

Abstract
Advances in understanding pediatric high-grade glioma (pHGG) genetics have revealed key differences between pediatric and adult high-grade gliomas (aHGGs), and have uncovered unique molecular drivers among subgroups within pHGG. The three core aHGG pathways, the receptor tyrosine kinase(RTK)/Ras/Phosphatidylinositide 3-kinase (PI3K), p53, and retinoblastoma (RB) networks, are also disrupted in pHGG, but they exhibit a different spectrum of effectors targeted by mutation. There are also similarities and differences in the genomic landscape of diffuse intrinsic pontine glioma (DIPG) and pediatric non-brainstem high-grade glioma (pNBS-HGG). In 2012, histone H3 mutations were identified in nearly 80% of DIPGs and ∼35% of pNBS-HGG. These were the first reports of histone mutations in human cancer, implicating novel biology in pediatric gliomagenesis. Additionally, DIPG and midline pNBS-HGG vary in the frequency and specific histone H3 amino acid substitution compared to pNBS-HGGs arising in the cerebral hemispheres, demonstrating a molecular difference among pHGG subgroups. The gene expression signatures as well as DNA methylation signatures of these tumors also carry distinctive signatures, reflecting a combination of the driving mutations and the developmental context from which they arise. These data collectively highlight unique selective pressures within the developing brainstem and solidify DIPG as a specific molecular and biological entity among pHGGs. Emerging studies continue to identify novel mutations that distinguish subgroups of pHGG. The molecular heterogeneity among pHGGs will undoubtedly have clinical implications moving forward. The discovery of unique oncogenic drivers is a critical first step in providing patients with appropriate, targeted therapies. Despite these insights, our vantage point has been largely limited to an in-depth analysis of protein coding sequences. Given the clear importance of histone mutations in pHGG, it will be interesting to see how aberrant epigenetic regulation contributes to tumorigenesis in the pediatric context. New mechanistic insights may allow for the identification of distinct vulnerabilities in this devastating spectrum of childhood tumors.
Opening remarks
The last several years mark a period of tremendous growth in our understanding of pediatric high-grade glioma (pHGG). Advances in genome-wide array-based and sequencing technologies, their precipitous drop in cost, and evaluation of increasingly larger cohorts, have all contributed to novel insights into the genetics of these devastating cancers, greatly extending earlier studies that evaluated candidate genes based on their involvement in adult high-grade glioma (aHGG). Our aim is to provide context to these studies and highlight their contribution to the current state of pHGG knowledge.
There are a number of biological features to suggest pediatric gliomas differ from those arising in adults. Most adult gliomas are high-grade supratentorial tumors. In contrast, the majority of childhood gliomas are low-grade, and both low-and-high-grade gliomas commonly arise within the posterior fossa, an area seldom affected in adults. Diffuse intrinsic pontine glioma (DIPG) is a brainstem HGG that occurs almost exclusively in children1,2. Additionally, the current standard of chemotherapeutic care for aHGG, temozolomide, has not been shown to improve long-term survival in pediatric trials3-5. Furthermore, malignant transformation, the process whereby a low-grade lesion progresses into a high-grade tumor, is a common event in adults but infrequent in children6. Genetic analyses have illuminated molecular differences driving pediatric and adult high-grade gliomagenesis.
The three core aHGG pathways show a different spectrum of alteration in pHGG
As the genomic landscape of aHGG came into view, it shaped initial work into the pediatric disease. The first pHGG studies focused primarily on investigating the involvement of high-frequency recurrent events found in adult tumors. For example, epidermal growth factor receptor (EGFR) is the most commonly altered receptor tyrosine kinase (RTK) in aHGG; with the corresponding gene locus undergoing amplification, intragenic deletion, or both in ∼50% of cases7-9. First identified in adult glioblastoma, EGFRvIII is the most common EGFR variant in aHGG and is formed by deletion of exons 2-7 resulting in a constitutively active kinase10-12. Accordingly, investigators early on sought to examine the degree of EGFR involvement in pediatric cases of HGG.
A number of studies found that EGFR alteration was less frequent in pHGG13-19, although gene amplification and EGFRvIII expression were detected in some pHGG20-23. Through genome-wide studies, PDGFRA, which encodes PDGFRα was identified as the most commonly targeted RTK in both DIPG and pNBS-HGG. Alterations in the gene itself include amplification, mutation, or both15-19,23-27. Experimentally, overexpression of wild type (WT) or mutant PDGFRα conferred a growth advantage to astrocytes, an effect that was diminished by introduction of the ATP-competitive inhibitors crenolanib or dasatinib27. PDGFRα mutants drive glioma formation in vivo27,28, with murine-derived HGGs recapitulating critical features of the human disease such as histopathologic characteristics and expression profiles27. In an effort to target PDGFR therapeutically, pediatric trials using dasatinib, crenolanib or imatinib have been launched29-31. Unfortunately, the benefit derived from selective RTK inhibitors may be marginal at best. pHGGs show evidence of intratumoral heterogeneity, with some cells co-amplifying multiple RTK genes or discrete cell populations within the same tumor amplifying different genes; suggesting that resistant populations are likely to be present even before treatment with targeted agents15,16,26.
Both PDGFRA and EGFR are part of the RTK/Ras/Phosphatidylinositide 3-kinase (PI3K) signaling cascade, which is altered in nearly 90% of aHGGs. Additionally, ∼80-90% of adult tumors show evidence of retinoblastoma (RB) and p53 pathway dysregulation7-9,32. For this reason, many of the first genetic pHGG studies focused on these same networks (Figure 1).
Figure 1. The p53/RB and RTK/Ras/PI3K pathways are dysregulated in pHGG.

a. The p53 and RB pathways regulate G1 cell cycle checkpoints. Mitogenic signaling activates the cyclin D-dependent kinases CKD4 or CKD6, coupled with cyclin D family members (CCND1/2/3). This complex phosphorylates pRB, releasing E2F and promoting transcription of genes responsible for G1/S cell cycle progression. Gene amplifications of CDK4, CDK6, or any of the three Cyclin D family members are found in pHGG, with greater frequency in DIPG. The tumor suppressor locus CDKN2A encodes two different proteins through translation of two different reading frames, p16INK4A and p19ARF. P16INK4A inhibits the activity of the cyclin D-dependent kinases CKD4 and CKD6. Oncogenic signals, DNA damage, or induction of P19ARF induce p53, leading to cell cycle arrest, apoptosis or senescence. Homozygous deletions of CDKN2A occur almost exclusively in NBS-HGGs; whereas TP53 mutations are common in both pNBS-HGG and DIPG.
b. Mutations in the RTK/RAS/PI3K pathway transduce unregulated signals for cell proliferation, growth and survival. RTK signaling begins when growth factor ligand binding leads to receptor dimerization. In pediatric HGG, PDGFRα is the RTK most frequently targeted by amplification and/or mutation. Upon dimerization, RTKs trans-phosphorylate one another at tyrosine residues in their cytosolic tails. p85, the regulatory subunit of PI3K, can then either directly bind to these phosphor-tyrosine residues or connect to RTKs through adaptor molecules and Ras. PI3K is comprised of catalytic (p110) and regulatory (p85) subunits, both of which are targeted by mutation, usually in a mutually exclusive pattern, in pHGG.
In adults the most commonly targeted components of the RTK/Ras/PI3K axis downstream of RTKs include activation of PI3K itself, or loss of function of PTEN, the main negative regulator of PI3K signaling, or NF1, a negative regulator of Ras-mediated signaling7-9. Activation of PI3K signaling caused by mutations of PIK3CA, encoding the catalytic p110α subunit of PI3K, or PIK3R1, encoding the regulatory subunit of PI3K, are usually present in mutually exclusive patterns, occurring in approximately 20% of aHGGs and a similar frequency of pHGG, including DIPG 9,25,33-39. The PTEN tumor suppressor is located on chromosome 10q. It remains unclear whether all tumors with loss of chromosome 10q are targeting PTEN loss of function when a wild-type PTEN allele is still retained. However, there are examples in experimental systems where PTEN haploinsufficiency contributes to tumorigenesis. Loss of heterozygosity of chromosome 10q, with or without concurrent PTEN mutation is very frequent in adult glioblastoma, with 10q LOH in approximately 80% and PTEN mutation in 25-40%, while the frequency is significantly lower in pHGGs, with 10q LOH in approximately 30% and PTEN mutation in less than 5-15% 9,13,15-19,23-26,35,36,40-42.
RB pathway dysregulation is common in both pNBS-HGGs and DIPG (Figure 1). The CDKN2A locus codes for two tumor suppressors, p16INK4a and ARF43. Notably, homozygous deletion of CDKN2A/B appears to be almost exclusive to pNBS tumors and largely absent in DIPG15-19,23,25,26. In contrast, amplification of CDK4/6 or CCND1/2/3 is found in approximately 30% of DIPG16,19,24. CDK4/6 code for cyclin D-dependent kinases that phosphorylate the retinoblastoma protein (pRb), facilitating G1/S cell cycle progression. To become active, these kinases must bind to cyclin D family members (encoded by CCND1/2/3), which themselves confer substrate specificity43. Therapeutic inhibition of this cyclin/CDK complex, using PD-0332991, a highly-selective non-ATP competitive CDK4/6 inhibitor, significantly increased survival in a murine model of DIPG, both as a single agent or following irradiation44.
TP53 mutations occur in up to 35% of pNBS-HGGs (range, 18-35%) and appear to be more common in DIPGs (40-50% of cases). When including alterations to other components of the pathway, such as MDM2 and/or ARF, those frequencies can reach 83% for both brainstem and NBS tumors14,25,32,35,40-42,45-47.
From the above data we can conclude that although the 3 main signaling pathways affected in aHGG are also affected in pHGG, pediatric and adult tumors differ with regard to the most frequently mutated effectors.
Copy number imbalances and gene expression profiling
Despite some common copy number imbalances such as 13q and 14q loss in approximately one third of HGG regardless of age or location, adult and pHGGs also exhibit a unique constellation of gains and losses that distinguish one from the other, and the same can be said for DIPGs and pNBS-HGGs15-19,23-26. This suggests that unique combinations of genetic drivers underlie adult and pediatric tumorigenesis, and among childhood HGG, DIPG and pNBS-HGG tumorigenesis.
Transcriptional analysis of tumors supports similar conclusions. Clustering of gene expression signatures from aHGGs identifies three to four major gene expression subgroups, with the most robust distinction between proneural and mesenchymal subgroups48-51. Unlike the WNT and sonic hedgehog subgroups of medulloblastoma52, the mutations associated with particular HGG subgroups are much less consistent. These same subgroups were identified in pHGGs by unsupervised comparisons, showing a clear relationship in the gene expression signatures of gliomas among different age groups and locations. However, supervised comparisons revealed expression signatures that distinguished adult from pediatric tumors, and within childhood glioma, DIPGs from pNBS-HGGs16,17,19,24,53,54.
Because biopsy on DIPG patients is not routinely performed in the US, most research material is acquired at autopsy from irradiated patients. The vast majority of these samples are designated WHO grade IV55. In contrast, clinicians in France regularly performed pre-treatment biopsies, and although most samples were HGGs, some were classified as low-grade56, raising the possibility that there may be a low-grade to high-grade malignant transformation in the genesis of DIPG. Importantly, there is a high degree of similarity in the copy number imbalances and expression signatures from DIPGs collected as biopsy samples prior to treatment and those collected at autopsy, which were treated by radiation in most cases, with or without chemotherapy 16,24. Furthermore, expression signatures of pediatric brainstem low-grade gliomas (LGGs) are much more closely related to non-brainstem LGG and not DIPGs, emphasizing different etiology underlying genesis of low-grade and high-grade gliomas arising in the brainstem16.
Histones make their mark
The work discussed above established the concept that oncogenic events driving pediatric HGG were different from those arising in adults. This appreciation, however, was not fully cemented until early 2012, with the discovery of recurrent histone mutations in pHGG (Figure 2). As the first reports of histone mutations in human cancer, these mutations implicated novel mechanisms in pHGG tumor biology that are not found to play a significant role in the adult disease.
Figure 2. Hotspot histone mutations occur in nearly 80% of DIPGs and ∼35% of NBS-HGGs.

a. The basic unit of chromatin is the nucleosome; DNA wrapped around a histone octamer consisting of two copies of H2A, H2B, H3, and H4. The N-terminal tails of histones undergo post-translational modifications (PTMs), represented by yellow and blue circles, which in turn alter chromatin accessibility and recruitment of effector proteins, together influencing transcriptional permissiveness. This is accomplished because PTMs can 1) themselves alter the strength of DNA-histone interactions 2) facilitate recruitment of chromatin remodeling complexes or histone PTM-binding effector proteins.
b. p.K27M substitutions occur in histone H3.1 and H3.3; p.G34R/V substitutions occur in H3.3. Histone H3.1/3.3 p.K27M exerts a dominant effect, preventing the accumulation of H3K27me2/3 on the wild-type histone H3 expressed in the same cell. p.G34R/V mutations do not exert the same effect on H3K27me3, but p.K27M and p.G34R/V histone mutations are associated with distinct genome-wide DNA methylation and gene expression tumor signatures.
c. Table summarizing the functional consequences of histone mutations.
Whole-genome sequencing of 7 DIPGs and matched germline DNA, as part of the St. Jude Children's Research Hospital-Washington University Pediatric Cancer Genome Project, identified somatic mutations in H3F3A leading to a p.K27M substitution in histone H3.3 in 4 of 7 cases. A fifth case contained an analogous mutation in HIST1H3B yielding a p.K27M alteration in histone H3.1. Subsequent targeted sequencing of all 16 genes encoding histone H3 in a larger cohort of pHGG found p.K27M somatic mutations in 78% of DIPGs; 60% were mutations in H3F3A and 18% in HIST1H3B57. Similar frequency of histone H3 mutation was found in an independent cohort; however, the proportion of H3F3A and HIST1H3B mutations varied between the groups, likely due to patient age, with HIST1H3B mutations arising in younger children57,58. In pNBS-HGG, H3F3A and HIST1H3B p.K27M substitutions were found in 19% and 3% of cases, respectively. Additionally, 14% of pNBS-HGGs harbored somatic mutations in H3F3A leading to p.G34R substitution, whereas no such alteration was identified in any DIPG. Most of the DIPG samples evaluated were collected at autopsy. However, of 8 DIPG samples collected from patients who had not received adjuvant therapy, 7 contained p.K27M substitutions. Hence, histone H3 alterations were not necessarily secondary to therapy57.
Collaborating groups in Canada and Germany performed whole-exome sequencing of 48 pNBS-HGGs and identified p.K27M, p.G34R, and p.G34V alterations in 19%, 10%, and 2% of cases, respectively, with all changes affecting H3F3A encoding the histone H3.3 variant. Additional targeted sequencing of the H3F3A locus in over 700 gliomas of various grades and patient ages revealed these mutations to be exclusive to high-grade tumors and significantly enriched in pediatric cases. Of the 11 adult cases harboring H3F3A mutation, all were missense substitutions of G34, and the majority was identified in young adults aged 20-30 years old. The specific histone mutation was associated with anatomical location. p.K27M mutations occured in tumors involving midline structures (such as the brainstem, cerebellum, and thalamus); while p.G34R/V mutations occurred in non-midline supratentorial lesions35,59 (Figure 3).
Figure 3. Histone mutations are strongly associated with anatomical location.
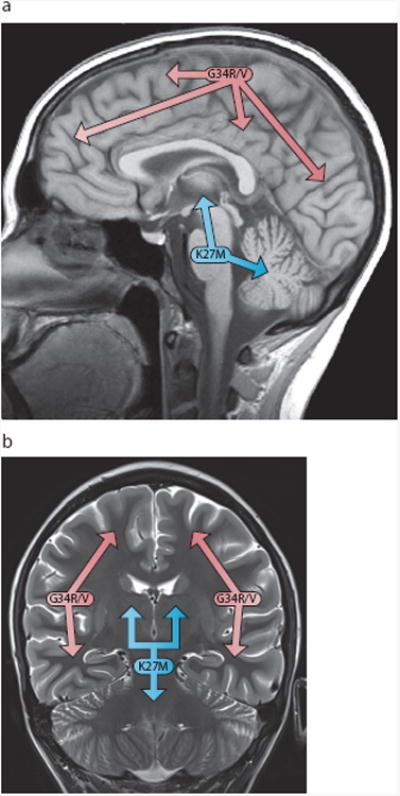
Midsagittal (a) and coronal (b) MR images demonstrating that p.K27M mutations occur predominantly in tumors involving midline structures (such as the brainstem, cerebellum, and thalamus); while p.G34R/V mutations mainly occur in tumors arising in the cerebral hemispheres.
Nucleosomes are the basic unit of chromatin, in which DNA is wrapped around a nucleosome core comprised of a histone octamer with two copies each of histones H2A, H2B, H3, and H4 (Figure 2). H3F3A and HIST1H3B code for the two histone H3 variant isoforms H3.3 and H3.1, respectively. There are three isoforms of histone H3. Histone H3.1 and H3.2 are encoded by 10 and 3 separate genes, respectively, and are synthesized during S phase of the cell cycle to package newly replicated DNA. Histone H3.3 is synthesized throughout the cell cycle and selectively incorporated into promoter regions of active genes, and through interactions with ATRX and DAXX, into pericentromeric heterochromatin and subtelomeric regions60. Notably, recurrent ATRX loss of function mutations were found in approximately one quarter of pHGG, which were also associated with alternative lengthening of telomeres (ALT). All tumors with H3F3A p.G34R/V mutation carried concomitant ATRX mutations, suggesting synergy between the two mutations35.
All identified histone mutations occur in the N-terminal tails of histones, unstructured regions that undergo extensive post-translational modification (PTM). These modifications in turn facilitate recruitment of effector proteins that regulate transcriptionally active or silent chromatin states61.
Understanding mutant histone gain-of-function
All histone H3 mutations in pHGG were heterozygous, and in any individual tumor, only one of 16 genes encoding histone H3 was mutated. This pattern clearly indicates a dominant gain-of-function effect.
Lysine 27 on histone H3 (H3K27) is a residue that can be acetylated or mono-, di-, or trimethylated (H3K27me3). Although mutant histone H3.1/3.3 make up a minority of the total cellular histone H3 pool62, p.K27M mutations led to loss of total H3K27me2/3 of the entire cellular H3 pool, most of which is wild-type62-65. This dominant negative effect appears to be caused by inhibition of the H3K27 methylase EZH2 due to interaction with p.K27M mutant histone H362,63. Globally, both p.K27M and p.G34R/V tumors exhibit DNA hypomethylation59,63. In an unsupervised comparison of genome-wide DNA methylation signatures, tumors with p.K27M and tumors with p.G34R/V form independent clusters based on histone mutation status. Many genes with differentially methylated promoters showed differential gene expression that was associated with the anatomic origin of the tumors, suggesting that both DNA methylation and gene expression may be a result of the origin of the tumor that may be further influenced by histone mutations59,63,64,66.
The dominant mechanism of action is less apparent for mutations affecting G34. While there is no clear dominant effect on modification of the nearby H3K36, there appears to be an altered genome-wide distribution of H3K36me3 binding. Interestingly, one of the most upregulated genes in such tumors is MYCN, a well-known oncogene66.
Additional genetic associations with pHGG subgroups
Genome-wide sequencing approaches revealed that 20-32% of DIPGs harbored somatic missense mutations in ACVR1, also known as ALK247,67-69 which encodes a receptor serine/threonine kinase mediating bone morphogenetic protein (BMP)-induced signal transduction70. These mutations frequently co-occur with histone H3.1 p.K27M substitutions. Both alterations tend to occur in younger DIPG patients, and were not found in pHGG arising outside the brainstem47,67-69. Thus, these mutations further clarify molecular subgroups within DIPG. Surprisingly, some of the somatic mutations found in DIPG were the same as previously reported ACVR1 germline mutations in fibrodysplasia ossificans progressiva (FOP), a disease characterized by heterotopic bone formation exacerbated by inflammation that is not associated with cancer predisposition71.
RNA-seq analysis allowed the identification of fusion genes that were generated by genomic rearrangements. Expression of fusion genes was common in pHGG, including DIPG, although most of the fusion genes were not recurrent. Strikingly, chimeric genes fusing N-terminal sequences from a number of different genes to the kinase domain of the neurotrophic tyrosine receptor kinase (NTRK) family members were recurrent gene fusions, found in 40% of infant NBS-HGGs, and at much lower frequency in pHGG overall47. The prognosis for pNBS-HGG in children younger than three is significantly better than for older children72. The NTRK fusion genes may provide a useful new therapeutic target for this patient population. NTRK fusions were also identified in adult glioblastoma as well as pLGG, but do not appear to be as enriched as in infant NBS-HGG73-75.
Closing remarks
Advances in microarrays and next-generation sequencing technologies have provided unprecedented insight into pHGG biology. But this insight is only a starting point. Researchers and clinicians must now endeavor to not only understand how this unique mutation spectrum contributes to tumorigenesis, but also more importantly, seek to exploit these genetic defects therapeutically. Recent discoveries should inform the generation of improved pre-clinical models that more faithfully resemble the pediatric disease, to be utilized for mechanistic studies as well as pre-clinical testing of new therapies.
We are grounded in knowing that our understanding of the protein coding genome far surpasses that of the remaining 98% of DNA sequence, which potentially hosts a vast network of regulatory elements. Furthermore, we are just beginning to piece together the role of epigenetics in normal and neoplastic contexts. The high frequency of histone mutations in pHGG strongly suggests that epigenetic dysregulation plays a major role in tumors arising within the pediatric setting. We must also recognize that our analyses yield snapshots of an ever-changing process. The genetics and epigenetics of tumors are dynamic, and the portrait of each cancer landscape evolves as a function of any selective pressure introduced, therapy included76.
The above studies, both large-and-small scale, genome-wide and targeted, have given today's researchers and clinicians a better understanding of pHGG than ever before. The biologies of DIPG, pNBS-HGG, and infant NBS-HGG are distinctly different, and these differences in turn require specific clinical consideration in terms of appropriate intervention. With a two-year survival rate of less than 20%, the prognosis for pHGG remains unacceptably poor77. However, there is a palpable excitement within the community that meaningful change is within reach.
Acknowledgments
We thank Dr. Zoltan Patay for the MRI images used in Figure 3. SJB is supported by NIH grant P01 CA096832 and ALSAC of St Jude Children's Research Hospital.
Footnotes
No potential conflicts of interest were disclosed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012;14 Suppl 5:v1–49. doi: 10.1093/neuonc/nos218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004;9:197–206. doi: 10.1634/theoncologist.9-2-197. [DOI] [PubMed] [Google Scholar]
- 3.Chassot A, et al. Radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. J Neurooncol. 2012;106:399–407. doi: 10.1007/s11060-011-0681-7. [DOI] [PubMed] [Google Scholar]
- 4.Cohen KJ, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol. 2011;13:317–23. doi: 10.1093/neuonc/noq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 6.Broniscer A, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol. 2007;25:682–9. doi: 10.1200/JCO.2006.06.8213. [DOI] [PubMed] [Google Scholar]
- 7.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphrey PA, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci U S A. 1990;87:4207–11. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong AJ, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89:2965–9. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N-and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89:4309–13. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pollack IF, et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children's Cancer Group 945 cohort. J Neurosurg. 2006;105:418–24. doi: 10.3171/ped.2006.105.5.418. [DOI] [PubMed] [Google Scholar]
- 14.Sung T, et al. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol. 2000;10:249–59. doi: 10.1111/j.1750-3639.2000.tb00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bax DA, et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res. 2010;16:3368–77. doi: 10.1158/1078-0432.CCR-10-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paugh BS, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29:3999–4006. doi: 10.1200/JCO.2011.35.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paugh BS, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061–8. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu HQ, et al. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol. 2010;12:153–63. doi: 10.1093/neuonc/nop001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zarghooni M, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28:1337–44. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 20.Gilbertson RJ, et al. ERBB1 is amplified and overexpressed in high-grade diffusely infiltrative pediatric brain stem glioma. Clin Cancer Res. 2003;9:3620–4. [PubMed] [Google Scholar]
- 21.Bax DA, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res. 2009;15:5753–61. doi: 10.1158/1078-0432.CCR-08-3210. [DOI] [PubMed] [Google Scholar]
- 22.Li G, et al. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J Neurooncol. 2012;108:395–402. doi: 10.1007/s11060-012-0842-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong KK, et al. Genome-wide allelic imbalance analysis of pediatric gliomas by single nucleotide polymorphic allele array. Cancer Res. 2006;66:11172–8. doi: 10.1158/0008-5472.CAN-06-2438. [DOI] [PubMed] [Google Scholar]
- 24.Puget S, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7:e30313. doi: 10.1371/journal.pone.0030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grill J, et al. Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer. 2012;58:489–91. doi: 10.1002/pbc.24060. [DOI] [PubMed] [Google Scholar]
- 26.Barrow J, et al. Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol. 2011;13:212–22. doi: 10.1093/neuonc/noq158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paugh BS, et al. Novel Oncogenic PDGFRA Mutations in Pediatric High-Grade Gliomas. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu KW, et al. SHP-2/PTPN11 mediates gliomagenesis driven by PDGFRA and INK4A/ARF aberrations in mice and humans. J Clin Invest. 2011;121:905–17. doi: 10.1172/JCI43690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baruchel S, et al. A Canadian paediatric brain tumour consortium (CPBTC) phase II molecularly targeted study of imatinib in recurrent and refractory paediatric central nervous system tumours. Eur J Cancer. 2009;45:2352–9. doi: 10.1016/j.ejca.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 30.Pollack IF, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro Oncol. 2007;9:145–60. doi: 10.1215/15228517-2006-031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broniscer A, et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin Cancer Res. 2013;19:3050–8. doi: 10.1158/1078-0432.CCR-13-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sturm D, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14:92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N. Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res. 1990;50:6683–8. [PubMed] [Google Scholar]
- 34.Gallia GL, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4:709–14. doi: 10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 35.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 36.Wu G, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014 doi: 10.1038/ng.2938. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broderick DK, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–50. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- 38.Hartmann C, Bartels G, Gehlhaar C, Holtkamp N, von Deimling A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005;109:639–42. doi: 10.1007/s00401-005-1000-1. [DOI] [PubMed] [Google Scholar]
- 39.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 40.Kraus JA, Felsberg J, Tonn JC, Reifenberger G, Pietsch T. Molecular genetic analysis of the TP53, PTEN, CDKN2A, EGFR, CDK4 and MDM2 tumour-associated genes in supratentorial primitive neuroectodermal tumours and glioblastomas of childhood. Neuropathol Appl Neurobiol. 2002;28:325–33. doi: 10.1046/j.1365-2990.2002.00413.x. [DOI] [PubMed] [Google Scholar]
- 41.Raffel C, et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res. 1999;5:4085–90. [PubMed] [Google Scholar]
- 42.Cheng Y, et al. Genetic alterations in pediatric high-grade astrocytomas. Hum Pathol. 1999;30:1284–90. doi: 10.1016/s0046-8177(99)90057-6. [DOI] [PubMed] [Google Scholar]
- 43.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 44.Barton KL, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One. 2013;8:e77639. doi: 10.1371/journal.pone.0077639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sure U, et al. Determination of p53 mutations, EGFR overexpression, and loss of p16 expression in pediatric glioblastomas. J Neuropathol Exp Neurol. 1997;56:782–9. [PubMed] [Google Scholar]
- 46.Antonelli M, et al. Prognostic significance of histological grading, p53 status, YKL-40 expression, and IDH1 mutations in pediatric high-grade gliomas. J Neurooncol. 2010;99:209–15. doi: 10.1007/s11060-010-0129-5. [DOI] [PubMed] [Google Scholar]
- 47.Wu G. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014 doi: 10.1038/ng.2938. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 49.Freije WA, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–10. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 50.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59:1190–9. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
- 52.Northcott PA, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12:818–34. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faury D, et al. Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. J Clin Oncol. 2007;25:1196–208. doi: 10.1200/JCO.2006.07.8626. [DOI] [PubMed] [Google Scholar]
- 54.Haque T, et al. Gene expression profiling from formalin-fixed paraffin-embedded tumors of pediatric glioblastoma. Clin Cancer Res. 2007;13:6284–92. doi: 10.1158/1078-0432.CCR-07-0525. [DOI] [PubMed] [Google Scholar]
- 55.Broniscer A, et al. Prospective collection of tissue samples at autopsy in children with diffuse intrinsic pontine glioma. Cancer. 2010;116:4632–7. doi: 10.1002/cncr.25405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roujeau T, et al. Stereotactic biopsy of diffuse pontine lesions in children. J Neurosurg. 2007;107:1–4. doi: 10.3171/PED-07/07/001. [DOI] [PubMed] [Google Scholar]
- 57.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–3. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–47. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 60.Elsaesser SJ, Goldberg AD, Allis CD. New functions for an old variant: no substitute for histone H3.3. Curr Opin Genet Dev. 2010;20:110–7. doi: 10.1016/j.gde.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24:567–74. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis PW, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–61. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bender S, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–72. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 64.Chan KM, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–90. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venneti S, et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013;23:558–64. doi: 10.1111/bpa.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bjerke L, et al. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-12-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taylor KR, et al. Recurrent activating ACVR1/ALK2 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014 doi: 10.1038/ng.2925. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buczkowicz P, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014 doi: 10.1038/ng.2936. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fontebasso AM, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014 doi: 10.1038/ng.2950. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Attisano L, et al. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell. 1993;75:671–80. doi: 10.1016/0092-8674(93)90488-c. [DOI] [PubMed] [Google Scholar]
- 71.Shore EM, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–7. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 72.Duffner PK, et al. Treatment of infants with malignant gliomas: the Pediatric Oncology Group experience. J Neurooncol. 1996;28:245–56. doi: 10.1007/BF00250203. [DOI] [PubMed] [Google Scholar]
- 73.Frattini V, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–9. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang J, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–12. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones DT, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–32. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gottardo NG, Gajjar A. Chemotherapy for malignant brain tumors of childhood. J Child Neurol. 2008;23:1149–59. doi: 10.1177/0883073808321765. [DOI] [PMC free article] [PubMed] [Google Scholar]