

Abstract
Ionizing radiation is a ubiquitous stress to which all life is continuously exposed, and thus complex mechanisms have evolved to regulate cellular responses to radiation, including cell cycle arrest, DNA repair, and programmed cell death. Changes in gene expression shape part of the response to radiation, and have historically provided insight into the underlying mechanisms of that response. However, the advent of microarrays, which can measure expression of all the genes in a cell simultaneously, has transformed the study of gene expression, and is beginning to have an impact on both basic mechanistic and clinical studies. This article provides an overview of concepts in gene expression and microarray technology, and highlights their impacts on the study of radiation biology.
Keywords: ionizing radiation, functional genomics, microarray, p53, oncology
Gamma rays (γ rays) and X rays are examples of ionizing radiations; that is, they have sufficient energy to remove electrons from atoms when they interact with matter. Soon after their discovery in 1895, X rays were enthusiastically put to use in medical diagnostics and treatment. It was quickly found, however, that radiation was a two-edged sword: it could shrink tumors, but it could also induce tumors and burn skin. Despite its potential dangers, the great benefits of radiation in medicine have inspired decades of studies aimed at understanding the mechanisms of response and defining conditions for which the positive effects significantly outweigh the negative effects of radiation. Use of radiation in medical diagnostics is now at an all-time high; the US population's exposure to medical radiation now exceeds its exposure to natural background radiation. With world reliance on nuclear power also growing, as are concerns about accidents or acts of radiological or nuclear terrorism, our need to understand the effects of radiation on life has never been greater.
The cellular response to ionizing radiation depends on physical factors such as dose and dose rate, and also on cell type and individual genotype. It is shaped by complex biochemical signaling pathways that are mediated in part by changes in gene expression. Unraveling these signals and under standing their interplay proceeded slowly by conventional reductionist approaches that studied one or a few genes at a time, but a hundred years after the discovery of X rays, the first cDNA (complementary deoxyribonucleic acid) microarray experiment (Schena et al. 1995) heralded the postgenomic era and a revolution in biology. With the completion of the sequencing of the human genome, microarrays provide the ability to measure expression levels of all genes in the genome in a single experiment, revealing for the first time the full complexity of transcriptional responses. The study of gene expression on the global, or whole-genome, level is referred to as “functional genomics,” “expression profiling,” or, occasionally, “transcriptomics.” Such global-scale experiments have driven a more integrative approach to thinking about biological systems and have provided insight into diverse aspects of radiation biology, offering even greater promise for the future.
Gene expression
Although all somatic cells in an organism have essentially the same DNA and hence the same genetic information, each cell uses only a part of that information at any given time. By regulating which genes are expressed and which are silent, cells can specialize, expressing different proteins and carrying out different functions. Gene expression is also a dynamic process that allows cells to respond to environmental changes such as temperature, nutrition, infection, or exposure to toxins or DNA-damaging agents (radiation, e.g.). Because of the dynamic nature of stress responses, it must be kept in mind that a single microarray can provide only a snapshot of global transcription at a specific time.
The general structure of a typical mammalian gene is illustrated in figure 1. Gene activity is regulated at multiple levels by complex mechanisms. The combined effects of multiple transcription factors bound to the upstream promoter can regulate the rate of gene transcription. Transcription factors can be regulated by binding to cofactors or inhibitors, or by post-translational modifications such as site-specific phosphorylation or acetylation, which can change a transcription factor's binding-site preference or its effect on transcription. Methylation of promoter regions or modification of chromatin structure can also inactivate gene expression. Expression and function can also be regulated after the transcription of a gene, such as by modification of mRNA (messenger ribonucleic acid) stability. The mRNA in turn serves as a template for the translation of proteins, which perform the biochemical work of the cell.
Figure 1.
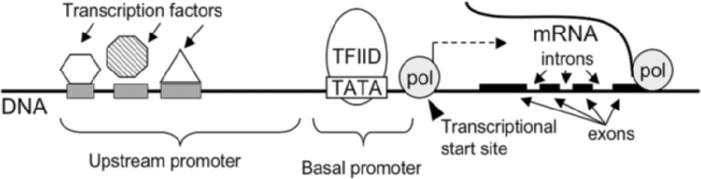
A region of DNA containing a protein-coding gene. A stretch of DNA that may extend several kilobases upstream (before the transcription start site) contains specific DNA sequences (represented by gray boxes on the DNA) where transcription factors, such as p53, NFκB (nuclear factor-kappa B), and AP1, can bind. This area is called the upstream promoter. Binding of transcription factor proteins can either enhance or repress transcription of a gene. The basal promoter is located within about 40 base pairs of the transcription start site, and contains a “TATA box,” a sequence present in all transcribed mammalian genes. The TATA box is bound by TFIID, a complex of many proteins that can recruit other proteins to the site of the gene. The transcription start site is where DNA-dependent RNA polymerase II (pol) binds to the gene and begins transcribing the DNA sequence into messenger RNA (mRNA). The mRNA will then be processed to remove introns (nonprotein coding regions) and the exons (coding sequences, represented by black boxes) will be spliced together. This message can then be translated into protein.
The amount of a protein present in a cell is still not the final determinant of function, as proteins can also be regulated by posttranslational modifications, including phosphorylation, acetylation, and ubiquitination. Such alterations can change their binding affinities for DNA or other proteins, and their catalytic activity, stability, or subcellular localization. For instance, the p53 transcription factor is phosphorylated on several sites in response to ionizing radiation. This increases the stability of the protein and alters its activity as a transcription factor, which can in turn change the rate of transcription of hundreds of target genes.
Although regulation at many levels contributes to overall cellular function, the study of gene expression in response to various stresses has provided insight into many aspects of cell biology. The availability of methods for global monitoring of gene expression has made this approach even more productive, while methods for global measurements of protein expression or modification still lag behind.
One of the earliest described gene-expression responses to stress was the SOS response of bacteria (Schlacher and Goodman 2007). Experiments with phage reactivation had indicated that Escherichia coli exposed to ultraviolet radiation (UV) could turn on an error-prone DNA repair system, later dubbed the SOS response. Decades of study of this inducible response eventually lead to the cloning of a set of coordinately regulated DNA damage-inducible genes (Kenyon and Walker 1980). More than 40 genes are now known to be part of the SOS DNA damage response in E. coli. Most of these genes are involved in error-free DNA repair and growth control, but some also contribute to recombination and mutation. Other gene-expression responses to stress are also known in bacteria, including heat shock, superoxide, hydrogen peroxide, and alkylating agent-specific responses. Stress-induced changes in gene expression have also been described in eukaryotes such as Drosophila (Akaboshi and Howard-Flanders 1989) and yeast, where an early estimate suggested that as much as 1% of the yeast genome might be involved in the response to DNA damage (Ruby and Szostak 1985). More recent whole-genome expression profiling studies now suggest that upward of a third of the yeast genome may be stress responsive (Jelinsky et al. 2000).
In contrast with bacteria, most mammalian DNA-repair pathways appear to be constitutively expressed rather than stress inducible. Nevertheless, contrary to early expectations, DNA damaging agents do alter gene expression in mammalian cells. Experiments in rodent cells used hybridization subtraction to enrich for transcripts expressed at higher levels after exposure to UV, and cloned more than 20 DNA damage-induced cDNAs (Fornace et al. 1988), only two of which matched known sequences. The list of radiation-regulated genes grew slowly, and included genes coding for cytokines, oncogenes, inflammation and transcription factors, cell-cycle checkpoint proteins, and proteins both promoting and protecting against apoptosis (programmed cell death). Radiation was shown to activate the transcription factors NFκB (nuclear factor-kappa B; Brach et al. 1991) and p53, which was associated with arrest in the G1 stage of the cell cycle (Kastan et al. 1991), and with apoptosis (Yonish-Rouach et al. 1991). Many of the early-identified radiation response genes were regulated by p53, such as GADD45A, CDKN1A, MDM2, BCL2, and BCL-X. Activated p53 could induce either cell-cycle arrest or apo ptotic cell death, contributing to an emerging picture of complex regulation, which was not well understood.
Microarray hybridization
The development of microarray technology brought the study of gene expression onto the cutting edge of biological science. The first array, which consisted of 45 Arabidopsis cDNAs robotically printed on a glass support (Schena et al. 1995), established the basic two-color fluorescent hybridization approach still widely used today (figure 2). It was quickly followed by an array of 1046 human genes and the demonstration of detection of both known and previously unknown heat-shock and phorbol ester gene-expression responses in T cells (Schena et al. 1996). As microarrays expanded to cover greater numbers of genes, large efforts were put into resequencing the cDNA libraries used. Early arrays were susceptible to clone misidentification and cross-contamination of wells during propagation and purification of clone DNA. Most spotted arrays now use long oligo-nucleotide libraries with optimized 60- or 70-mer probes to streamline protocols and minimize such problems. Long oligo probes can also be synthesized in situ on the arrays using ink-jet printing technology (Hughes et al. 2001), as exemplified by the Agilent platform.
Figure 2.

Two-color microarray hybridization. Ribo-nucleic acid (RNA) from two different samples of interest is labeled by carrying out a reverse transcription reaction incorporating a different fluorochrome into each sample. In the example, the control sample is labeled with cyanine-5 (Cy5) and the irradiated sample with cyanine-3 (Cy3). The two samples are hybridized together to the same micro array. After washing and scanning, the brightness of each fluorescent wavelength in the scanned image is compared for each feature (a spot representing an individual gene). In the composite image, genes such as CDKN1A, which are upregulated by radiation exposure, appear as red spots, reflecting the fact that there are more copies of these genes present in the RNA pool that was labeled with the red flurochrome. Similarly, down-regulated genes, such as MYC, appear as green spots, and genes that do not change, such as GAPDH, have equal amounts of both colors and appear yellow.
The cyanine dyes Cy3 and Cy5 were used in the first micro-array experiment, and are still the most commonly used fluorochrome pair for two-color or spotted microarrays. They are not without drawbacks, however. The incorporation of Cy5 tends to be less efficient than that of Cy3 because of greater steric hindrance, and Cy5 is susceptible to rapid degradation by atmospheric ozone at levels present in most laboratories. This can produce a shift away from the theoretical ratio of 1.0 for genes that are not differentially expressed in the two samples being compared. Such ratio bias can also vary across a microarray as a function of the overall intensity of hybridization, or in a sequence-dependent manner. Some experimental designs use a fluorochrome-switching or “dye-swap” approach to control for some of these biases. In this case, each pair of samples being compared is hybridized with two micro arrays. In one hybridization, the control sample is labeled with Cy3 and the test sample with Cy5, and in the second hybridization, the dyes will be swapped so that the control is labeled with Cy5 and the test sample with Cy3. Mathematical ratio normalization (Quackenbush 2002) is also important for meaningful interpretation of two-color micro-array data. Many approaches have been developed to correct for ratio bias due to small systematic, regional, and intensity-dependent ratio variations.
An alternative technology, based on single-color hybridization and comparison across separate arrays (figure 3), was rapidly commercialized as the Affymetrix platform and adopted by many institutions. This technology uses hybridization to short oligonucleotides that are synthesized in situ on the microarray using a photolithographic process. Although array-to-array variation in early spotted cDNA arrays made a single-color approach with comparisons between independently hybridized arrays impractical, high reproducibility and quality control of current commercial platforms now also yield equivalent quality data from both oneand two-color protocols applied to the long oligonucleotide platforms (Patterson et al. 2006). Although the single-color approach eliminates the problem of dye bias, normalization of hybridization intensities across all arrays in an experiment is still needed before experiments can be compared, and multiple approaches for this have been developed. Recent studies indicate that the choice of normalization method in single-color experiments may affect the outcome of later data analysis (Harr and Schlötterer 2006, Qin et al. 2006, Shippy et al. 2006), and so must be selected with care.
Figure 3.

Single-color microarray hybridization (photolithographic platform). Ribonucleic acid from the samples to be compared is reverse transcribed to complementary DNA (cDNA), then in vitro transcription is carried out in the presence of labeled (in this case bio-tinylated) nucleotides to produce labeled cRNA. The labeled complementary RNA (cRNA) is fragmented to facilitate sequence-specific hybridization, and each sample is hybridized to a separate array. After washing, staining, and scanning, the fluorescent intensity of each feature is compared between arrays, making normalization across experiments extremely important. In the illustrated experiment, features representing genes upregulated by radiation exposure, such as CDKN1A, will have a greater intensity on the array hybridized to the irradiated sample. Downregulated genes, such as MYC, will show a greater intensity in control samples, and unchanged genes, such as GAPDH, will have equal intensity on the two arrays.
Microarray data analysis
For any microarray experiment, the first goal is to identify all the genes with different expression, either between phenotypic classes or following exposure to a stress such as ionizing radiation. Some early experiments used an arbitrary cutoff for the ratio of expression between two samples or classes—for instance, defining any gene with a ratio greater than 2 as upregulated. Differential expression is now defined statistically, often by applying a cutoff p value following a t test or F test. However, microarrays entail tens of thousands of multiple statistical comparisons, and sufficient numbers of genes may be identified as differentially expressed through chance alone to undermine the usefulness of conventional p values. Those false positives can be limited by simply lowering the p value used to declare a gene significantly changed. This approach does not entirely solve the problem, however, and will also increase the proportion of false negatives (genes that really are expressed at different levels, but that do not meet stringent statistical requirements). The most commonly adopted approach to this problem of multiple hypothesis testing is application of a false discovery rate (FDR) (Pounds 2006). The FDR can be used to estimate the proportion of false positives at a selected p-value level. One of the most widespread applications of FDR is that implemented by the significance analysis of microarrays (Tusher et al. 2001), which provides an easy-to-use interface for data analysis. Many other approaches are available, such as the local FDR, which can estimate the probability that an individual gene is a false positive (Aubert et al. 2004).
Once a list of differentially expressed genes has been obtained with some level of confidence, the results must be interpreted. Early studies tended to approach this daunting task by focusing on a few of the most changed genes, or on several with known roles in the process under study, and ignoring the rest. For instance, in the first microarray study of ionizing radiation response, 48 genes were significantly differentially expressed four hours after exposure to γ rays, and only 18 of these had previously been described to be radiation responsive (Amundson et al. 1999). Nine newly identified and three previously known radiation response genes were selected for further study, and their responses to UV radiation, an alkylating agent, and γ rays were compared by quantitative single-probe hybridization in six p53 wild-type and six p53 mutant cell lines. The patterns of response suggested that two of the newly identified radiation response genes, ATF3 and FOSL1, might be regulated by p53, and this was confirmed in isogenic cell lines and in vivo irradiated wild-type and p53 knockout mice. Such an approach does not come close to taking full advantage of the information generated by whole genome studies, however, and more powerful approaches are continually being developed to assist in the analysis of complex data sets.
One of the earliest analysis tools applied to microarray data was hierarchical clustering (Eisen et al. 1998). In cluster analysis, every pair of genes is tested for the degree of similarity between their expression intensities or ratios across all experiments. The genes are then arranged so that those with the most similarity in expression are closest together. The experimental samples can be sorted the same way. There are other mathematical approaches to clustering, including K-means clustering and self-organizing map approaches. The result of cluster analysis is generally visualized as a heat map (figure 4), with a range of colors used to represent high to low expression ratios or intensities. In this way, similarities and differences between experiments can be seen more readily than by reading through large tables of numbers. Clusters within the heat map may indicate genes with coordinate regulation or genes that are part of the same process, although this is not always the case. In the example shown in figure 4, for instance, two cell lines have been exposed to 13 different toxic stresses (Amundson et al. 2005), and patterns of gene expression that vary with the type of exposure can be readily distinguished. The metal-responsive genes in the “A” cluster nearly all code for metallothioneins, proteins that bind and regulate metals. The “B” cluster contains genes coding for TNF (tumor necrosis factor), chemokines, and interleukins, which are preferentially induced by exposure to 12-O-tetradecanoylphorbol 13-acetate (TPA). Another cluster of genes strongly responsive to TPA (“C”) contains a high proportion of MAPK (mitogen-activated protein kinase) phosphatases and transcription factors. Although some insight may be gained from exploring such patterns, clustering is often most useful for illustrating a result rather than for primary interpretation of full data sets.
Figure 4.

Heat map generated by clustering gene-expression ratio data (Amundson et al. 2005). Gene-expression ratios (treated/control) are represented as colors according to the scale at the bottom of the figure, with brighter red indicating more induction of expression, and brighter green indicating more suppression of expression after treatment. Each horizontal row represents the expression pattern of an individual gene across all experiments, and each vertical column represents the expression pattern of all genes within an individual experiment. Clustering has been used to arrange the experiments so that those producing the most similar pattern of gene-expression changes across all genes are closest together. Thus, for instance, all samples treated with metals (arsenite [As] and cadmium chloride [Cd]) appear next to each other, as do all samples treated with 12-O-tetradecanoylphorbol 13-acetate (TPA). The genes have also been clustered, so that those with the most similar response across all experiments are again placed next to each other. This reveals some specific patterns, such as those that have been marked with yellow boxes and discussed in the text. For instance, the genes in cluster A are mostly metallothioneins, which clearly respond much more strongly to metal exposure than to the other stresses studied.
Gene-expression profiling in radiation biology
As might be anticipated, genomewide gene-expression analysis has greatly increased the number of genes reported to respond at the mRNA level to ionizing radiation, with some studies suggesting the response of upward of a thousand genes. The true potential of whole-genome expression profiling experiments, however, does not lie simply in the generation of lists of genes, but rather in using transcriptional patterns to understand the transcription factors involved, the signaling pathways regulating gene expression, and the downstream actions of the expressed gene products. The increase in known radiation-response genes has not supplanted the apparent position of the p53 signaling pathway as a major regulator of gene expression in response to radiation both in vitro (Amundson et al. 2000, 2005, 2008, Kis et al. 2006) and in vivo (Knoops et al. 2007), but is starting to provide insight into the roles of p53. The transcriptional effects of several p53 mutations commonly found in human tumors have also been studied on a global level, by expressing tumor-derived mutant p53 proteins in cells lacking wild-type p53, resulting in regulation of a set of genes involved in cell growth (Scian et al. 2004). Such studies can provide insight into mechanisms underlying the “gain-of-function” phenotype associated with specific p53 mutations in cancer.
Many studies have focused on specific aspects of the p53 response, attempting to understand, for example, what regulates the decision between apoptosis and cell-cycle arrest in cells with activated p53. Such studies have examined the role of specific protein modifications (phosphorylation or acetylation) or extracellular factors on global gene expression. One of the two transactivation domains in p53 has been linked both to apoptosis and to the induction of many apoptosis-related genes in one such study (Ohki et al. 2007), and acetylation of a specific lysine in p53 has been shown to dampen p53-dependent apoptosis and the response of several apoptosis-related genes (Chao et al. 2006). Activation of p53 by different stresses also results in a different spectrum of gene-expression response, especially when DNA-damaging and non-DNA-damaging stresses are compared (Amundson et al. 2005, Hammond et al. 2006). For instance, activation of p53 by hypoxia, a non-DNA-damaging stress, resulted mainly in the repression of gene expression (Hammond et al. 2006), in contrast with the response to DNA damage, in which most p53-regulated genes are upregulated.
An increasing focus on genes that are repressed by radiation exposure has been emerging from genomewide studies. A small number of genes, such as MYC and TOP2A, were previously known to be downregulated by p53 in response to ionizing radiation, but most early gene-expression studies focused on upregulation. Microarray studies suggest, however, that downregulation of specific genes in response to radiation exposure may be much more strongly conserved than gene upregulation among cancer cell lines. A recent global gene-expression profiling study has identified a set of genes involved in cell-cycle regulation that appears to be downregulated by overexpression of p53 or by DNA damage (Spurgers et al. 2006), suggesting a previously uncharacterized role for p53 in cell cycle regulation. In another study, an increase in the number of repressed genes was found to coincide with the onset of apoptosis (Mirza et al. 2003), further emphasizing the potential importance of p53 transcriptional repression. Although all the intricacies of the p53 pathway regulation of the response to ionizing radiation are not yet fully understood, whole-genome profiling studies are rapidly supplying more pieces of the puzzle.
Microarray analysis has also been applied to other aspects of radiation response, with investigation of effects specific to low-dose or low-dose-rate exposures being of notable interest. Despite the fact that most radiation exposures of human populations are low doses delivered at a low dose rate (that is, protracted in time), the effects of such exposures have been difficult to study directly. The potential health effects of such radiation exposures have generally been guessed at by extrapolating from results of experiments using large doses and acute exposures (with the dose delivered nearly instantaneously). Functional genomics has opened a window onto some of the unique responses that may result from such exposures.
For instance, the first study of the effect of dose rate on gene expression reported clusters of genes with radiation responses that either decreased with decreasing dose rate (e.g., GADD45A and CDKN1A) or were independent of dose rate (e.g., MDM2, PHLDA3, and BTG2) (Amundson et al. 2003). A protective effect of low-dose-rate exposure was found for the induction of apoptosis, corresponding to nearly half of the genes in the dose-rate dependent cluster having known roles in apoptosis. In contrast, no dose-rate effect was found for cell cycle delay, consistent with the majority of genes in the dose-rate independent cluster, which had known roles in cell cycle and proliferation. In a different cell line, lowering the dose rate even further was reported to result in a higher level of MDM2 induction than the same dose delivered at high dose rate (Sugihara et al. 2004). A more recent study in primary human fibroblasts reported 780 genes responding to 1 gray (Gy) γ rays delivered by high, but not low, dose rate, and 641 genes responding to low, but not high, dose rate (Sokolov et al. 2006).
This finding lends support to the idea that distinct responses that cannot be directly predicted by extrapolation from studies of acute exposure may occur at low dose rates. The protraction of low dose-rate exposure in the latter two studies over several days greatly complicates the interpretation of the results, however, since it requires the comparison of gene expression at several days after the start of exposure with gene expression at several hours after the start of exposure. It is currently unknown how much of the observed differences in gene expression between acute and protracted irradiation may be due to the evolution of the responses over time, and how much can be attributed uniquely to the effect of dose rate. It is clear, however, that significant alterations in global gene expression do occur during protracted radiation exposures. The appearance of some unique gene-expression changes in response to protracted irradiation may have a direct bearing on human health, and deserves further study.
Another phenomenon with relevance to low-dose and low-dose-rate exposures is the bystander effect. This refers to responses occurring in unirradiated cells that were in close proximity to irradiated cells. Irradiated cells appear able to communicate with other cells, at least in vitro, through both direct cell-to-cell contact and through diffusible factors, and can induce DNA damage, apoptosis, and mutation (Little 2006). Application of microarray analysis has begun to unravel some of the mechanisms behind the observed bystander effect, including the identification of connexin43 (GJA1) (Azzam et al. 2003) and cyclooxygenase 2 (PTGS2) (Zhou et al. 2005) as genes upregulated in bystander cells that were demonstrated to play important roles in relaying the bystander signal. Another study using medium transfer, a technique in which the culture medium from irradiated cells is placed on unirradiated cells, reported that 37 genes were upregulated in response to medium from irradiated cells, but no genes were downregulated (Chaudhry 2006). The upregulated genes coded for growth factors and their receptors, and for other proteins with known roles in extracellular signaling, consistent with a signal being transmitted by an extracellular diffusible factor. Many details of the bystander effect remain to be explained, but global profiling experiments are uncovering important clues to the mechanisms involved.
Expression of some genes has also been reported to occur in response to low doses of direct radiation, but not in response to high doses (Yin et al. 2003, Ding et al. 2005). In one such study, the majority of genes sensitive to low- but not high-dose radiation responded only at 48 hours after treatment, but not at earlier or later times (Franco et al. 2005). This underscores the importance of temporal kinetics in gene-expression studies.
In another study, the idea that the timing of each part of a radiation response cascade could vary slightly among individuals was a breakthrough for the analysis of in vivo gene expression in response to low doses of radiation. Gene expression was profiled in skin biopsies that had been exposed to 0.01, 0.1, or 1 Gy during radiotherapy, but, using standard statistical methods, no genes were found to respond significantly to the lower exposures (Goldberg et al. 2006). The authors of this study hypothesized that the lack of statistically significant responses could be due to slight fluctuations in the timing of signal transduction cascades among the eight individuals biopsied. If this was the case, the microarray analysis from each patient may show regulation of different genes from within the same response pathways. By grouping the genes on the microarray into common pathways and molecular classes, then testing the gene groups rather than individual genes for statistically significant regulation (Rocke et al. 2005), coherent response patterns could be identified. The response detected by this analysis suggested that pathways involved in inflammation, prosurvival, and DNA remodeling were activated by low-dose irradiation in vivo. This study also illustrates the growing concept of analyzing the response of gene sets or pathways rather than individual genes. Gene-set enrichment analysis (GSEA) is a different approach that is being widely adopted, and it may also assist in comparing data from different laboratories and across different microarray platforms (Subramanian et al. 2005).
Gene-expression profiling in radiation oncology
Beyond the elucidation of signaling pathways and basic mechanisms of radiation response, functional genomics studies are also beginning to have an impact on radiation oncology. One of the first uses of human microarrays was the characterization of differences between normal tissue and cancers, and the search for gene-expression signatures that predict an individual tumor's response to therapy. In an early study in this area that compared primary tumors of patients with breast cancer that later metastasized with tumors that did not metastasize, researchers identified 231 differentially expressed genes. A 70-gene signature derived from this work predicted cancer recurrence (van ‘t Veer et al. 2002). This has become one of the first major success stories of functional genomics in medicine: the “MammaPrint” test based on this work has been accepted for clinical use in Europe, and in early 2007 it became the first gene-expression cancer prognostic test approved by the Food and Drug Administration for use in the United States. Profiling studies of many other tumor types have been conducted, and more are ongoing.
There is also great interest in being able to predict the response of a tumor to therapy, so that therapy can be tailored to the individual. A growing number of microarray studies have searched for genes that are differentially expressed in radiation-sensitive and radiation-resistant cell lines; of particular interest are the intrinsic differences in radiation sensitivity and radioresistance induced after multiple fractions of radiation, as a typical treatment regimen would entail. Most of these studies have focused on a specific type of cancer, and have used only a small number of cell lines, often only one sensitive and one resistant line, which makes it difficult to generalize the findings.
A more broadly based study used gene-expression profiles of 35 cancer cell lines representing nine different tissues of origin to define a profile that could predict the surviving fraction of a cell line after a dose of 2 Gy (Torres-Roca et al. 2005). In this study, three novel genes were identified that had expression levels that correlated with cell survival at 2 Gy. Expression of these genes was confirmed by real-time PCR (polymerase chain reaction), and transfection and overexpression of one of these genes, RbAp48, increased the radiation sensitivity of cells. Another study compared radiation sensitivity of 60 tumor cell lines with both basal and radiation-induced gene expression, identifying 22 radiation-responsive genes and 175 genes with basal expression levels associated with low survival after exposure to 2 Gy γ rays (Amundson et al. 2008). It is not known, however, how well the radiation sensitivity of the tumor cell lines predicts the radio sensitivity of the original tumors from which they were derived.
Other studies have used gene-expression profiling directly on pretreatment biopsies from radiation-sensitive or radiation-resistant tumors. As cancer cell lines undergo many changes in culture and may not be exactly representative of all aspects of tumor biology, this may be a more informative approach. Studies on cervical cancer have shown distinct gene-expression profiles for different stages of disease, and large numbers of genes differentially expressed in radiation-sensitive and radiation-resistant tumors. In one study, 171 genes were differentially expressed in tumors from 19 patients, and a set of 62 of these genes correctly predicted response to radiotherapy in 18 of the 19 tumors (Kitahara et al. 2002). In another study, more than 300 genes were differentially expressed between 13 radiation-sensitive or radiation-resistant patients, and could predict the response to therapy with similar accuracy (Wong et al. 2003). More recently, two slightly larger studies of rectal cancer reported similar findings. In a study of 30 patients, a set of 54 genes was identified that predicted response to radiochemotherapy with 78% sensitivity and 86% specificity (Ghadimi et al. 2005); in a study of 52 patients, 33 genes were found that could predict response with 82.4% accuracy (Watanabe et al. 2006).
There is, however, no overlap of any of the genes predicting tumor radiation sensitivity in these four studies. This disappointing finding is actually consistent with results of studies defining gene-expression signatures to predict tumor recurrence. For instance, the van ‘t Veer signature (van ‘t Veer et al. 2002) underlying the MammaPrint was tested on 295 patients, but it has only three genes in common with another signature predicting breast cancer metastasis (Wang et al. 2005), which used a cohort of 286 patients. Other researchers have shown that there tends not to be a single unique signature predicting a complex disease process like tumor metastasis, even within a single study (Ein-Dor et al. 2005). This is because, at least in part, a great many genes are correlated with patient survival and there is not a lot of difference in magnitude between these correlations, and the correlation of an individual gene with patient survival is quite variable among different subsets of patients.
Ein-Dor and colleagues (2005) showed that multiple gene-expression signatures, which have very few genes in common, could predict metastatic breast cancer in the patients in the van ‘t Veer study with the same or even better accuracy as the original MammaPrint signature. The same researchers later calculated that for two breast cancer metastasis predictive signatures to have 50% genes in common, each study would have to use gene-expression profiles of several thousand patients (Ein-Dor et al. 2006). Although such numbers are somewhat daunting, it does not mean that useful predictive signatures cannot be determined from smaller-size studies. It is likely, however, that the frustration of nonoverlapping gene sets in predictive signatures will remain a reality in this field for some time. Such lack of overlap should not be interpreted as an experimental failure.
Another important consideration in radiation therapy is the damage normal tissue incurs. Because the sensitivity of patients to normal tissue damage and late effects varies, therapeutic doses of radiation are limited by the responses of the most sensitive members of the population. Several studies have tried to develop gene-expression signatures that could predict normal tissue damage and late effects, as opposed to the response of the tumor. Differences in the expression of genes for cytokine receptors were found in a small-scale study of fibro blasts from breast cancer patients with and without severe fibrosis (Quarmby et al. 2002). Because this study used a targeted cytokine microarray and enrolled only six patients, it was too small to test the predictive ability of the genes with different responses, however.
Another study focused on severe early toxicity (Rieger et al. 2004). Epstein-Barr virus was used to develop immortalized cell lines from lymphocytes of 14 patients who had a range of severe adverse reactions within a month of radiotherapy, and 13 controls with low toxicity. The immortalized cell lines were exposed to 5 Gy of ionizing radiation and gene expression was profiled four hours later. Twenty-four genes were identified with responses to in vitro radiation that differed between the patients with and without severe early toxicity. This 24-gene radiation response signature correctly predicted adverse responses in 9 of the 14 patients with severe toxicity, and produced no false positives when tested against a larger set of 43 control lymphoblastoid cell lines. This study demonstrated that the degree of radiation sensitivity of an individual could be detected in cells of a type different from the tissues suffering the adverse effect. This is a very important finding, as lymphocytes provide a much less invasive target for biopsy than do many of the tissues at direct risk of radiation injury. It also demonstrated that it is not always necessary to use primary cells to obtain information relevant to patient therapy, and that individual radiation sensitivity seems to be maintained in immortalized cell lines.
In a similarly designed study (Svensson et al. 2006), peripheral blood lymphocytes were obtained from 21 prostate cancer patients with late radiation toxicity and 17 patients free from toxicity two years after radiotherapy. The lymphocytes were stimulated to divide and treated with 2 Gy X rays, and global gene-expression profiles were measured 24 hours later. Common p53-regulated genes responded to irradiation and showed variations in response between patients, but variations in these genes did not correlate with late toxicity. However, the radiation induction of other genes did, and a set of 62 genes could predict late toxicity with 63% accuracy. Researchers next grouped all the genes on their arrays by gene ontology functions or biological processes, and used expression responses within groups rather than expression responses of individual genes. Interestingly, the gene groups improved the predictive ability of the signatures to 86% accuracy. This finding is reminiscent of the study of low-dose in vivo skin irradiation, in which coherent radiation response patterns emerged only when genes were grouped into pathways (Goldberg et al. 2006).
Gene-expression profiling has also recently provided the first potential signature of individual susceptibility to radiation-induced cancer (Detours et al. 2007). The study examined gene expression in papillary thyroid cancers from the Chernobyl Tissue Bank, which were presumed to have been induced by documented radiation exposure, and compared this to gene expression in similar tumors from French patients with no history of radiation exposure. A set of 118 genes previously shown to be induced in lymphoblasts by both γ rays and H2O2 (hydrogen perioxide) (Amundson et al. 2005) was found to predict the origin (exposed to Chernobyl radiation or not) of the thyroid tumors with 85% to 73% accuracy. It is not yet clear if this signature is indicative of tumors caused by radiation, or of individuals with heightened susceptibility to radiation-induced carcinogenesis, as suggested by the authors of the study. Confirmation of either would represent a considerable advance over our present understanding.
Putting together the big picture
Global gene-expression profiling holds great promise for unraveling the mechanisms that control the response to ionizing radiation and individual variations in that response. The hope is that such an understanding could lead to personalized medicine, including individually tailored radiotherapy for cancer patients. Unraveling the mechanisms underlying individual radiation sensitivity or susceptibility to radiation-induced cancer could provide individualized risk estimates and better radiation protection. For example, gene-expression profiles are being developed as biomonitors of radiation exposure for use in triage after large-scale radiological accidents or terrorist events (Amundson et al. 2000, 2004, Dressman et al. 2007). Much work is still needed to confirm the usefulness of such an approach, but one potential advantage is that in addition to providing a dose estimate, a gene-expression profile may be able to detect individuals with unusual sensitivities to specific radiation injuries, or predict those at most risk for late effects such as carcinogenesis after the initial crisis has passed.
The introduction of easily accessible methods for making rapid global measurements of gene expression is driving a paradigm shift in radiation biology, and in the biological sciences in general. In classical reductionist biology, one or a very few genes would be studied in relative isolation, and understanding of molecular pathways was accumulated slowly. Signal transduction from radiation exposure started with DNA damage, which activated ATM (ataxia telangiectasia mutated), which activated p53, which in turn drove the transcription of a handful of genes of interest, with everything else necessarily being ignored. In the postgenomic era, however, there are suddenly no simple linear pathways, and no gene or protein can be considered in isolation. Rather, the new focus is on mapping and understanding the myriad interactions that combine to regulate the response to radiation in a cell, tissue, or individual. Methods are being developed for making global measurements of protein expression and modifications, transcription-factor binding, and DNA methylation and copy number changes. The next big challenge will be the integration of information from all these sources to develop a fuller understanding of biological systems. The challenges are immense, but so are the potential payoffs, as we enter the era of systems biology.
Acknowledgments
Thanks go to Lubomir Smilenov for critical reading of the manuscript. This work was supported by the Office of Science (Biological and Environmental Research), US Department of Energy, grant no. DE-FG02-07ER46336, and by National Institutes of Health grants CA 49062 and U19 AI067773.
References cited
- Akaboshi E, Howard-Flanders P. Proteins induced by DNA-damaging agents in cultured Drosophila cells. Mutation Research. 1989;227:1–6. doi: 10.1016/0165-7992(89)90059-6. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Bittner M, Chen Y, Trent J, Meltzer P, Fornace AJ., Jr cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene. 1999;18:3666–3672. doi: 10.1038/sj.onc.1202676. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Shahab S, Bittner M, Meltzer P, Trent J, Fornace AJ., Jr Identification of potential mRNA markers in peripheral blood lympho -cytes for human exposure to ionizing radiation. Radiation Research. 2000;154:342–346. doi: 10.1667/0033-7587(2000)154[0342:iopmbi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Lee RA, Koch-Paiz CA, Bittner M, Meltzer P, Trent J, Fornace AJ., Jr Differential responses of stress genes to low dose-rate gamma irradiation. Molecular Cancer Research. 2003;1:445–452. [PubMed] [Google Scholar]
- Amundson SA, Grace M, McLeland C, Epperly M, Yeager A, Zhan Q, Greenberger J, Fornace AJ., Jr Human in vivo radiation-induced bio-markers: Gene expression changes in radiotherapy patients. Cancer Research. 2004;64:6368–6371. doi: 10.1158/0008-5472.CAN-04-1883. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Do KT, Vinikoor LC, Koch-Paiz CA, Bittner M, Trent J, Meltzer P, Fornace AJ., Jr Stress-specific signatures: Expression profiling of p53 wild-type and -null human cells. Oncogene. 2005;24:4572–4579. doi: 10.1038/sj.onc.1208653. [DOI] [PubMed] [Google Scholar]
- Amundson SA, et al. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute anticancer drug screen. Cancer Research. 2008;68:415–424. doi: 10.1158/0008-5472.CAN-07-2120. [DOI] [PubMed] [Google Scholar]
- Aubert J, Bar-Hen A, Daudin J-J, Robin S. Determination of the differentially expressed genes in microarray experiments using local FDR. BMC Bioinformatics. 2004;5:125. doi: 10.1186/1471-2105-5-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam EI, de Toledo SM, Little JB. Expression of CONNEXIN43 is highly sensitive to ionizing radiation and other environmental stresses. Cancer Research. 2003;63:7128–7135. [PubMed] [Google Scholar]
- Brach MA, Hass R, Sherman ML, Gunji H, Weichselbaum R, Kufe D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. Journal of Clinical Investigations. 1991;88:691–695. doi: 10.1172/JCI115354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, Wang JY, Anderson CW, Appella E, Xu Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Molecular and Cellular Biology. 2006;26:6859–6869. doi: 10.1128/MCB.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry MA. Bystander effect: Biological endpoints and microarray analysis. Mutation Research. 2006;597:98–112. doi: 10.1016/j.mrfmmm.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Detours V, Delys L, Libert F, Weiss Solis D, Bogdanova T, Dumont JE, Franc B, Thomas G, Maenhaut C. Genome-wide gene expression profiling suggests distinct radiation susceptibilities in sporadic and post-Chernobyl papillary thyroid cancers. British Journal of Cancer. 2007;97:818–825. doi: 10.1038/sj.bjc.6603938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding LH, Shingyoji M, Chen F, Hwang JF, Burma S, Lee C, Cheng JF, Chen DJ. Gene expression profiles of normal human fibroblasts after exposure to ionizing radiation: A comparative study of low and high doses. Radiation Research. 2005;164:17–26. doi: 10.1667/rr3354. [DOI] [PubMed] [Google Scholar]
- Dressman HK, Muramoto GG, Chao NJ, Meadows S, Marshall D, Ginsburg GS, Nevins JR, Chute JP. Gene expression signatures that predict radiation exposure in mice and humans. PLoS Medicine. 2007;4:e106. doi: 10.1371/journal.pmed.0040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics. 2005;21:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- Ein-Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proceedings of the National Academy of Sciences. 2006;103:5923–5928. doi: 10.1073/pnas.0601231103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornace AJ, Alamo I, Hollander MC. DNA damage-inducible transcripts in mammalian cells. Proceedings of the National Academy of Sciences. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco N, Lamartine J, Frouin V, Le Minter P, Petat C, Leplat JJ, Libert F, Gidrol X, Martin MT. Low-dose exposure to gamma rays induces specific gene regulations in normal human keratinocytes. Radiation Research. 2005;163:623–635. doi: 10.1667/rr3391. [DOI] [PubMed] [Google Scholar]
- Ghadimi BM, et al. Effectiveness of gene expression profiling for response prediction of rectal adenocarcinomas to preoperative chemoradiotherapy. Journal of Clinical Oncology. 2005;23:1826–1838. doi: 10.1200/JCO.2005.00.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg Z, Rocke DM, Schwietert C, Berglund SR, Santana A, Jones A, Lehmann J, Stern R, Lu R, Hartmann Siantar C. Human in vivo dose-response to controlled, low-dose low linear energy transfer ionizing radiation exposure. Clinical Cancer Research. 2006;12:3723–3729. doi: 10.1158/1078-0432.CCR-05-2625. [DOI] [PubMed] [Google Scholar]
- Hammond EM, Mandell DJ, Salim A, Krieg AJ, Johnson TM, Shirazi HA, Attardi LD, Giaccia AJ. Genome-wide analysis of p53 under hypoxic conditions. Molecular and Cellular Biology. 2006;26:3492–3504. doi: 10.1128/MCB.26.9.3492-3504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B, Schlötterer C. Comparison of algorithms for the analysis of affymetrix microarray data as evaluated by co-expression of genes in known operons. Nucleic Acids Research. 2006;34:e8. doi: 10.1093/nar/gnj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nature Biotechnology. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- Jelinsky SA, Estep P, Church GM, Samson LD. Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes. Molecular and Cellular Biology. 2000;20:8157–8167. doi: 10.1128/mcb.20.21.8157-8167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Research. 1991;51:6304–6311. [PubMed] [Google Scholar]
- Kenyon CJ, Walker GC. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proceedings of the National Academy of Sciences. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kis E, Szatmari T, Keszei M, Farkas R, Esik O, Lumniczky K, Falus A, Safrany G. Microarray analysis of radiation response genes in primary human fibroblasts. International Journal of Radiation Oncology Biology and Physics. 2006;66:1506–1514. doi: 10.1016/j.ijrobp.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Kitahara O, Katagiri T, Tsunoda T, Harima Y, Nakamura Y. Classification of sensitivity or resistance of cervical cancers to ionizing radiation according to expression profiles of 62 genes selected by cDNA micro array analysis. Neoplasia. 2002;4:295–303. doi: 10.1038/sj.neo.7900251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops L, et al. In vivo p53 response and immune reaction underlie highly effective low-dose radiotherapy in follicular lymphoma. Blood. 2007;110:1116–1122. doi: 10.1182/blood-2007-01-067579. [DOI] [PubMed] [Google Scholar]
- Little JB. Cellular radiation effects and the bystander response. Mutation Research. 2006;597:113–118. doi: 10.1016/j.mrfmmm.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Mirza A, et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–3654. doi: 10.1038/sj.onc.1206477. [DOI] [PubMed] [Google Scholar]
- Ohki R, Kawase T, Ohta T, Ichikawa H, Taya Y. Dissecting functional roles of p53 N-terminal transactivation domains by microarray expression analysis. Cancer Science. 2007;98:189–200. doi: 10.1111/j.1349-7006.2006.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson TA, et al. Performance comparison of one-color and two-color platforms within the Microarray Quality Control (MAQC) project. Nature Biotechnology. 2006;24:1140–1150. doi: 10.1038/nbt1242. [DOI] [PubMed] [Google Scholar]
- Pounds SB. Estimation and control of multiple testing error rates for microarray studies. Briefings in Bioinformatics. 2006;7:25–36. doi: 10.1093/bib/bbk002. [DOI] [PubMed] [Google Scholar]
- Qin LX, Beyer RP, Hudson FN, Linford NJ, Morris DE, Kerr KF. Evaluation of methods for oligonucleotide array data via quantitative real-time PCR. BMC Bioinformatics. 2006;7:23. doi: 10.1186/1471-2105-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nature Genetics. 2002;32(suppl.):496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Quarmby S, West C, Magee B, Stewart A, Hunter R, Kumar S. Differential expression of cytokine genes in fibroblasts derived from skin biopsies of patients who developed minimal or severe normal tissue damage after radiotherapy. Radiation Research. 2002;157:243–248. doi: 10.1667/0033-7587(2002)157[0243:deocgi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Rieger KE, Hong W-J, Tusher VG, Tang J, Tibshirani R, Chu G. Toxicity from radiation therapy associated with abnormal transcriptional responses to DNA damage. Proceedings of the National Academy of Sciences. 2004;101:6635–6640. doi: 10.1073/pnas.0307761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocke DM, Goldberg Z, Schweitert C, Santana A. A method for detection of differential gene expression in the presence of inter-individual variability in response. Bioinformatics. 2005;21:3990–3992. doi: 10.1093/bioinformatics/bti667. [DOI] [PubMed] [Google Scholar]
- Ruby SW, Szostak JW. Specific Saccharomyces cerevisiae genes are expressed in response to DNA-damaging agents. Molecular and Cellular Biology. 1985;5:75–84. doi: 10.1128/mcb.5.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: Microarray-based expression monitoring of 1000 genes. Proceedings of the National Academy of Sciences. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Goodman MF. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nature Reviews in Molecular Cell Biology. 2007;8:587–594. doi: 10.1038/nrm2198. [DOI] [PubMed] [Google Scholar]
- Scian MJ, Stagliano KER, Ellis MA, Hassan S, Bowman M, Miles MF, Deb SP, Deb S. Modulation of gene expression by tumor-derived p53 mutants. Cancer Research. 2004;64:7447–7454. doi: 10.1158/0008-5472.CAN-04-1568. [DOI] [PubMed] [Google Scholar]
- Shippy R, et al. Using RNA sample titrations to assess microarray platform performance and normalization techniques. Nature Biotechnology. 2006;24:1123–1131. doi: 10.1038/nbt1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov MV, Smirnova NA, Camerini-Otero RD, Neumann RD, Panyutin IG. Microarray analysis of differentially expressed genes after exposure of normal human fibroblasts to ionizing radiation from an external source and from DNA-incorporated iodine-125 radionuclide. Gene. 2006;382:47–56. doi: 10.1016/j.gene.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE, Logothetis CJ, McDonnell TJ. Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. Journal of Biological Chemistry. 2006;281:25134–25142. doi: 10.1074/jbc.M513901200. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara T, Magae J, Wadhwa R, Kaul SC, Kawakami Y, Matsumoto T, Tanaka K. Dose and dose-rate effects of low-dose ionizing radiation on activation of Trp53 in immortalized murine cells. Radiation Research. 2004;162:296–307. doi: 10.1667/rr3223. [DOI] [PubMed] [Google Scholar]
- Svensson JP, Stalpers LJA, Esveldt-Van Lange REE, Franken NAP, Haveman J, Klein B, Turesson I, Vrieling H, Giphart-Gassler M. Analysis of gene expression using gene sets discriminates cancer patients with and without late radiation toxicity. PLoS Medicine. 2006;3:1904–1914. doi: 10.1371/journal.pmed.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Roca JF, et al. Prediction of radiation sensitivity using a gene expression classifier. Cancer Research. 2005;65:7169–7176. doi: 10.1158/0008-5472.CAN-05-0656. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ‘t Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Watanabe T, et al. Prediction of sensitivity of rectal cancer cells in response to preoperative radiotherapy by DNA microarray analysis of gene expression profiles. Cancer Research. 2006;66:3370–3374. doi: 10.1158/0008-5472.CAN-05-3834. [DOI] [PubMed] [Google Scholar]
- Wong Y, et al. Expression genomics of cervical cancer: Molecular classification and prediction of radiotherapy response by DNA microarray. Clinical Cancer Research. 2003;9:5486–5492. [PubMed] [Google Scholar]
- Yin E, Nelson DO, Coleman MA, Peterson LE, Wyrobek AJ. Gene expression changes in mouse brain after exposure to low-dose ionizing radiation. International Journal of Radiation Biology. 2003;79:759–775. doi: 10.1080/09553000310001610961. [DOI] [PubMed] [Google Scholar]
- Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- Zhou H, Ivanov VN, Gillespie J, Geard CR, Amundson SA, Brenner DJ, Yu Z, Lieberman HB, Hei TK. Mechanism of radiation-induced bystander effect: Role of the cyclooxygenase-2 signaling pathway. Proceedings of the National Academy of Sciences. 2005;102:14641–14646. doi: 10.1073/pnas.0505473102. [DOI] [PMC free article] [PubMed] [Google Scholar]