

Abstract
Airway epithelial cells form a barrier to the outside world, and are at the frontline of mucosal immunity. Epithelial apical junctional complexes are multi-protein subunits that promote cell-cell adhesion and barrier integrity. Recent studies in the skin and GI tract suggest that disruption of cell-cell junctions is required to initiate epithelial immune responses, but how this applies to mucosal immunity in the lung is not clear. Increasing evidence indicates that defective epithelial barrier function is a feature of airway inflammation in asthma. One challenge in this area is that barrier function and junctional integrity are difficult to study in the intact lung, but innovative approaches should provide new knowledge in this area in the near future. In this article, we review the structure and function of epithelial apical junctional complexes, emphasizing how regulation of the epithelial barrier impacts innate and adaptive immunity. We discuss why defective epithelial barrier function may be linked to Th2 polarization in asthma, and propose a rheostat model of barrier dysfunction that implicates the size of inhaled allergen particles as an important factor influencing adaptive immunity.
Keywords: Airway epithelium, Asthma, Barrier Defect, Mucosal Immunity, Tight Junction, Adherens Junction, Innate immunity, Allergy
Introduction
Airway epithelial cells are an important part of the innate immune system in the lung. In addition to establishing mucociliary clearance, epithelial cells produce anti-microbial peptides, chemokines, and cytokines that recruit and activate other cell types and promote pathogen clearance 1. Recent studies have emphasized the importance of epithelial derived cytokines in promoting Th2 immune responses, at least in part by conditioning local dendritic cells (DC) 2, 3. Epithelial cells also form a barrier to the outside world, comprised of airway surface liquids, mucus, and apical junctional complexes (AJC) that form between neighboring cells. AJC consist of the apical tight junctions and underlying adherens junctions that bind together via homotypic and heterotypic interactions (Figure 1). Epithelial tight junctions and adherens junctions establish cell-cell contact, cell polarity, and also regulate the paracellular movement of ions and macromolecules. Recent studies have documented the presence of dysfunctional epithelial AJC in the asthmatic airway, although the precise mechanisms involved and consequences for airway inflammation are not clear. Interestingly, inhaled allergens, pollution particles, and respiratory viruses can disrupt barrier integrity, which may represent a risk factor for allergen sensitization. Certain inflammatory cytokines can also cause barrier dysfunction, potentially creating a positive feedback loop. In addition to allowing better penetration of inhaled allergens and particles, airway barrier dysfunction likely initiates signal transduction cascades affecting epithelial activation and differentiation. Therefore, regulation of airway epithelial barrier function is emerging as an important checkpoint in asthma immunology. Before considering the mechanisms and consequences of barrier dysfunction for allergic airway inflammation, a brief overview of junctional structure is in order.
Figure 1.
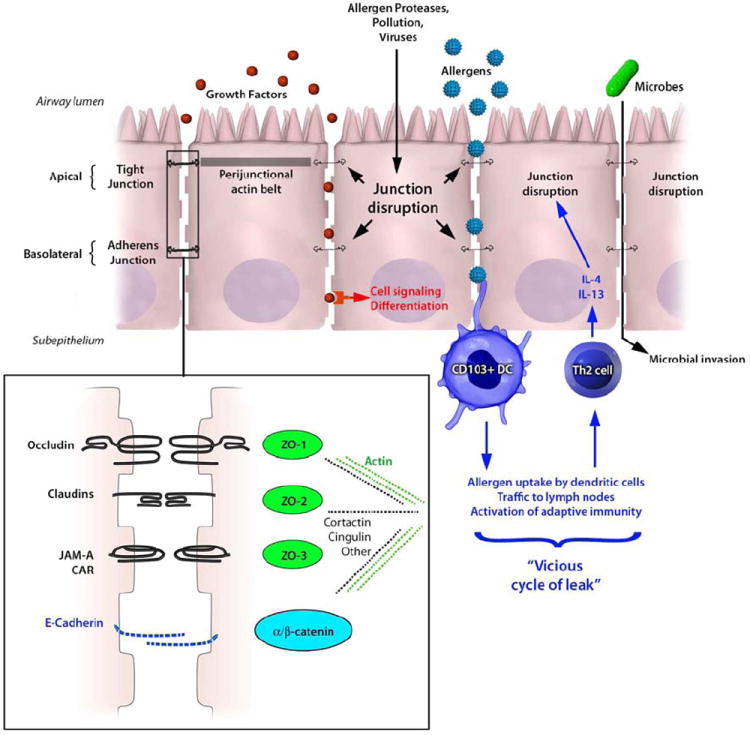
Cartoon diagram of airway epithelial cells indicating junctional structures including tight junctions (black) and adherens junctions (blue), which are intimately linked with perijunctional actin filaments. The inset shows an enlarged schematic of protein-protein interactions in tight junctions (black text) and adherens junctions (blue text), including ability of ZO proteins to interact with intracytoplasmic domains. The inset also indicates that junctional proteins are linked to the actin cytoskeleton (green dashed line) via several potential adaptor proteins (black dashed line). Inhaled allergens, air pollutants, and respiratory viruses can cause to dysfunction of epithelial junction resulting in greater outside/in permeability (see text, and Table 2). Barrier dysfunction can lead to epithelial cell signaling or differentiated, since it will allow apical growth factors constitutively present in epithelial lining fluids (red dots) to interact with their basolateral receptors. In the presence of intact epithelial junctions, these ligand/receptor interactions are prevented. Barrier dysfunction will also allow greater sampling of luminal allergens (blue stars) by intra-epithelial dendritic cells (DC), including CD103+ DC which interdigitate in the epithelium. Allergen-induced Th2 responses can induce a vicious cycle of leak, since Th2 cytokines perpetuate junctional dysfunction (see text and Table 3). Another consequence of leaky epithelial barriers is increased microbial invasion (green oval), which might predispose susceptible asthmatics to exacerbations or lung infections.
1. Apical junctional complexes: basic structure and function
Junctions between neighboring cells were first discovered using electron microscopy and appear as apposing strands that eliminate the intercellular space 4. Junctional complexes contain the most apical tight junctions (TJ) and underlying adherens junctions (AJ), which are both linked to perijunctional actin filaments 5, 6 (Figure 1). TJ regulate paracellular transport of ions and certain small molecules, whereas AJ are important for initiation and maintenance of cell-cell adhesion 7, 8. TJ and AJ interact to establish apical vs. basolateral membrane domains (i.e. cell polarity), and also regulate each other’s structure. Both TJ and AJ are involved in numerous signal transduction cascades 9. A current model proposes that there are two pathways for paracellular movement of molecules across TJ. The claudin-containing “pore” controls movement of ions in a charge and size-selective manner, whereas the “leak” pathway allows limited movement of larger macromolecules 8. The precise molecular basis for size and charge discrimination by different junctional components is currently under active investigation, and several reviews are available on this topic 8, 10, 11.
Our current understanding of junctional structure and function comes largely from studies of epithelial monolayers in vitro, and a current challenge is to understand how findings in model epithelia extrapolate to the multicellular epithelium in real-world conditions. In vitro studies typically grow epithelial monolayers to confluence on semipermeable membranes, and compare epithelial barrier function with junctional structure as determined using microscopy. Different functional assays can be used to study barrier integrity. Transepithelial electrical resistance (TEER) is easy to measure and commonly used to assess junctional integrity, since intact junctions will be relatively impermeable to ion flux (i.e. high TEER). However, low TEER does not always imply higher macromolecular permeability (discussed in 12), and consequently multiple approaches should be used to provide a complete picture of junctional integrity. In later sections of this review, we discuss additional assays that have been used to study outside/in airway barrier function in living organisms.
TJ and AJ are macromolecular complexes that bind together in the intercellular space, and also make numerous intracytoplasmic protein-protein interactions. Table 1 summarizes the major families of junctional complex proteins, including the three TJ families. New junctional components and protein interactions are being discovered regularly, and this Table is meant to be illustrative rather than comprehensive. First, claudins are a large family of tetraspanning transmembrane proteins that are expressed in a tissue- and cell-type selective manner, and interact in a homotypic or heterotypic fashion in the extracellular space. Claudins can be either barrier-promoting or barrier-disrupting (or “leaky”). For example, Claudin-1, the founding family member 13, is necessary and sufficient for junction formation function 14. Claudin-1 deficient mice die soon after birth, and suffer from excessive transepidermal water loss 15. This study established a key role for keratinocyte TJ in skin barrier function. Interestingly, defective expression of epidermal Claudin-1 was observed in the skin of subjects with atopic dermatitis 16, where it may serve as a risk factor for viral infection and allergen sensitization 17, 18. Claudin-2, in contrast, is an example of a leaky claudin associated with increased permeability in the intestine, where it is induced by interleukin-13 (IL-13) in a Stat6-dependent manner 19. Although IL-4 and IL-13 also enhance airway epithelial permeability and barrier dysfunction, they do so without inducing claudin-2 in 16HBE airway epithelial cells 20. These studies indicate that Th2 cytokine-induced epithelial barrier dysfunction can occur in the intestine and airway by different mechanisms. Other claudins expressed in the respiratory tract include Claudins-1, 3, 4, 7, and 18, where their expression and function is under active study 21 (reviewed in 10-12, 22).
Table 1.
Major Tight Junction, Adherens Junction and Plaque proteins
| Tight Junctions | Adherens Junctions | Cytosolic plaque proteins |
|---|---|---|
|
| ||
Claudin family
|
Cadherin family
|
Zonula occludens (ZO) family
|
Tight junction associated Marvel protein (TAMP) family
|
Nectin family | Catenin family
|
Others
|
||
Immunoglobulin family
|
||
The second group of TJ proteins is the Tight Junction-Associated Marvel Protein (TAMP) family, which has three members: occludin, tricellulin and MarvelD3 23. In contrast to claudins, TAMP family members are not essential for normal epithelial development and barrier function, although they appear to play a role in barrier regulation during inflammation. The intracytoplasmic tails of occludin and other TAMP family members are subject to numerous post-translational modifications, which are thought to affect interactions with scaffold components, signaling molecules, and the actin cytoskeleton 24. The observation that occludin-null mice were viable and have intact TJ in numerous epithelia indicated that this molecule was not essential for mucosal barrier function 25, 26. However, more research needs to be done to study the role of these proteins under conditions of epithelial stress or inflammation. Several recent studies indicate that occludin may have other functions separate from maintenance of epithelial integrity. For example, Huber et al. found that the migration of neutrophils across an MDCK monolayer was regulated by the occludin N-terminus independently of effects on TEER or paracellular permeability to mannitol 27. MDCK cells are derived from a canine kidney tumor, and are a widely used model of TJ structure and function. Recently, Edulblum et al. used intravital imaging with novel transgenic mice and found that γδ-intra-epithelial lymphocytes expressed occludin and appeared to migrate within the epithelium via homotypic interactions with occludin-expressing epithelial cells 28. This interesting study suggested a potential role for occludin in immune surveillance. Less is known about the expression and function of Tricellulin or MARVELD3 in the lung, and more research is needed into the role of epithelial occludin and the TAMP family in airway inflammation and asthma.
The third group of airway TJ is the Immunoglobulin-like family, specifically junctional adhesion molecule (JAM) and the coxsackie adenovirus receptor (CAR). These tight junction components are receptors for several important viruses 29, 30 Airway epithelial cells express multiple CAR isoforms, which promote the entry of viral particles. A pioneering study demonstrated that adenovirus binding to CAR caused disassembly of junctional complexes and enhanced epithelial permeability 31. Since adenoviral particles are shed into the basolateral space, junction dysfunction allows inside/out paracellular leak and escape of virus into the airway lumen, thus promoting infectivity of neighboring cells. Consequently, junction dysfunction may represent a strategy used by viruses to enhance their replication. JAM-A has a tissue specific role in regulating epithelial integrity, since JAM-A deficiency in mice did not alter steady-state or LPS-induced lung permeability 32, but did enhance intestinal permeability 33. JAM and CAR proteins regulate cutaneous immune responses following epidermal injury by affecting interactions between keratincoytes and skin γδ-T cells 34, 35. Taken together with other recent studies 36, 37, it is apparent that some T cell subsets in the skin sense and respond to subtle perturbations of barrier structure even in the absence of microbial invasion or overt inflammation. It will be interesting to determine whether similar events occur in the airway epithelium.
Adherens junctions are the second component of the AJC, and are located below TJ in the lateral membrane. Adherens junctions are especially important for maintenance of cell-cell adhesion, and are comprised of the cadherin and nectin families. In epithelial cells, E-cadherin binds to intracellular catenins including p120 and β-catenin, thus linking adherens junctions with the Wnt signaling pathway. By regulating the stability and nuclear import of β-catenin, this evolutionarily conserved pathway regulates gene expression and chromatin structure implicated in epithelial wound repair responses and differentiation 38. Sustained loss of E-cadherin leads to epithelial differentiation into a mesenchymal phenotype, a process known as epithelial-mesenchymal transition (EMT). The molecular basis of EMT is complex, and occurs during both embryogenesis as well as in epithelial neoplasia 39. Emerging data suggest that EMT is also feature of epithelial cells in asthma, where it likely contributes to airway remodeling 40, 41. Interestingly, house dust mite (HDM) extracts were shown to induce features of EMT in 16HBE epithelial cells in vitro, especially in concert with TGF-β1 42. In a separate study, chronic HDM administration in mice resulted in airway remodeling and features of EMT in large airways, including loss of E-cadherin and occludin 43. Support for the idea that loss of E-cadherin occurs in asthma comes from the observation that sputum E-cadherin levels correlated with asthma severity 44. Although reduced junctional protein expression during EMT might result in greater epithelial cell permeability, the net effect on airway leakiness will also be affected by sub-epithelial fibrosis and other compensatory structural changes that might occur over time. Consequently, more research is needed to understand how EMT and other changes in epithelial differentiation affect airway barrier properties.
Insights into the role of adherens junctions in intestinal epithelial barrier function were obtained using conditional deletion approaches in mice. For example, Smalley-Freed et al. generated mice lacking p120 catenin in the intestinal epithelium, which resulted in disrupted AJ and TJ 45. Interestingly, partial loss of p120 catenin resulted in spontaneous intestinal inflammation and GI bleeding probably due to translocation of luminal microbes 45. In lung endothelial cells, p120 is degraded by lipopolysaccharide (LPS), and also negatively regulates TLR4 signaling 46. This important study demonstrated that junctional structures can cross-talk with innate immune signal transduction.
Tight junctions and adherens junctions bind to numerous cytoplasmic proteins and link to the actin cytoskeleton, forming the ‘cytosolic plaque’. Key among the plaque proteins is the zonula occludens (ZO) family that links the intracellular domains of TJ and AJ with actin-binding proteins (e.g. cortactin, α-catenin, vinculin and α-actinin) and other cytoskeletal components. ZO proteins (which include ZO-1, -2, and -3) are expressed in a tissue specific manner, and contain numerous domains capable of protein-protein interactions with signaling molecules (reviewed in 12, 47).
2. Models of inducible barrier dysfunction
Increased epithelial permeability is a hallmark of mucosal inflammation, and can occur through multiple mechanisms. Any insult that results in epithelial cell death or detachment from the basement membrane will lead to increased permeability. More subtle exposures likely also increase leakiness of the epithelial barrier by affecting junctional complex structure and function without causing cell death. In the airway, enhanced outside/in permeability will result in greater penetration of inhaled allergens and particles into the subepithelial space, facilitating antigen sampling and innate and adaptive immune responses, and might activate epithelial signal transduction (Figure 1, see below). Epithelial junctional complexes can be disrupted directly by inhaled substances that penetrate the mucus layer, or indirectly by cytokines and other inflammatory mediators. Examples of environmental exposures implicated in airway epithelial barrier dysfunction include respiratory viruses (e.g. coxsackie virus, rhinovirus and respiratory syncytial virus 48-51), air pollution components (e.g. ozone and ambient particulate matter 52-54), cigarette smoke 55, 56, and allergens (considered below). Viruses appear to cause junction dysfunction by different mechanisms. For example, coxsackie virus causes occludin macropinocytosis driven by Rab GTPases 48, whereas rhinovirus reduces epithelial occludin gene expression in an NADPH-oxidase dependent manner 49, 50. We recently showed that respiratory syncytial virus (RSV) disrupts apical junctional complex structure and function without markedly affecting the expression of individual junctional components 51. Rather RSV infection leads to disassembly of junctional complexes from the cell surface, in association with actin remodeling and phosphorylation of the actin binding protein cortactin. The barrier disruptive effects of RSV were inhibited by antagonists of protein kinase D, similar to our previous work using the viral mimetic polyI:C 57. It will be interesting in future studies to determine if other viruses converge on PKD to cause junctional disruption, which would suggest that antagonists of this versatile signaling molecule might have therapeutic potential. We recently reviewed the mechanisms and consequence of barrier disruption induced by respiratory viruses and air pollution components 12. In the following section, we cover the effects of allergens and cytokines on epithelial junctional structure and function.
Dangerous allergens: protease-dependent epithelial barrier dysfunction
A central tenet of the “protease hypothesis” is that protease-containing allergens have the potential to directly cleave epithelial tight junctions and disrupt barrier structures 58. One of the first examples of this phenomenon was reported in 1995 using extracts of house dust mites 59 (see Table 2, top section). A follow-up study demonstrated that aged dust mite fecal pellets or purified Der p 1 increased permeability in MDCK monolayers and also decreased cell surface occludin expression in 16HBElo- cells 60. Because occludin fragments were detected in Der p 1-treated cells using Western blot, and Der p 1 was able to cleave peptide fragments of occludin and claudin in vitro, the authors concluded that Der p 1 directly targeted cell surface TJ. More recently, Heijink et al. used a commercially available house dust mite extract and found no effects on surface E-cadherin or β-catenin at concentrations up to 50 μg/ml 61, although a transient 20% reduction in TEER was detectable using a cell impedance sensing 62, 63. Subsequently, Post and Heijink found that protease activity of different HDM extracts correlated poorly with their ability to induce barrier dysfunction in vitro or lung inflammation after inhalation in mice 64. Interestingly, however, the ability to cause barrier disruption in model epithelia in vitro predicted inflammatory potential in vivo, showing that these two properties are closely aligned 64. Thus even independently of protease activity per se, these authors concluded that barrier disruption is a key feature of pro-inflammatory allergen extracts.
Table 2.
Dangerous Allergens: Models of airway epithelial barrier disruption
| Allergen | Cells | TEER | Permeability (Tracer) | AJC expression | Notes | Reference |
|---|---|---|---|---|---|---|
|
| ||||||
| Dust mite extracts and allergens | ||||||
|
| ||||||
| Der p growth medium | Bovine bronchial sheets | ND | Increased (BSA) | ND | In presence of 1.5 mM DTT | 59 |
| Affinity purified Der p 1 (300-3000 μg/ml) | ||||||
|
| ||||||
| Aged fecal pellets (~30 pellets/cm2) | MDCK | ND | Increased (mannitol) | Decreased occludin and ZO-1 | Occludin cleavage | 60, 120 |
| Purified Der p 1 | 16HBElo- | Blocked by E-64 | ||||
|
| ||||||
| HDM extract (50 μg/ml) | 16HBElo- | 20% fall in resistance measured by ECIS | ND | No effect on E-cadherin or β-catenin | Dependent on EGFR and ATP | 61-63 |
| Barrier recover by 1 hour | ||||||
|
| ||||||
| Der p 1 | PNEC (n=4) | ND | ND | Decreased JAM-A Claudin-1 | 70 | |
|
| ||||||
| Pollen extracts | ||||||
|
| ||||||
| Giant Ragweed (Ambrosia trifida), White Birch (Betula pendula), Kentucky Blue Grass (Poa pratensis) [Protein concentration: 6.25 to 100 mg/ml] | MDCK | ND | ND | Decreased occludin, Claudin-1, ZO-1 | Blocked by protease inhibitors | 65 |
| Calu-3 | ||||||
|
| ||||||
| Olive (Olea europaea), Orchard Grass (Dactylis glomerata), Italian Cypress (Cupressus sempervirens), and Scots Pine (Pinus sylvestris) [Protein concentration: ~0.1 to 5 mg/ml] | Calu-3 at ALI | ND | Increased (RITC dextran) | Decreased E-cadherin, claudin-1, occludin in allergen-specific manner | Especially pine Blocked by AEBSF | 66 |
| No effect on ZO-1 | ||||||
|
| ||||||
| Timothy Grass (Phleum pratense) [equivalent to 1 mg of pollen grains] | PBEC | No effect | ND | No effect on ZO-1 | Pollen-induced chemokine production depending on donor severity | 68 |
|
| ||||||
| Japanese hop (Hop J) [100 μg/ml] | Calu-3 | ND | Increased (FITC dextran) | Decreased occludin | Occludin degradation blocked by NAC | 67 |
ALI = air-liquid interface
EGFR = epidermal growth factor receptor
MDCK = Madin-Darby Canine kidney
NAC = N-acetyl cysteine
ND = not determined
PBEC = primary bronchial epithelial cells
PNEC = primary nasal epithelial cells
In addition to dust mite extracts, several studies have demonstrated that extracts of pollens can disrupt junctional structure or function using model epithelia in vitro (Table 2, bottom section). For example, diffusates of Giant Ragweed (Ambrosia trifida), White Birch (Betula pendula), and Kentucky Blue Grass (Poa pratensis) decreased expression of different TJ components in MDCK and Calu-3 cells, although junction function was not specifically investigated in that report 65. Calu-3 cells are airway epithelial cells derived from a patient with lung adenocarcinoma. In a detailed study, Vinhas et al. applied diffusates of Olive (Olea europaea), Orchard grass (Dactylis glomerata), Italian cypress (Cupressus sempervirens), and Scots pine (Pinus sylvestris) to Calu-3 cells grown at air-liquid interface, and studied the effects on epithelial permeability and junctional structure 66. Extracts of each allergen induced variable degrees of junctional disruption, which could be blocked using protease inhibitors. Interestingly, protease activity as assessed using model substrates in vitro did not correlate with barrier disruption (e.g. pine extracts with modest specific activity potently enhanced permeability), and there were allergen-specific effects on both junction function and structure. Combinations of allergens were not additive or synergistic, but instead protective effects emerged (e.g. cypress extracts tending to neutralize pine extracts) 66. Consequently, this study demonstrates the difficulties in predicting bioactivity of allergens based on biochemical properties, and underscores the need for more empirical research.
Lee and colleagues recently studied the effect of extracts of Japanese hop on permeability of Calu-3 cells 67. These extracts increased epithelial permeability and occludin degradation in a ROS-dependent manner that was blocked by the anti-oxidant NAC. In contrast to these studies, Blume and Davies found that an extract of Timothy grass extract (Phleum pratense) had no effect on TEER or ZO-1 expression of primary human bronchial epithelial cells (PBEC) derived from asthmatic donors, although pollen exposure did stimulate chemokine secretion 68. Another report from the Davies lab demonstrated that exposure of 16HBE cells to extracts of the allergenic fungus Alternaria alternata reduced TEER in a dose-dependent manner (e.g. ~50% reduction at 24 hrs using 100 μg/ml), although PBEC from healthy donors appeared to be resistant to these effects 69. Interestingly, PBEC derived from severe asthmatics were more susceptible to Alternaria-induced reductions in TEER, although this effect was transient and only apparent using high extract concentrations (>100 μg/ml).
Taken together, these studies highlight some of the challenges of working with extracts of real-world allergens in models of inducible barrier dysfunction 59-70. First, allergen extracts are extremely heterogeneous and vary in protease activity, LPS content, and other danger signals that can activate target cells. Consequently, it is difficult to compare results from different groups using different allergen preparations. Standardization of allergen extracts has been important for clinical allergy diagnostics and immunotherapy, but we need more sustained efforts to standardize allergen extracts used in basic science research. Another challenge is comparing data using cell lines to primary cultures of differentiated epithelia. Epithelial cell lines do not recapitulate the pseudostratified airway epithelium, with multiple cell types and overlying mucus and surface liquids. It is also challenging to estimate the concentration of allergen extracts to use in tissue culture experiments. In order to directly contact airway epithelial cell surfaces after depositing in the airway, inhaled allergens must penetrate the mucus layer and escape neutralization by anti-proteases or anti-oxidants in airway surface liquids. There may be “hotspots” of allergen deposition within the airway, and some subjects with defective anti-protease or anti-oxidant defenses might be particularly susceptible to allergen-induced barrier dysfunction 71. But it seems likely that the local concentration at the apical cell surface is likely extremely low and below concentrations used in many in vitro studies.
Epithelial barrier dysfunction induced by cytokines
It is currently thought that intestinal permeability in inflammatory bowel diseases is caused by cytokine-induced barrier dysfunction in the absence of apoptosis 72, 73, and it is becoming clear that similar pathways likely operate in the airway. Inflammatory cytokines including IL-4, IL-13, IFN-γ, and TNF-α have been shown to disrupt airway epithelial barrier function via diverse mechanisms (see Table 4). In PBEC cultured at air-liquid interface, IFN-γ and TNF-α synergistically disrupt barrier function in association with reduced ZO-1 and JAM expression 74. Using chemical inhibitors and Western blot assays, these investigators implicated a role for atypical PKC family members. More recently, Hardyman et al. reported that TNF-α alone induced PBEC barrier dysfunction without affecting AJC protein expression per se 75. Rather, these investigators found that TNF-α causes marked AJC disassembly in a Src-dependent manner. We found that both IL-4 and IL-13 induced barrier disruption in 16HBElo- cells by inhibiting surface expression of ZO-1, occludin, E-cadherin and β-catenin 20. In contrast to the Th2-depdendent induction of leaky claudin-2 observed in intestinal epithelia, neither IL-4 nor IL-13 induced claudin-2 expression in 16HBElo- cells. The innate type 2 cytokines TSLP, IL-25, IL-33 also had no effect on airway barrier integrity in our model. Interestingly, Soyka et al. recently reported that IL-4 and IFN-γ disrupted junctional structure and function in primary nasal epithelial cells from subjects with chronic rhinosinusitis, whereas IL-17A had no effect 76. By immunofluorescence microscopy, cytokine treatment disrupted the integration of ZO-1 and occludin into membrane junctions, without affecting their gene expression. Parker and colleagues grew bronchial epithelial cells from asthmatic and non-asthmatic children in vitro at air liquid interface, and studied the effects of IL-9 and IL-13 (alone or in combination) on epithelial differentiation and TEER 77. The presence of IL-13 in particular affected the cellular composition of epithelial monolayers, with fewer ciliated and more goblet cells detected at the end of culture, which translated into slight reductions in TEER 77. This study highlights the importance of considering epithelial plasticity and differentiation state when analyzing barrier structure and function 78, 79. In addition to cytokines, basophil/mast cell-derived mediators have been shown to disrupt AJC structure and function. Histamine induces transient disruption of barrier function in PBEC and loss of E-cadherin expression, leading to greater infectivity of adenovirus 80. JAM-A is targeted by mast cell derived tryptase in intestinal epithelial cells 81, but whether this same pathway operates in the airway requires further study. Taken together, these studies demonstrate that cytokines and mediators associated with allergic airway inflammation induce barrier disruption often by interfering with junctional complex assembly at the apical membrane rather than by interfering with junctional protein expression per se. AJC disassembly in the intestine occurs by endocytosis of surface molecules involving complex interactions with cytoskeletal machinery 6, 82, and future studies will be needed to determine the precise mechanisms involved in airway barrier dysfunction during allergic inflammation. The induction of barrier dysfunction by Th2 cytokines raises the possibility of a vicious cycle in the airway (Figure 1, right side). After mucosal allergen sensitization leading to Th2 polarization, if local secretion of IL-4 and IL-13 causes airway epithelial leakiness, then greater penetration of inhaled allergens and noxious particles could perpetuate the allergic immune response. It will be interesting to determine whether this “cycle of leak” operates in human asthmatics and whether apical junctions disrupted by allergens or inflammatory mediators can be restored.
Table 4.
Evidence for airway barrier dysfunction in asthma
| Author | Tissue source and subject characteristics | Key findings | Reference |
|---|---|---|---|
| Xiao | Biopsies and brushings from mild, moderate and severe asthmatics (see text) |
|
101 |
| De Boer | Bronchial biopsies (14 NA, 22 mild A, 25 atopic NA) |
|
97 |
| Parker | Bronchial brushings (9 NA, 7 A) (children) |
|
77, 98 |
| Post | Bronchial brushings (6 NA, 5 mild A) |
|
63 |
| Blume | Bronchial brushings (21 NA, 15 severe A) |
|
68 |
| Hackett | Cadaveric lungs (6 NA, 6 A) Bronchial brushings (6 NA and 5 mild A) |
|
99, 100 |
A: asthma, NA: non-asthma
ALI: epithelial cells from brushings or lung digests propagated in vitro in defined culture medium for several weeks
ECIS: electrical cell impendence sensing
3. Evidence for epithelial barrier dysfunction in asthma
The presence of a skin barrier defect in atopic dermatitis is well established, and is now known to involve defects not only in the stratum corneum (e.g. filaggrin 83-87) but also in keratinocyte tight junctions 17, 18, 88. The structure and function of the epidermal barrier can be studied in skin biopsies or explants from affected subjects, and monitored non-invasively by measuring transepidermal water loss (TEWL). TEWL is a measure of inside/out barrier function, and increased TEWL reflects defective function of claudins and other TJ components. There is no surrogate of TEWL to monitor inside/out barrier function of the airway in asthma. Measuring exhaled breath condensate volume is one potential approach, but this does not appear to be enhanced in asthmatics compared to control subjects after normalizing for minute ventilation. Clara cell secretory product (CCSP) is normally secreted apically into the airway lumen by airway epithelial cells, and increased serum or urine CCSP concentrations have been used to infer the presence of enhanced outside/in epithelial permeability 89. For example, serum or urinary CCSP increase after exposure to ozone 90, cold-dry air challenge 91, and also following RSV infection in children 92. In a population study, increased urinary CCSP was used as a biomarker of permeability caused by ultrafine particulate air pollution 93. However, CCSP is produced by other glandular epithelia and potentially affected by corticosteroids and lung inflammation 94-96, and remains an indirect measure of barrier integrity. Until other non-invasive measurements of airway permeability are developed, direct analysis of tissue biopsies, explants, or epithelial cell monolayers will be needed to investigate the presence of barrier defects in asthma.
Several investigators have recently studied airway biopsies and/or epithelial cells propagated in vitro at air-liquid interface (ALI) and uncovered evidence for defects in apical junctional complex structure and function in the asthmatic airway epithelium. Table 5 summarizes research to-date, noting that some studies had small sample sizes and were exploratory in nature 63, 68, 77, 97-100. Three studies documented reduced expression of tight junction components in the asthmatic epithelium using immunohistochemistry (IHC) 97, 99, 101. Reduced TEER in epithelial cells obtained from asthmatic donors propagated in vitro was observed in some studies 63, 68, 101, but not others 77, 99, 100. The most comprehensive analysis to-date was conducted by Xiao and Davies 101. These investigators obtained bronchial biopsies or epithelial brushings from healthy controls and subjects with varying degrees of asthma severity and studied both apical junctional structure using immunofluorescence microscopy, as well as barrier function with both TEER and permeability assays. Junctional structure was perturbed in both bronchial biopsies as well as epithelial cells propagated in vitro at ALI, as determined by reduced or patchy expression of cell surface ZO-1 (and trends for reduced occludin). The expression of mRNA for ZO-1 or occludin was not different between groups, arguing for post-transcriptional alterations in AJC formation. Importantly, barrier function was also reduced with lower TEER and higher permeability detected in cells propagated in vitro from asthmatic subjects, especially those with moderate and severe disease. Treatment with epidermal growth factor restored barrier function towards normal indicating that defective airway barrier function in asthma is potentially reversible 101.
One remarkable aspect of this study is that defects in barrier structure and function were observed in airway epithelial monolayers propagated in vitro for several weeks at ALI (as also noted by 63). Similar results were observed in nasal epithelial obtained from subjects with chronic rhinosinusitis and nasal polyposis, which demonstrated reduced TEER and higher permeability than tissues or cells from control subjects 76. This indicates that reduced barrier function is a stable property of these cells, or at least can be perpetuated in vitro under defined culture conditions. The molecular basis for this “hard wiring” of leaky epithelial cells remains to be determined, but one possibility is that is encoded in epithelial stem cells 102. Taken together with the now well-established role for barrier defects in atopic dermatitis, these exciting studies indicate that dysregulation of epithelial junctional complex structure and function may be a unifying feature of allergic diseases.
4. Consequences of barrier dysfunction for allergic airway inflammation and asthma immunology
Although there is growing evidence for disruption of epithelial barrier structures in airway diseases, the pathophysiological significance of these observations is currently unknown. Three general downstream effects of relevance to asthma immunology and allergic airway inflammation can be envisioned, which are not mutually exclusive (Figure 1). First, AJC dysfunction could promote outside/in permeability of inhaled particles and allergens into the sub-epithelial space. By subtly altering epithelial structure and facilitating sampling of luminal contents by intra-epithelial dendritic cells (DC), AJC dysfunction likely promotes innate and adaptive immune responses. Growing evidence suggests the junction disruption may even be required to initiate mucosal immunity in naïve hosts. Second, a leaky barrier could be a risk factor for infections by facilitating the penetration of microbes or viruses beyond the epithelial surface. Third, by affecting the polarized distribution of cell surface receptors and exposing the basolateral membrane to apical mediators (and vice versa), epithelial AJC disruption can cause intracellular signaling, secretory activity, and affect differentiation. We consider these possibilities below. An additional potential consequence of barrier dysfunction may actually be beneficial. If junctional disruption allows better penetration of inhaled medications into the airway, then local concentrations reaching target cells (e.g. beta-agonists and subepithelial smooth muscle cells) may actually be enhanced.
Enhanced antigen sampling
It seems logical to speculate that defective barriers will allow greater penetration of inhaled allergens or particles into the subepitheilal space, where they will encounter intra-epithelial DC and other immune targets. There is a rich network of intra-epithelial DC including the CD103+ subset that express αeβ7 and bind directly to E-cadherin on epithelial cells 103-105. Dendrites from these DC interdigitate between epithelial cells and express integrins and other TJ molecules (e.g. claudin-1) that maintain barrier integrity 103. It is currently not known if DC dendrites extend beyond TJ in the steady-state. This is an active area of investigation, but technically challenging because imaging techniques used to visualize epithelial:DC interactions in the airway can perturb tissue architecture and/or activate cells. In the skin, confocal microscopy demonstrated that antigen uptake by intradermal Langerhans cells involved subtle protrusion of dendrites with reorganization of keratinocyte TJ 106. A recent study used minimally-invasive intravital two-photon microscopy with exteriorized bowel loops, and concluded that at steady-state intestinal goblet cells (and not intraepithelial CD103+ DC) constitutively sampled luminal contents 107. These studies raise the possibility that disruption of epithelial junctional complexes may be required for effective sampling of inhaled antigens by intra-epithelial DC. A corollary of this hypothesis is that substances that disrupt barrier function may prove to be effective mucosal adjuvants.
Direct evidence supporting a role for airway leakiness in mucosal allergen sensitization is currently lacking. Future models of outside/in allergen translocation will need to consider that the ability of inhaled particles to penetrate the epithelium depends on their shape, size and surface chemistry. These properties are being exploited therapeutically to enhance the efficacy of mucosal vaccines 108, but how the physical properties of inhaled allergens impacts their immunogenicity is largely an unexplored area. What is clear is that in addition to affecting deposition within the respiratory tract 109, particle size affects translocation across lung epithelial barriers. In a rat model, inhaled particles <34 nanometers (nm) were rapidly detected in lung draining lymph nodes, likely reflecting direct outside/in translocation into afferent lymph channels 110. In order to determine the site of translocation, von Garnier and colleagues used confocal microcopy and flow cytometry and found that the vast majority of inhaled particles were taken up by alveolar macrophages regardless of size, but a few 20 – 50 nm nanoparticles were detected in intra-tracheal DC 24 hours after inhalation 111. Similarly, Zoltan Veres et al. used two-photon imaging of thick-cut lung slices, and found that 1 micron particles were entirely taken up by alveolar macrophages and not-intra-epithelial DC 112. Taken together, it seems that at steady-state, uptake of airway lumenal antigens by intra-epithelial DC is uncommon, and unlikely to occur with particles greater than ~50 nm in diameter. However, future research will be needed to determine how surface chemistry (e.g. allergen proteases) or host susceptibility (e.g. virus-induced tight junction defects) influence allergen sampling and immunogenicity in the respiratory epithelium. Exposure models using low-level aerosol exposure should be particularly insightful because they will mimic physiologically relevant conditions.
Microbial infection
Defective junctional complexes might also facilitate outside/in translocation of lumenal microbes or viruses across the airway epithelium. As opposed to inert allergens, bacteria actively penetrate epithelia and secrete toxins that by themselves are barrier disruptive 113, 114. Consequently, even micron-sized bacteria can translocate across epithelial cells, but AJC still provide a first line of defense. The role of epithelial barriers in maintaining immune homeostasis is a topic of great interest, since defects in this regard are linked to microbial dissemination and inflammation, especially in the intestine 115. But “barrier defects” in this context usually refer to diminished function of epithelial cells or intra-epithelial lymphocytes, and few studies have investigated the immunological consequences of junctional dysfunction per se. One exception is the conditional deletion of p120 catenin discussed above, which led to spontaneous intestinal inflammation 45. Since the microbial burden is lower in the lung, deletion of p120 (and other tight and adherens junction components) in the airway epithelium should be better tolerated, but might affect the lung microbiome or represent a risk factor for mucosal allergen sensitization. Another exception is the recent observation that Myd88-adapter like (Mal) signaling is required for the expression of occludin, ZO-1, and claudin-3 in intestinal epithelial cells. This helps explain the observation that Mal-deficiency predisposes to Salmonella Typhomuirum infection, and links TLR signaling directly with AJC integrity 116. In addition to microbial invasion, AJC dysfunction may be a risk factor for respiratory viral infection, since basolateral receptors will be more accessible 29-31. Because virsues can also cause junctional dysfunction 48-51, this indicates the potential for a positive feed-back loop resulting in susceptibility to subsequent viral infections or bacterial superinfection. In fact, Sajan and Hershenson formally demonstrated that rhinovirus infection markedly increased the translocation of bacteria across epithelial monolayers 49. In future studies, it will be important to determine if asthmatics with leaky airways are particularly prone to respiratory infections and pathogen-induced exacerbations.
Epithelial Signaling
The idea that tight junction disruption can have intrinsic signaling properties is best supported in the case of epithelial growth factors, which are constitutively expressed in apical airway surface liquids but separated from their basolateral receptors by intact junctions (Figure 1). When epithelial integrity is compromised, ligand/receptor binding can rapidly initiate a wound repair response 117. One intriguing idea is that junctional disruption could be a Th2-promoting signal in the airway. A study by Heijink and colleagues supports this possibility, since these authors showed that siRNA knock-down of E-cadherin led to the production of Th2 promoting cytokines TARC and TSLP by airway epithelial cells in an EGFR dependent manner 118. This possibility would help explain the association of epithelial barrier dysfunction with Th2-driven allergic diseases, and is consistent with the hypothesis that Th2-immunity may have evolved to restore mucosal integrity after parasitic infections 119. Since Th2 responses can promote wound healing and fibrosis, persistent junction dysfunction in the airway may also be a risk factor for airway remodeling.
5. Concluding remarks
We propose that epithelial barrier dysfunction is not “all or none”, but rather a graded phenomenon with consequences for allergen uptake and processing that may impact subsequent adaptive immune responses (Figure 2). Inducible barrier dysfunction caused by environmental exposures can vary in severity and will affect the penetration and fate of inhaled particles, depending on their size and other physical characteristics. Inhaled allergens themselves may be capable of promoting transient barrier disruption, but sustained dysfunction is likely more common following inhalation of toxic air pollutants and respiratory viral infections. Inducible barrier dysfunction is a strategy used by viruses to promote their replication, but likely represents a risk factor for allergen sensitization. Future studies of the mechanisms and consequences of airway epithelial barrier dysfunction in asthma should enhance our understanding of asthma heterogeneity.
Figure 2.
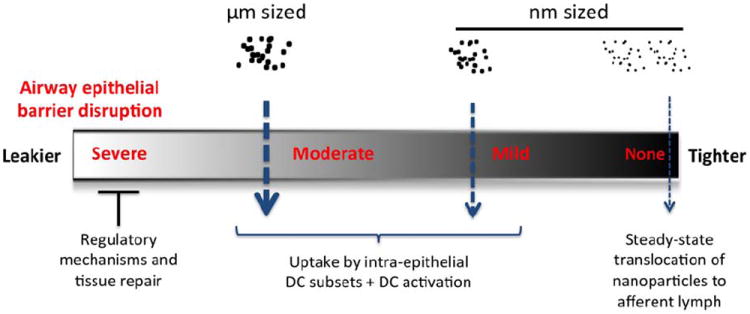
Rheostat model of airway epithelial barrier function. At steady-state (far right), airway epithelial cells normally exclude particles greater than ~30-50 nM in diameter. In the presence of dysfunctional barriers, progressively larger particles will traverse apical junctions (right to left). Barrier dysfunction likely facilitates sampling of luminal contents by DC dendrites (not shown), thus promoting the outside/in translocation of inhaled allergen particles that deposit on the cell surface. In addition to surface properties, the size of inhaled allergen particles might influence their uptake by intra-epithelial DC subsets, which in turn could influence quality and intensity of subsequent adaptive immune responses. The possibility that sustained and persistent barrier disruption leads to reparative responses or increased regulatory tone is indicated on the far left.
Table 3.
Cytokines implicated in airway epithelial barrier disruption
| Stimulus | Cells | TEER | Permeability (Tracer) | AJC expression | Notes | Reference |
|---|---|---|---|---|---|---|
|
| ||||||
| IFN-γ, IL-1β, TNF-α (10-100 ng/ml) | PBEC (normal and CF) | Decreased | Increased (2 kDa dextran) | Decreased ZO-1 and JAM | Synergy between IFN-γ+TNF-α | 74 |
| CF more sensitive Atypical PKC i/l | ||||||
|
| ||||||
| IL-4, IFN-γ, TNF-α | Calu-3 | Decreased | ND | Decreased ZO-1, occludin | Involvement of EGFR and MAPK | 121 |
|
| ||||||
| IL-4, IL-13, TSLP, IL-25, IL-33 (0.5-50 ng/ml) | 16HBElo- | Decreased | Increased (3 kDa dextran) | Decreased ZO-1, occludin, | Jak dependent | 20 |
| E-cadherin, β-catenin | No induction of claudin-2 | |||||
| No effect of innate type 2 cytokines | ||||||
|
| ||||||
| IL-4, IFN-γ (10 ng/ml) | PNEC | Decreased | Increased | Decreased ZO-1, occludin | No effect of IL-17 | 76 |
| More pronounced in CRSwNP | ||||||
|
| ||||||
| IL-13 (20 ng/ml) | PBEC | Decreased | ND | ND | Long-term exposure | 77 |
|
| ||||||
| TNF-α (10 ng/ml) | PBEC | Decreased | Increased (4 kDa dextran) | Decreased occludin, | Src kinase | 75 |
| Claudin-3, 4, 8 by IF (not WB) | ||||||
CF = cystic fibrosis
CRSwNP = chronic rhinosinusitis with nasal polyposis
IF = immunofluorescence
MAPK = mitogen associated protein kinases
ND = not determined
PBEC = primary bronchial epithelial cells
PNEC = primary nasal epithelial cells
PKC = protein kinase C
WB = Western blot
List of abbreviations
- ALI
air-liquid interface
- CAR
Coxsackie adenovirus receptor
- CF
cystic fibrosis
- CRSwNP
chronic rhinosinusitis with nasal polyposis
- ECIS
electrical cell impendence sensing
- EGFR
epidermal growth factor receptor
- IF
immunofluorescence
- JAM-A
Junctional adhesion molecule-A
- MAPK
mitogen associated protein kinases
- MDCK
Madin-Darby Canine kidney
- NAC
N-acetyl cysteine
- PBEC
primary bronchial epithelial cells
- PNEC
primary nasal epithelial cells
- PKC
protein kinase C
- WB
Western blot
- ZO
Zonula occludens
Glossary
- Tight Junction
A multi-subunit complex of transmembrane proteins that interact in the intercellular space to promote epithelial apposition. Tight junctions are comprised of different family members (e.g. claudins, occludin), and link to the actin cytoskeleton.
- Adherens Junction
These junctional structures form below tight junctions and help establish barrier function and epithelial polarity.
- Actin
A protein found especially in microfilaments and active in cellular movement and maintenance of cell shape. A “belt” of actin below the plasma membrane helps maintain the integrity of cellular junctions.
- Transepithelial electrical resistance (TEER)
The opposition of the epithelium to the passage of a steady electrical current, which measures instantaneous ion flux. High TEER implies low ion flux and a tight epithelial barrier.
- IL-13
A cytokine produced by Th2 and ILC2 cells capable of inducing the IgE isotype switch. It’s receptor is not found on mast cells (as is the case for IL-4), but IL-13 is more widely produced than IL-4. IL-13 contributes to airway mucus hypersecretion and airway hyperreactivity in mouse models.
- γδ-T Cells
A subset of T cells whose T cell antigen receptors (TCRs) have γ and δ chains. These cells express a restricted repertoire of TCRs. They are capable of responding to non-peptide and non-processed antigens, such as lipids, and appear to recognize antigens directly (independent of class I or class II MHC).
- Epithelial-Mesenchymal Transition (EMT)
A biologic process in which polarized epithelial cells assume a more mesenchymal phenotype characterized by migration and invasiveness. An early event in EMT is loss of junctional protein expression including E-cadherin.
- TLR4
Toll-like receptor 4. The first TLR identified. TLR4 binds to bacterial endotoxin (a lipopolysaccharide in the cell membrane of gram negative bacteria) and viral coat proteins. Binding to TLR4 activates signal transduction via the MyD88 adaptor protein.
- Protease-containing allergens
Cysteine and seine proteases are found in many common allergens including fungal and insect extracts (e.g. dust mite, cockroach). Allergen-associated proteases may promote allergic sensitization by disrupting epithelial junctional structures.
- Alternaria alternata
An aeroallergen of the Ascomycota phyla. Its spores have characteristic, elongated, beak-like chains. Spores are capable of traveling hundreds of miles and are found in grain-growing regions of temperate climates with a peak in the late summer and fall. It is one of the most common spores found in dust from North American homes.
- Transepidermal water loss (TEWL)
A non-invasive measurement that utilizes vapor pressure gradient estimation. Humidity and temperature affect its measurement. TEWL is elevated in subjects with atopic dermatitis, reflecting defective skin barrier properties.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–84. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol. 2012;130:845–52. doi: 10.1016/j.jaci.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J Allergy Clin Immunol. 2013;131:856–65. doi: 10.1016/j.jaci.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2:a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–28. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 6.Ivanov AI, Naydenov NG. Dynamics and regulation of epithelial adherens junctions: recent discoveries and controversies. Int Rev Cell Mol Biol. 2013;303:27–99. doi: 10.1016/B978-0-12-407697-6.00002-7. [DOI] [PubMed] [Google Scholar]
- 7.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127:2525–32. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight Junction Pore and Leak Pathways: A Dynamic Duo. Annu Rev Physiol. 2010 doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matter K, Balda MS. Signalling to and from tight junctions. Nat Rev Mol Cell Biol. 2003;4:225–36. doi: 10.1038/nrm1055. [DOI] [PubMed] [Google Scholar]
- 10.Frank JA. Claudins and alveolar epithelial barrier function in the lung. Ann N Y Acad Sci. 1257:175–83. doi: 10.1111/j.1749-6632.2012.06533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koval M. Claudin heterogeneity and control of lung tight junctions. Annu Rev Physiol. 2013;75:551–67. doi: 10.1146/annurev-physiol-030212-183809. [DOI] [PubMed] [Google Scholar]
- 12.Rezaee F, Georas SN. Breaking Barriers: New Insights Into Airway Epithelial Barrier Function in Health and Disease. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2013-0541RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–50. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or - 2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143:391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuse M, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156:1099–111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Benedetto A, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2010;127:773–86. e1–7. doi: 10.1016/j.jaci.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Benedetto A, et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J Allergy Clin Immunol. 2011;128:242–246 e5. doi: 10.1016/j.jaci.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol. 2012;132:949–63. doi: 10.1038/jid.2011.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosen MJ, et al. STAT6 deficiency ameliorates severity of oxazolone colitis by decreasing expression of claudin-2 and Th2-inducing cytokines. J Immunol. 2013;190:1849–58. doi: 10.4049/jimmunol.1201373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saatian B, et al. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1:e24333. doi: 10.4161/tisb.24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G, et al. Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2013-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soini Y. Claudins in lung diseases. Respir Res. 2011;12:70. doi: 10.1186/1465-9921-12-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raleigh DR, et al. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol Biol Cell. 2010;21:1200–13. doi: 10.1091/mbc.E09-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cummins PM. Occludin: one protein, many forms. Mol Cell Biol. 2012;32:242–50. doi: 10.1128/MCB.06029-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saitou M, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–42. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulzke JD, et al. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669:34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Huber D, Balda MS, Matter K. Occludin modulates transepithelial migration of neutrophils. J Biol Chem. 2000;275:5773–8. doi: 10.1074/jbc.275.8.5773. [DOI] [PubMed] [Google Scholar]
- 28.Edelblum KL, et al. Dynamic migration of gammadelta intraepithelial lymphocytes requires occludin. Proc Natl Acad Sci U S A. 2012;109:7097–102. doi: 10.1073/pnas.1112519109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Excoffon KJ, et al. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J Infect Dis. 2008;197:1189–97. doi: 10.1086/529515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Excoffon KJ, et al. Isoform-specific regulation and localization of the coxsackie and adenovirus receptor in human airway epithelia. PLoS One. 2010;5:e9909. doi: 10.1371/journal.pone.0009909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters RW, et al. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell. 2002;110:789–99. doi: 10.1016/s0092-8674(02)00912-1. [DOI] [PubMed] [Google Scholar]
- 32.Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol. 2012;303:L758–66. doi: 10.1152/ajplung.00107.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monteiro AC, et al. JAM-A associates with ZO-2, Afadin and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell. 2013 doi: 10.1091/mbc.E13-06-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verdino P, Witherden DA, Havran WL, Wilson IA. The molecular interaction of CAR and JAML recruits the central cell signal transducer PI3K. Science. 2010;329:1210–4. doi: 10.1126/science.1187996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witherden DA, et al. The junctional adhesion molecule JAML is a costimulatory receptor for epithelial gammadelta T cell activation. Science. 329:1205–10. doi: 10.1126/science.1192698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chodaczek G, Papanna V, Zal MA, Zal T. Body-barrier surveillance by epidermal gammadelta TCRs. Nat Immunol. 2012;13:272–82. doi: 10.1038/ni.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Jong A, et al. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol. 2014;15:177–85. doi: 10.1038/ni.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fagotto F. Looking beyond the Wnt pathway for the deep nature of beta-catenin. EMBO Rep. 2013;14:422–33. doi: 10.1038/embor.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–76. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 40.Hackett TL, et al. Induction of epithelial-mesenchymal transition in primary airwa649 y epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. 2009;180:122–33. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 41.Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12:53–9. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- 42.Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol. 2010;42:69–79. doi: 10.1165/rcmb.2008-0449OC. [DOI] [PubMed] [Google Scholar]
- 43.Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS One. 2011;6:e16175. doi: 10.1371/journal.pone.0016175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuyama K, et al. Sputum E-cadherin and asthma severity. J Allergy Clin Immunol. 2003;112:208–9. doi: 10.1067/mai.2003.1526. [DOI] [PubMed] [Google Scholar]
- 45.Smalley-Freed WG, et al. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J Clin Invest. 2010;120:1824–35. doi: 10.1172/JCI41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang YL, et al. Innate immune function of the adherens junction protein p120- catenin in endothelial response to endotoxin. J Immunol. 2011;186:3180–7. doi: 10.4049/jimmunol.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez-Mariscal L, Quiros M, Diaz-Coranguez M. ZO proteins and redox667 dependent processes. Antioxid Redox Signal. 2011;15:1235–53. doi: 10.1089/ars.2011.3913. [DOI] [PubMed] [Google Scholar]
- 48.Coyne CB, Shen L, Turner JR, Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe. 2007;2:181–92. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sajjan U, Wang Q, Zhao Y, Gruenert DC, Hershenson MB. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178:1271–81. doi: 10.1164/rccm.200801-136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Comstock AT, et al. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol. 2011;85:6795–808. doi: 10.1128/JVI.02074-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rezaee F, et al. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J Virol. 2013;87:11088–95. doi: 10.1128/JVI.01573-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu XY, Takahashi N, Croxton TL, Spannhake EW. Modulation of bronchial epithelial cell barrier function by in vitro ozone exposure. Environ Health Perspect. 1994;102:1068–72. doi: 10.1289/ehp.941021068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bayram H, Rusznak C, Khair OA, Sapsford RJ, Abdelaziz MM. Effect of ozone and nitrogen dioxide on the permeability of bronchial epithelial cell cultures of non683 asthmatic and asthmatic subjects. Clin Exp Allergy. 2002;32:1285–92. doi: 10.1046/j.1365-2745.2002.01435.x. [DOI] [PubMed] [Google Scholar]
- 54.Sidhaye VK, Chau E, Breysse P, King LS. Septin-2 Mediates Airway Epithelial Barrier Function in Physiologic and Pathologic Conditions. Am J Respir Cell Mol Biol. 2011 doi: 10.1165/rcmb.2010-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petecchia L, et al. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest. 2009;135:1502–12. doi: 10.1378/chest.08-1780. [DOI] [PubMed] [Google Scholar]
- 56.Schamberger AC, et al. Cigarette Smoke-Induced Disruption of Bronchial Epithelial Tight Junctions is Prevented by Transforming Growth Factor-Beta. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2013-0090OC. [DOI] [PubMed] [Google Scholar]
- 57.Rezaee F, et al. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J Allergy Clin Immunol. 2011;128:1216–1224 e11. doi: 10.1016/j.jaci.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Georas SN, Rezaee F, Lerner L, Beck L. Dangerous allergens: why some allergens are bad actors. Curr Allergy Asthma Rep. 2010;10:92–8. doi: 10.1007/s11882-010-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herbert CA, et al. Augmentation of permeability in the bronchial epithelium by the house dust mite allergen Der p1. Am J Respir Cell Mol Biol. 1995;12:369–78. doi: 10.1165/ajrcmb.12.4.7695916. [DOI] [PubMed] [Google Scholar]
- 60.Wan H, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104:123–33. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heijink IH, et al. Characterisation of cell adhesion in airway epithelial cell types using electric cell-substrate impedance sensing. Eur Respir J. 2009;35:894–903. doi: 10.1183/09031936.00065809. [DOI] [PubMed] [Google Scholar]
- 62.Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J. 2010;36:1016–26. doi: 10.1183/09031936.00125809. [DOI] [PubMed] [Google Scholar]
- 63.Post S, et al. House dust mite-induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. Allergy. 2013 doi: 10.1111/all.12202. [DOI] [PubMed] [Google Scholar]
- 64.Post S, et al. The composition of house dust mite is critical for mucosal barrier dysfunction and allergic sensitisation. Thorax. 2012;67:488–95. doi: 10.1136/thoraxjnl-2011-200606. [DOI] [PubMed] [Google Scholar]
- 65.Runswick S, Mitchell T, Davies P, Robinson C, Garrod DR. Pollen proteolytic enzymes degrade tight junctions. Respirology. 2007;12:834–42. doi: 10.1111/j.1440-1843.2007.01175.x. [DOI] [PubMed] [Google Scholar]
- 66.Vinhas R, et al. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy. 2011;66:1088–98. doi: 10.1111/j.1398-9995.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- 67.Lee SI, Pham LD, Shin YS, Suh DH, Park HS. Environmental changes could enhance the biological effect of Hop J pollens on human airway epithelial cells. J Allergy Clin Immunol. 2014 doi: 10.1016/j.jaci.2014.01.034. [DOI] [PubMed] [Google Scholar]
- 68.Blume C, et al. Barrier responses of human bronchial epithelial cells to grass pollen exposure. Eur Respir J. 2013;42:87–97. doi: 10.1183/09031936.00075612. [DOI] [PubMed] [Google Scholar]
- 69.Leino MS, et al. Barrier disrupting effects of alternaria alternata extract on bronchial epithelium from asthmatic donors. PLoS One. 2013;8:e71278. doi: 10.1371/journal.pone.0071278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henriquez OA, et al. House dust mite allergen Der p 1 effects on sinonasal epithelial tight junctions. Int Forum Allergy Rhinol. 2013;3:630–5. doi: 10.1002/alr.21168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tomazic PV, et al. Nasal mucus proteomic changes reflect altered immune responses and epithelial permeability in patients with allergic rhinitis. J Allergy Clin Immunol. 2014;133:741–50. doi: 10.1016/j.jaci.2013.09.040. [DOI] [PubMed] [Google Scholar]
- 72.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 73.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–71. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coyne CB, et al. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13:3218–34. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hardyman MA, et al. TNF-alpha-mediated bronchial barrier disruption and regulation by src-family kinase activation. J Allergy Clin Immunol. 2013 doi: 10.1016/j.jaci.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 76.Soyka MB, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-gamma and IL-4. J Allergy Clin Immunol. 2012;130:1087–1096 e10. doi: 10.1016/j.jaci.2012.05.052. [DOI] [PubMed] [Google Scholar]
- 77.Parker JC, et al. Chronic IL9 and IL-13 exposure leads to an altered differentiation of ciliated cells in a well-differentiated paediatric bronchial epithelial cell model. PLoS One. 2013;8:e61023. doi: 10.1371/journal.pone.0061023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knight DA, Stick SM, Hackett TL. Defective function at the epithelial junction: a novel therapeutic frontier in asthma? J Allergy Clin Immunol. 2011;128:557–8. doi: 10.1016/j.jaci.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 79.Avila PC. Plasticity of airway epithelial cells. J Allergy Clin Immunol. 2011;128:1225–6. doi: 10.1016/j.jaci.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zabner J, et al. Histamine alters E-cadherin cell adhesion to increase human airway epithelial permeability. J Appl Physiol. 2003;95:394–401. doi: 10.1152/japplphysiol.01134.2002. [DOI] [PubMed] [Google Scholar]
- 81.Wilcz-Villega EM, McClean S, O’Sullivan MA. Mast cell tryptase reduces junctional adhesion molecule-A (JAM-A) expression in intestinal epithelial cells: implications for the mechanisms of barrier dysfunction in irritable bowel syndrome. Am J Gastroenterol. 2013;108:1140–51. doi: 10.1038/ajg.2013.92. [DOI] [PubMed] [Google Scholar]
- 82.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–24. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marenholz I, et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol. 2006;118:866–71. doi: 10.1016/j.jaci.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 84.Palmer CN, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 85.Weidinger S, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214–9. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 86.Howell MD, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2009;124:R7–R12. doi: 10.1016/j.jaci.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 87.McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol. 2013;131:280–91. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- 88.Kuo IH, Yoshida T, De Benedetto A, Beck LA. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131:266–78. doi: 10.1016/j.jaci.2012.12.1563. [DOI] [PubMed] [Google Scholar]
- 89.Hermans C, Bernard A. Lung epithelium-specific proteins: characteristics and potential applications as markers. Am J Respir Crit Care Med. 1999;159:646–78. doi: 10.1164/ajrccm.159.2.9806064. [DOI] [PubMed] [Google Scholar]
- 90.Broeckaert F, et al. Serum clara cell protein: a sensitive biomarker of increased lung epithelium permeability caused by ambient ozone. Environ Health Perspect. 2000;108:533–7. doi: 10.1289/ehp.00108533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bolger C, et al. Effect of inspired air conditions on exercise-induced bronchoconstriction and urinary CC16 levels in athletes. J Appl Physiol (1985) 2011;111:1059–65. doi: 10.1152/japplphysiol.00113.2011. [DOI] [PubMed] [Google Scholar]
- 92.Johansson S, Kristjansson S, Bjarnarson SP, Wennergren G, Rudin A. Clara cell protein 16 (CC16) serum levels in infants during respiratory syncytial virus infection. Acta Paediatr. 2009;98:579–81. doi: 10.1111/j.1651-2227.2008.01083.x. [DOI] [PubMed] [Google Scholar]
- 93.Timonen KL, et al. Daily variation in fine and ultrafine particulate air pollution and urinary concentrations of lung Clara cell protein CC16. Occup Environ Med. 2004;61:908–14. doi: 10.1136/oem.2004.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beier HM. The discovery of uteroglobin and its significance for reproductive biology and endocrinology. Ann N Y Acad Sci. 2000;923:9–24. doi: 10.1111/j.1749-6632.2000.tb05516.x. [DOI] [PubMed] [Google Scholar]
- 95.Berg T, Cassel TN, Schwarze PE, Nord M. Glucocorticoids regulate the CCSP and CYP2B1 promoters via C/EBPbeta and delta in lung cells. Biochem Biophys Res Commun. 2002;293:907–12. doi: 10.1016/S0006-291X(02)00319-4. [DOI] [PubMed] [Google Scholar]
- 96.Arsalane K, et al. Clara cell specific protein (CC16) expression after acute lung inflammation induced by intratracheal lipopolysaccharide administration. Am J Respir Crit Care Med. 2000;161:1624–30. doi: 10.1164/ajrccm.161.5.9812157. [DOI] [PubMed] [Google Scholar]
- 97.de Boer WI, et al. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol. 2008;86:105–12. doi: 10.1139/y08-004. [DOI] [PubMed] [Google Scholar]
- 98.Parker J, et al. A 3-D well-differentiated model of pediatric bronchial epithelium demonstrates unstimulated morphological differences between asthmatic and nonasthmatic cells. Pediatr Res. 2009;67:17–22. doi: 10.1203/PDR.0b013e3181c0b200. [DOI] [PubMed] [Google Scholar]
- 99.Hackett TL, et al. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol. 2011;45:1090–100. doi: 10.1165/rcmb.2011-0031OC. [DOI] [PubMed] [Google Scholar]
- 100.Hackett TL, et al. Caveolin-1 Controls Airway Epithelial Barrier Function: Implications for Asthma. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2013-0124OC. [DOI] [PubMed] [Google Scholar]
- 101.Xiao C, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128:549–556 e12. doi: 10.1016/j.jaci.2011.05.038. [DOI] [PubMed] [Google Scholar]
- 102.Tata PR, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–23. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sung SS, et al. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161–72. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 104.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–24. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 105.van Helden MJ, Lambrecht BN. Dendritic cells in asthma. Curr Opin Immunol. 2013;25:745–54. doi: 10.1016/j.coi.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 106.Kubo A, Nagao K, Yokouchi M, Sasaki H, Amagai M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J Exp Med. 2009;206:2937–46. doi: 10.1084/jem.20091527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McDole JR, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. 2012;483:345–9. doi: 10.1038/nature10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith DM, Simon JK, Baker JR., Jr Applications of nanotechnology for immunology. Nat Rev Immunol. 2013;13:592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 2005;113:823–39. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Choi HS, et al. Rapid translocation of nanoparticles from the lung airspaces to the body. Nat Biotechnol. 2010;28:1300–3. doi: 10.1038/nbt.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blank F, et al. Size-dependent uptake of particles by pulmonary antigen-presenting cell populations and trafficking to regional lymph nodes. Am J Respir Cell Mol Biol. 2013;49:67–77. doi: 10.1165/rcmb.2012-0387OC. [DOI] [PubMed] [Google Scholar]
- 112.Zoltan Veres T, Voedisch S, Spies E, Tschernig T, Braun A. Spatiotemporal and functional behavior of airway dendritic cells visualized by two-photon microscopy. Am J Pathol. 2011;179:603–9. doi: 10.1016/j.ajpath.2011.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Soong G, et al. Staphylococcus aureus protein A mediates invasion across airway epithelial cells through activation of RhoA GTPase signaling and proteolytic activity. J Biol Chem. 2011;286:35891–8. doi: 10.1074/jbc.M111.295386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45:189–201. doi: 10.1165/rcmb.2011-0011RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–53. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 116.Corr SC, et al. MyD88 adaptor-like (Mal) functions in the epithelial barrier and contributes to intestinal integrity via protein kinase C. Mucosal Immunol. 2014;7:57–67. doi: 10.1038/mi.2013.24. [DOI] [PubMed] [Google Scholar]
- 117.Vermeer PD, et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422:322–6. doi: 10.1038/nature01440. [DOI] [PubMed] [Google Scholar]
- 118.Heijink IH, et al. Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J Immunol. 2007;178:7678–85. doi: 10.4049/jimmunol.178.12.7678. [DOI] [PubMed] [Google Scholar]
- 119.Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. 2013;13:607–14. doi: 10.1038/nri3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wan H, et al. Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen Der p 1. Clin Exp Allergy. 2000;30:685–98. doi: 10.1046/j.1365-2222.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- 121.Petecchia L, et al. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab Invest. 2012;92:1140–8. doi: 10.1038/labinvest.2012.67. [DOI] [PubMed] [Google Scholar]