubMed] [Google Scholar]
- 20.Clarke S. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int J Pept Protein Res. 1987;30:808–21. doi: 10.1111/j.1399-3011.1987.tb03390.x. [DOI] [PubMed] [Google Scholar]
- 21.Xiao G, Bondarenko PV, Jacob J, Chu GC, Chelius D. 18O labeling method for identification and quantification of succinimide in proteins. Anal Chem. 2007;79:2714–21. doi: 10.1021/ac0617870. [DOI] [PubMed] [Google Scholar]
- 22.Wakankar AA, Borchardt RT, Eigenbrot C, Shia S, Wang YJ, Shire SJ, et al. Aspartate isomerization in the complementarity-determining regions of two closely related monoclonal antibodies. Biochemistry. 2007;46:1534–44. doi: 10.1021/bi061500t. [DOI] [PubMed] [Google Scholar]
- 23.Valliere-Douglass J, Wallace A, Balland A. Separation of populations of antibody variants by fine tuning of hydrophobic-interaction chromatography operating conditions. J Chromatogr A. 2008;1214:81–9. doi: 10.1016/j.chroma.2008.10.078. [DOI] [PubMed] [Google Scholar]
- 24.Huang HZ, Nichols A, Liu D. Direct identification and quantification of aspartyl succinimide in an IgG2 mAb by RapiGest assisted digestion. Anal Chem. 2009;81:1686–92. doi: 10.1021/ac802708s. [DOI] [PubMed] [Google Scholar]
- 25.Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl. 2001;752:233–45. doi: 10.1016/S0378-4347(00)00548-X. [DOI] [PubMed] [Google Scholar]
- 26.Chu GC, Chelius D, Xiao G, Khor HK, Coulibaly S, Bondarenko PV. Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm Res. 2007;24:1145–56. doi: 10.1007/s11095-007-9241-4. [DOI] [PubMed] [Google Scholar]
- 27.Huang L, Lu J, Wroblewski VJ, Beals JM, Riggin RM. In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal Chem. 2005;77:1432–9. doi: 10.1021/ac0494174. [DOI] [PubMed] [Google Scholar]
- 28.Doyle HA, Zhou J, Wolff MJ, Harvey BP, Roman RM, Gee RJ, et al. Isoaspartyl post-translational modification triggers anti-tumor T and B lymphocyte immunity. J Biol Chem. 2006;281:32676–83. doi: 10.1074/jbc.M604847200. [DOI] [PubMed] [Google Scholar]
- 29.Doyle HA, Gee RJ, Mamula MJ. Altered immunogenicity of isoaspartate containing proteins. Autoimmunity. 2007;40:131–7. doi: 10.1080/08916930601165180. [DOI] [PubMed] [Google Scholar]
- 30.Young GW, Hoofring SA, Mamula MJ, Doyle HA, Bunick GJ, Hu Y, et al. Protein L-isoaspartyl methyltransferase catalyzes in vivo racemization of Aspartate-25 in mammalian histone H2B. J Biol Chem. 2005;280:26094–8. doi: 10.1074/jbc.M503624200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Martinez T, Woodruff B, Goetze A, Bailey R, Pettit D, et al. Hydrophobic interaction chromatography of soluble interleukin I receptor type II to reveal chemical degradations resulting in loss of potency. Anal Chem. 2008;80:7022–8. doi: 10.1021/ac800928z. [DOI] [PubMed] [Google Scholar]
- 32.Boyd D, Kaschak T, Yan B. HIC resolution of an IgG1 with an oxidized Trp in a complementarity determining region. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:955–60. doi: 10.1016/j.jchromb.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Gong B, Wang L, Wang C, Geng X. Preparation of hydrophobic interaction chromatographic packings based on monodisperse poly(glycidylmethacrylate-co-ethylenedimethacrylate) beads and their application. J Chromatogr A. 2004;1022:33–9. doi: 10.1016/j.chroma.2003.09.063. [DOI] [PubMed] [Google Scholar]
- 34.Kato Y, Nakatani S, Nakamura K, Kitamura T, Moriyama H, Hasegawa M, et al. Hydrophobicity gradient columns for the separation of trypsin inhibitor by hydrophobic interaction chromatography at low salt concentration. J Chromatogr A. 2003;986:83–8. doi: 10.1016/S0021-9673(02)01997-0. [DOI] [PubMed] [Google Scholar]
- 35.Valliere-Douglass J, Jones L, Shpektor D, Kodama P, Wallace A, Balland A, et al. Separation and characterization of an IgG2 antibody containing a cyclic imide in CDR1 of light chain by hydrophobic interaction chromatography and mass spectrometry. Anal Chem. 2008;80:3168–74. doi: 10.1021/ac702245c. [DOI] [PubMed] [Google Scholar]
- 36.Yan B, Steen S, Hambly D, Valliere-Douglass J, Vanden Bos T, Smallwood S, et al. Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J Pharm Sci. 2009;98:3509–21. doi: 10.1002/jps.21655. [DOI] [PubMed] [Google Scholar]
- 37.Yin S, Pastuskovas CV, Khawli LA, Stults JT. Characterization of therapeutic monoclonal antibodies reveals differences between in vitro and in vivo time-course studies. Pharm Res. 2013;30:167–78. doi: 10.1007/s11095-012-0860-z. [DOI] [PubMed] [Google Scholar]
- 38.Yu XC, Joe K, Zhang Y, Adriano A, Wang Y, Gazzano-Santoro H, et al. Accurate determination of succinimide degradation products using high fidelity trypsin digestion peptide map analysis. Anal Chem. 2011;83:5912–9. doi: 10.1021/ac200750u. [DOI] [PubMed] [Google Scholar]
- 39.Zhao H, Graf O, Milovic N, Luan X, Bluemel M, Smolny M, et al. Formulation development of antibodies using robotic system and high-throughput laboratory (HTL) J Pharm Sci. 2010;99:2279–94. doi: 10.1002/jps.22008. [DOI] [PubMed] [Google Scholar]
- 40.Sukumar M, Doyle BL, Combs JL, Pekar AH. Opalescent appearance of an IgG1 antibody at high concentrations and its relationship to noncovalent association. Pharm Res. 2004;21:1087–93. doi: 10.1023/B:PHAM.0000032993.98705.73. [DOI] [PubMed] [Google Scholar]
- 41.Hawe A, Hulse WL, Jiskoot W, Forbes RT. Taylor dispersion analysis compared to dynamic light scattering for the size analysis of therapeutic peptides and proteins and their aggregates. Pharm Res. 2011;28:2302–10. doi: 10.1007/s11095-011-0460-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hawe A, Friess W, Sutter M, Jiskoot W. Online fluorescent dye detection method for the characterization of immunoglobulin G aggregation by size exclusion chromatography and asymmetrical flow field flow fractionation. Anal Biochem. 2008;378:115–22. doi: 10.1016/j.ab.2008.03.050. [DOI] [PubMed] [Google Scholar]
- 43.Bermudez O, Forciniti D. Aggregation and denaturation of antibodies: a capillary electrophoresis, dynamic light scattering, and aqueous two-phase partitioning study. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;807:17–24. doi: 10.1016/j.jchromb.2004.01.029. [DOI] [PubMed] [Google Scholar]