

Abstract
Background: To maintain normal function, the lymphatic endothelium is regulated by cell–cell junctions. There have been few studies of lymphatic endothelial cell junctions using standard cell biological methods. This study had two purposes: to characterize cell junctions in cultured lymphatic endothelial cells and to investigate the effects of the inflammatory cytokine TNF-α on altered cell–cell junctions.
Methods and Results: Cultured human dermal lymphatic endothelial cells (HDLEC) were immunostained with the tight junction marker, ZO-1, and adherens junction markers, VE-cadherin and PECAM-1. In TNF-α-treated HDLEC, we evaluated changes in endothelial cell junctions by immunostaining and through the use of transendothelial electrical resistance (TER). Immunofluorescence staining of HDLEC revealed heterogeneity among the endothelial cell junctions, which could be classified into continuous and discontinuous junctions. In these cell junctions, ZO-1 and VE-cadherin were co-localized. Double immunofluorescence staining revealed the broad distribution of VE-cadherin at the cell periphery, where VE-cadherin and PECAM-1 were co-localized. TNF-α treatment decreased TER, caused a predominance in the appearance of discontinuous junctions with a reduction in the broad distribution of VE-cadherin at the cell periphery in HDLEC.
Conclusions: The results indicate a heterogeneous distribution of cell junctions in HDLEC involving continuous and discontinuous junctions. Our data also suggest that TNF-α alters the normal distribution of cell junctions and affects the endothelial barrier of cultured lymphatic endothelial cells. The broad distribution of VE-cadherin at the cell periphery may reflect the lymphatic permeability.
Introduction
The lymphatic vasculature is essential for fluid homeostasis and the immune response. Recently, the importance of lymphatic vessels in various pathological conditions, such as tumor metastasis and chronic inflammation, has been recognized.1 Both in quiescent conditions and in activated situations such as inflammation, the lymphatic endothelium is regulated by cell–cell junctions. These junctions play important roles in maintaining normal lymphatic function and are important in recovering homeostasis during pathological processes.2
Endothelial cells are joined via cell–cell junctions called tight junctions and adherens junctions, both of which play crucial roles in the organization and maintenance of vascular integrity.3,4 Tight junctions regulate paracellular permeability whereas adherence junctions are principally responsible for mechanical adhesion. While tight and adherens junctions in epithelial cells are spatially distinct, these junctions in endothelial cells are frequently intermingled.5
Membrane proteins that form the core structure of tight junctions are from the claudin family of proteins, in which claudin-5 is specific to endothelial cells.6 Claudins bind to intracellular components, such as the zonula occludens-1 (ZO-1) protein localized at endothelial cell–cell junctions.7 In adherens junctions, vascular endothelial (VE)-cadherin is a major adhesion molecule in endothelial cells.3,8 Similar to other classical cadherins, the cytoplasmic tail of VE-cadherin associates with various intracellular proteins including β-catenin and p120 catenin.3 Furthermore, the connection between adherens junctions and actin filaments mediated by VE-cadherin is believed to be crucial for the regulation of blood vascular endothelial functions, including cellular reactions to various endothelial permeability factors and angiogenic growth factors.9
The lymphatic endothelium has a unique cell–cell junctional organization that is different than blood vascular endothelium.10 Each of the different lymphatic vascular components, such as capillaries, pre-collecting ducts, and collecting ducts, have specialized cellular junctions between their endothelial cells.11 This diversity reflects the dual roles of the lymphatic endothelium in terms of fluid and macromolecule absorption and lymph transport.
To maintain fluid homeostasis, the lymphatic vessels have a two-valve system for unidirectional entry and the movement of fluid and cells.12 In lymphatic capillaries, oak leaf-shaped endothelial cells are connected by discontinuous button-like junctions without mural cells.11 Fluid flows unidirectionally along hydrostatic pressure gradients from the interstitial space to the initial lymphatic ducts via openings between these button-like junctions. This structure is considered as the primary valve. Secondary valves are found in collecting lymphatic vessels intraluminally to ensure the unidirectional flow of lymphatic fluid. The endothelial cells in these collecting lymphatic vessels are elongated and connected by continuous zipper-like junctions that are similar to those in the blood vasculature, covered with a continuous basement membrane and smooth muscle cells.11 This structure prevents leakage of lymph during its transport. Both discontinuous button-like and continuous zipper-like junctions are composed of the same junctional components as other endothelial junctions, including VE-cadherin, claudin-5, ZO-1, and endothelial cell adhesion molecule-1 (PECAM-1; also known as CD31).
Lymphatic cell junctions have a certain degree of plasticity to allow the vessels to grow and remodel during development and repair. In primitive lymphatic endothelium of mice at embryonic day, intercellular junctions are of the continuous zipper-like type. However, in tracheal initial lymphatics, the number of these zipper-like junctions decreases rapidly just before birth, followed by an increase in the proportion of button-like junctions during postnatal development.2 This junctional transformation coincides with birth, and is considered to be required for the efficient clearance of fluid from the lungs after the onset of breathing. In contrast, during periods of airway inflammation from Mycoplasma pulmonis infection of the respiratory tract, zipper-like junctions replace button-like junctions in airway lymphatics; this substitution can be reversed by the use of dexamethasone.2 These results indicate the dynamics of intercellular junctions under physiological and pathological situations.
After the isolation of human dermal lymphatic endothelial cells (HDLEC) by Nguyen et al.,13 cell biological studies using cultured human lymphatic endothelial cells have been conducted.14–17 In contrast to blood endothelium, a detailed description of endothelial junctional complex in cultured lymphatic endothelial cells is still insufficient. The primary purpose of the current study was to characterize cell junctions in HDLEC. Numerous studies have reported that alterations to cellular junctions during cytoskeletal remodeling occur in response to various cytokine stimuli in blood vascular endothelial cells. Some vasoactive agents, such as histamine or thrombin, act very rapidly, and the effect is quickly reversible once these stimuli are removed.9 By contrast, inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), induce long-term changes to cellular junctions and require several hours to increase permeability in human umbilical vein endothelial cells (HUVEC).18
As a second part of this study, we sought to investigate how endothelium is altered following TNF-α stimulation using a detailed assessment of the changes to cellular junctions and by measuring transendothelial electrical resistance (TER). We describe the characteristic distribution of VE-cadherin at the cell periphery, in addition to the presence of continuous and discontinuous junctions in HDLEC. We also show that TNF-α treatment alters cell junctions, causing a disturbance in barrier function. This study contributes to the characterization of cell junctions in cultured lymphatic endothelial cells and the elucidation of change of lymphatic intercellular junctions under inflammation.
Materials and Methods
Cell culture
Adult human dermal lymphatic endothelial cells (HDLEC) were obtained from ScienCell Research Laboratories (Carlsbad, CA). HDLEC were routinely maintained on fibronectin-coated cell cultureware surfaces (Sekiyarika, Tokyo, Japan) in Endothelial Cell Medium (ScienCell Research Laboratories) supplemented with co-delivered mixed additives that resulted in a final concentration of 1% ECGS (endothelial cells growth supplement), 5% fetal bovine serum, and 1% of penicillin/streptomycin. Cells used in the experiments were between passages 2 to 10 counting from the stage of primary culture, for which the stable expression of investigated proteins had been confirmed.
Reagents and antibodies
Goat anti-VE-cadherin antibody (C-19) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-human CD31 (JC/70A) monoclonal antibody was purchased from Covance Inc. (Princeton, NJ). Mouse anti-β-catenin monoclonal antibody was purchased from BD Transduction Laboratory (San Jose, CA). Mouse anti-ZO-1 monoclonal antibody (T8-754) was characterized as described previously.19 Actin filaments were labeled with Alexa 568-Phalloidin (Invitrogen Molecular Probes, Karlsruhe, Germany). Secondary antibodies conjugated with Alexa 488, Cy3, and Cy5 were purchased from Invitrogen and Jackson Laboratories (Sacramento, CA). Recombinant human TNF-α was purchased from R&D Systems (Minneapolis, MN).
Immunofluorescence staining and microscopy
For immunofluorescence microscopy, cells were grown on micro cover glass that was coated with fibronectin stock solution (Sigma-Aldrich, St Louis, MI) in 35×10 mm-style polystyrene tissue culture dishes. Cells were fixed with 1% formaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature, treated with 0.2% Triton X-100 in PBS for 5 min, and washed with PBS. The cells were blocked with 1% bovine serum albumin in PBS overnight and then incubated with primary antibodies overnight. They were then rinsed three times with PBS and incubated with appropriate secondary antibodies for 30 min. After rinsing with PBS, the specimens were embedded in FluorSave (Calbiochem, Darmstadt, Germany) and observed with an IX71 fluorescence microscope (Olympus, Hamburg, Germany). Image acquisition was performed using a combined ORCA-ER cooled CCD camera (Hamamatsu Photonics K.K., Shizuoka, Japan). For the TNF-α experiment, immunofluorescence staining was performed 24 h after the addition of 10 ng/mL TNF-α to confluent HDLEC.
Measurement of TER
Aliquots containing 1×105 cells/cm2 were plated onto Transwell filters (12 mm in diameter; six filters for each cell line) and cultured in fresh medium for 3 days until a confluent layer was formed. TER measurements were made with Millicell-ERS (electrical resistance meter; Millipore, Schwalbach, Germany) just before and after the addition of 10 ng/mL TNF-α at given time points (12, 24, 36, and 48 h). This concentration of TNF-α was as previously reported.20 Resistance (delta TER) of HDLEC layers under the different conditions was calculated as the mean of at least four replicates from which the mean resistance of control inserts (without cells, four replicates) was subtracted. To compare independent experiments, normalized delta TER values were calculated in relation to the delta TER measured in low serum medium just before the addition of effectors.
Quantification of cell number and VE-cadherin-positive broad area
Cells were immunostained after TNF-α stimulation for 24 h. For quantification of the VE-cadherin-positive broad area, which is described in detail in the Results, the number of cells involved the cell periphery in which only the broad distribution of VE-cadherin could be observed and ZO-1 expression was absent was counted in five, randomly selected, independent microscopic images (20×). The results are representative of two independent experiments. Data were processed and statistical significance determined using Student's t-test with SPSS ver. 15.0 (SPSS, Chicago).
Western blot analysis
Cells were lysed with lysis buffer containing 0.5 M Tris-HCl (pH 6.8), 10% SDS, and glycerol; 1 M dithiothreitol was added to cell lysate before loading. Equal amounts of protein were loaded for SDS-PAGE in 10% acrylamide gels. Mobility was assessed using Dual Color Standards (Bio-Rad, Hercules, CA). All Western blotting with goat anti-VE-cadherin antibody (C-19) and mouse anti-actin monoclonal antibody (Chemicon, Temecula, CA) followed with by appropriate HRP-conjugated secondary antibodies was performed on Immnobilon-P PVDF membrane (Millipore, Schwalbach, Germany), which were developed using an enhanced chemiluminescence system (GE Healthcare, Buckinghamshire, England). Blots were scanned with a LAS-3000 mini imaging system (Fujifilm, Tokyo, Japan).
Results
As an initial step toward the characterization of endothelial cell junctions in HDLEC, we first confirmed the expressions of two lymphatic markers, VEGFR-3 and podoplanin, and three adherens junction markers, VE-cadherin, β-catenin and PECAM-1, and a tight junction marker, ZO-1. The expressions of the lymphatic markers were detected in HDLEC (data not shown). VE-cadherin and β-catenin were completely co-localized in HDLEC, by which we considered that the localization of VE-cadherin could reflect the one of β-catenin (Supplementary Fig. S1; supplementary material is available online at www.liebertpub.com). ZO-1, which is a tight junction-associated cytoplasmic plaque protein, was an excellent marker for endothelial cell junctions in HDLEC, as shown in Figure 1. The expression of ZO-1 revealed that two types of cell junctions in HDLEC could be distinguished: continuous linear junctions and discontinuous zigzag junctions showing a perpendicular orientation of ZO-1 toward cell–cell contacts (Fig. 1). In both the continuous and discontinuous junctions, ZO-1 and VE-cadherin were co-localized. Although VE-cadherin expression was also detected broadly near the cell periphery (Fig. 1), ZO-1 expression was absent in those areas.
FIG. 1.
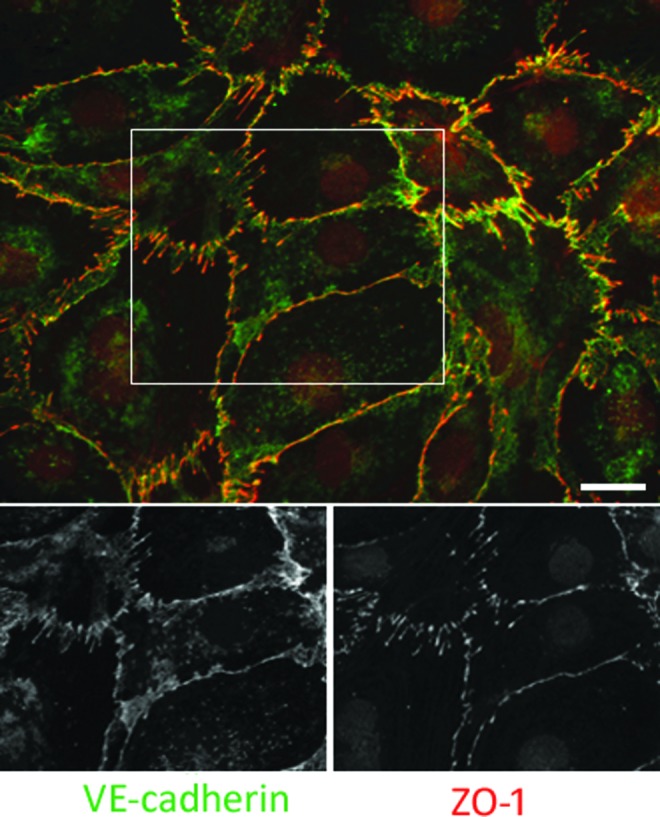
Double immunofluorescence staining for VE-cadherin and ZO-1. Cell junctions of HDLEC involved two distinct types: continuous linear junctions and discontinuous zigzag junctions. Although VE-cadherin (green) and ZO-1 (red) were co-localized in both cell junctions, the ZO-1 expression was absent in the VE-cadherin broadly distributing areas near the cell periphery and at the cell–cell contacts. Bar=20 μm. A color version of this figure is available in the online article at www.liebertpub.com/lrb
Like VE-cadherin, PECAM-1 is a major component of endothelial adherens junction, but with functions distinct to that of VE-cadherin. We next performed double immunofluorescence staining for VE-cadherin and PECAM-1. While PECAM-1 was co-localized with VE-cadherin in the continuous linear junctions (Fig. 2B and B′), the expression level of PECAM-1 was weak in some parts of the discontinuous junctions (Fig. 2C and C′). The broad expression of PECAM-1 was also detected with VE-cadherin near the cell periphery, and the fluorescence intensity of PECAM-1 in those areas was relatively higher than that for VE-cadherin (Fig. 2D and D′). These results indicate that cell junctions in HDLEC involve two distinct types (continuous and discontinuous) even at a confluent state, as recognized by the co-distribution of VE-cadherin and ZO-1. The areas in which VE-cadherin and ZO-1 do not co-localize can be recognized through the intense expression of PECAM-1. Herein we term this region the “VE-cadherin-positive broad area.”
FIG. 2.
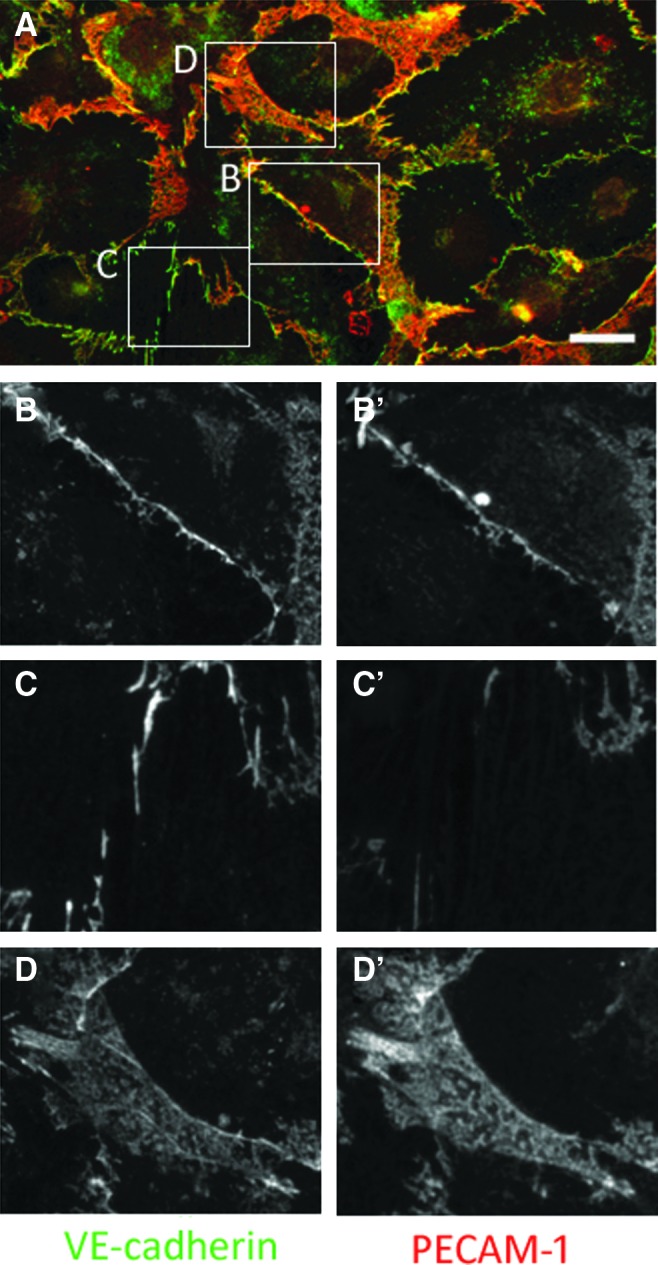
Double immunofluorescence staining for VE-cadherin and PECAM-1 in HDLEC. (A) Merged image. PECAM-1 (red) did not completely co-localize with VE-cadherin (green). Bar=20 μm. Enlarged images of continuous (B, B′) and discontinuous (C, C′) junctions. While VE-cadherin and PECAM-1 were co-localized in continuous linear junctions (B, B′), PECAM-1 was absent in the VE-cadherin-positive discontinuous zigzag junctions (C, C′). (D) and (D′) show the ‘VE-cadherin-positive broad areas’ at the cell periphery. In those areas, the fluorescence intensity of PECAM-1 was relatively higher than that for VE-cadherin. A color version of this figure is available in the online article at www.liebertpub.com/lrb
We next sought to investigate alterations to cell junctional distribution during pathological states using TNF-α stimulation. We performed triple immunofluorescence staining for VE-cadherin, ZO-1 (cell junction marker), and F-actin, a major component of the cytoskeleton. In control cells, continuous junctions were aligned by the circular actin bundles expanding toward the cell periphery (Fig. 3). This pattern of expression is similar but not completely the same as that seen during the middle stage of epithelial cell polarization,21 as the circumferential F-actin ring does not overlap with VE-cadherin (Fig. 3). In control cells, the assembly of stellar actin fibers radiating from several foci was also observed in spots (Fig. 3). Totsukawa et al. previously described the assembly of stellar actin stress fibers in the center of fibroblasts caused by an elevated phosphorylation of light chain of myosin, a motor protein responsible for actin-based cell motility.22 In the discontinuous junctions of control cells, zipper-like discontinuous VE-cadherin signals were attached to the radial actin bundles originating from adjacent cells at cell–cell contacts (Fig. 3). In stimulated cells, TNF-α induced endothelial cell elongation and the cells were aligned in parallel with actin stress fibers. Furthermore, in TNF-α-treated HDLEC, the discontinuous junctions that showed some co-localization with the tips of these elongated actin bundles were more dominant than those in the continuous linear junctions (Fig. 3). Moreover, TNF-α treatment reduced the extent of the VE-cadherin-positive broad area.
FIG. 3.
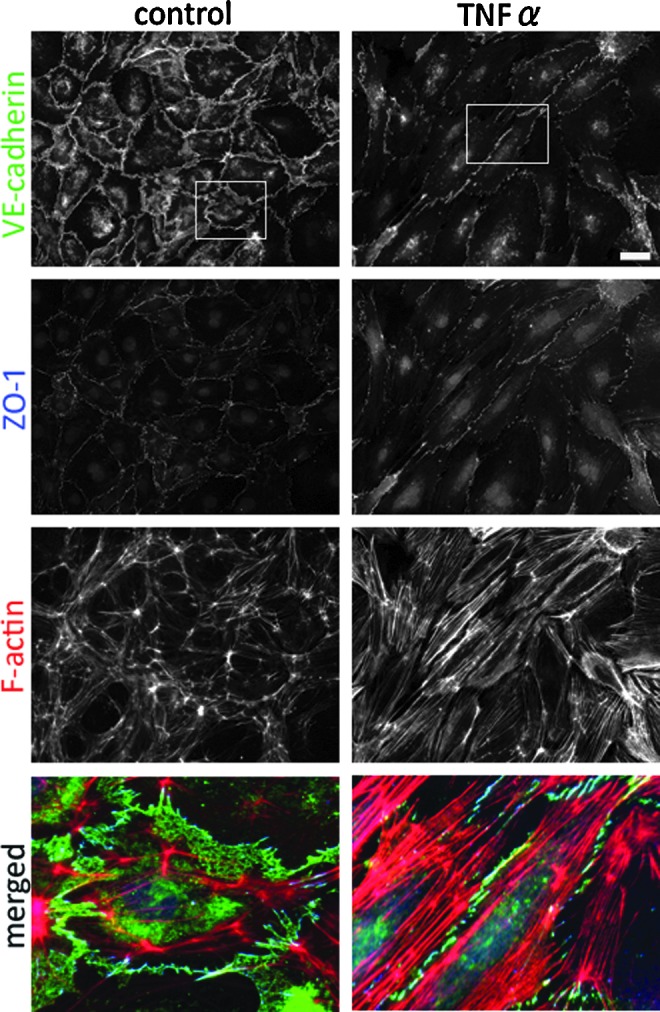
Triple immunofluorescence staining for VE-cadherin (green), ZO-1 (blue), and F-actin (red) in control (left) and TNF-α treated HDLEC (right). Bar=40 μm. Enlarged images (below) show the alterations to cell junctions and the cytoskeleton. Right panels demonstrate the induction of discontinuous junctions and the reduction in the VE-cadherin-positive broad areas by TNF-α. A color version of this figure is available in the online article at www.liebertpub.com/lrb
Finally, we investigated TER as an index of barrier function in HDLEC. TNF-α caused a steady decline in TER (Fig. 4A)and significantly reduced the extent of the VE-cadherin-positive broad area (P<0.05) in HDLEC (Fig. 4B). We further determined the change of the protein level of VE-cadherin in TNF-α-treated HDLEC. There were no significant differences between control and TNF-α-treated cells (Supplementary Fig. S2).
FIG. 4.

(A) Time-dependent transendothelial electrical resistance (TER) reduction by TNF-α. (B) A strong reduction of cells involved VE-cadherin-positive broad areas at the cell periphery in HDLEC treated with 10 ng/mL TNF-α stimulation for 24 h (p<0.01). Control (n=5 images) and TNF-α treated HDLEC (n=5 images). The results are representative of two independent experiments. Values are the mean±SEM (error bars). P values were calculated with a two-tailed Student's t test.
Discussion
Within the current literature, there are fewer cell biology studies using cultured lymphatic endothelial cells than those reporting on blood vascular endothelial cells. Although previous reports have focused on barrier function or on the tubule formation ability of cultured lymphatic endothelial cells,14,15,23 the detailed descriptions about cell junctions are insufficient. The primary purpose of this study was to characterize cell junctions in cultured lymphatic endothelial cells. Our findings revealed the heterogeneity of cell junctions in HDLEC, which could be distinguished into two distinct types (continuous and discontinuous). Continuous linear junctions were similar to stable and mature endothelial cell junctions, as previously reported.20 In contrast, discontinuous junctions seemed to resemble remodeling junctions previously reported in epithelial, endothelial cells and fibroblasts by three other groups: Yonemura et al.21 referred to them as ‘spot-like adherens junctions;’ Vasioukhin et al.24 referred to them as ‘zipper-like junctions’; and Millán et al.,25 also using HUVEC, referred to them as ‘discontinuous adherens junctions.’ Additionally, Hordijk et al. reported that VE-cadherin was distributed in a striped or ‘jagged’ fashion because of its co-localization with the endpoints of actin stress fibers, which terminated at the site of cell–cell contact in HUVEC.26 We found that continuous and discontinuous junctions were intermingled, even at confluent states in HDLEC; this was different to that observed with cultured epithelial cells, where tight and adherens junctions mature and become stabilized and polarized, with separation of the junctions upon reaching a confluent state.21
We found a characteristic expression of VE-cadherin near the cell periphery, which we referred to as “VE-cadherin-positive broad area.” A similar broad localization of VE-cadherin at cell–cell contacts was previously reported by Birukova et al.27 In their study, human pulmonary artery endothelial cells were treated with iloprost, a stable prostacyclin analog, which has been shown to elevate intracellular cAMP concentrations and has been implicated in endothelial barrier enhancement and acceleration of endothelial monolayer recovery after stimulation with edemagenic agonists.28–30 They showed that iloprost prominently increased the VE-cadherin-positive areas near the regions of the cell–cell interface, seemingly to tighten the endothelial monolayer and to enhance the endothelial barrier.27 From this, we can assume that VE-cadherin-positive areas near cell–cell contacts are related to endothelial barrier integrity.
A previous important in vivo study by Baluk et al.11 reported the presence of two types of junctions between endothelial cells of lymphatics: continuous zipper-like junctions and discontinuous button-like junctions. Continuous zipper-like junctions in collecting lymphatics are similar to those in blood vessels. However, the overlapping flaps at the borders of endothelial cells of initial lymphatics lack the junctions at the tips but are anchored on the sides of cells by discontinuous button-like junctions. Button-like junctions are considered to be functionally specialized junctions that allow fluid entry. Both types of junctions are composed of the cell–cell junctional proteins VE-cadherin, ZO-1, and PECAM-1.
VE-cadherin is essential in blood vascular structure assembly, as VE-cadherin-null mice are embryonically lethal owing to an extensive angiogenic defect.31 By comparison, PECAM-1 null mice are viable and have a normal vasculature.32 PECAM-1 is considered to contribute to the maintenance of vascular endothelial integrity during pathological states, because PECAM-1-null mice have decreased leukocyte transmigration, increased bleeding times and vascular leakages,33,34 and greater susceptibility to endotoxic shock.35 β-Catenin is also essential for the endothelial integrity,36 and the stabilization of cadherins is known to be dependent on β-catenin.37 VE-cadherin and β-catenin co-localized in HDLEC, while the spatial and temporal appearance of VE-cadherin and PECAM-1 are different. Spatial analysis using HUVEC has revealed that VE-cadherin is located at adjacent (usually more apical) yet clearly distinct domains of the lateral membrane to PECAM-1.38 Temporal analysis revealed that VE-cadherin is first organized into adherens junctions, while PECAM-1 becomes associated with surface adhesions significantly later, with its progressive association with cadherin-containing adhesions in HUVEC.38 Moreover, PECAM-1 does not co-localize with actin stress fibers, but consistently localizes to a broad area between the cells.37
PECAM-1 is known to be expressed in lymphatic endothelial cells from immunohistochemical observation in vivo40 and in culture.41 While the importance of VE-cadherin in the organization of specialized junctions in lymphatic endothelial cells is well established, the role of PECAM-1 in lymphatic endothelial cells is still unclear.11 Initial lymphatics are composed of oak-leaf shaped endothelial cells, and flaps of adjacent cells have complementary shapes with overlapping edges. VE-cadherin and ZO-1 are restricted to button-like junctions at the sides of cells. In contrast, PECAM-1 is distributed across the entire tip of the flaps. Taken together, the broad distribution of PECAM-1 near cell–cell contacts in HDLEC may reflect the properties of initial lymphatics, because HDLEC are derived from human dermal lymphatic capillaries. It is unclear whether the VE-cadherin-positive broad area near the cell periphery described in our study is the same as the VE-cadherin-positive areas near the cell–cell interface reported by Birukova et al.27 Our results show that the expression level of PECAM-1 is more intense than that of VE-cadherin in the VE-cadherin-positive broad area (Fig. 2A). We consider that the broad distribution of VE-cadherin at the cell periphery in HDLEC represents the overlapping areas between adjacent cells. Normal HDLEC are probably able to overlap at cell–cell contacts in addition to forming continuous and discontinuous cell junctions. Previous reports indicate that the localization of PECAM-1 to a broad area between the cells represents the overlapping cell–cell contacts in blood endothelial cells.39 Therefore, blood vascular endothelial cells certainly have the ability to overlap with other blood vascular endothelial cells. Previous in vivo studies noted the specialty of cell junctions in initial lymphatics, which comprises overlapping flaps at cell borders, anchored by discontinuous button-like junctions. We conclude that HDLEC have the features to cause an overlap between adjacent cells at the cell periphery, forming cell–cell junctions.
We show that TNF-α altered the cell junctions and disputed barrier function in HDLEC (Fig. 4). Although it has been previously reported that <50 ng/mL TNF-α is not significantly cytotoxic in epithelial cells,42 TNF-α has been shown to reduce proliferation of lymphatic endothelial cells at concentrations as low as 5 ng/mL.43–45 We treated cells with 10 ng/mL TNF-α for 24 hours. In a previous report using bovine pulmonary artery endothelial cells, TNF-α induced apoptosis in a dose-dependent fashion.46 A previous in vivo study revealed that lymphatic vessels were markedly enlarged and leaky in inflamed skin.47 We found that TNF-α caused a switch toward an increase in the presence of discontinuous junctions and reduced the VE-cadherin-positive broad area, which affects the overlapping edges between adjacent cells. TNF-α also caused an enlargement of cells. Finally, the steady decline in TER in TNF-α-treated HDLEC was probably caused by these alterations to cell junctions, including the dominance of discontinuous cell junctions, the reduction in cell–cell overlapping areas.
Yao et al. reported that zipper-like junctions replace button-like junctions in sustained inflammation, and the reversal by steroid of the zipper-to-button conversion could improve fluid clearance and contribute and mucosal edema.2 In addition to their findings, we indicate the possibility that the cell–cell overlapping areas may reflect the permeability of HDLEC. Because TNF-α stimulation almost diminished the VE-cadherin positive areas in our study, and in vascular endothelial cells, VE-cadherin-positive areas near the regions of the cell–cell interface may be related to endothelial barrier integrity.27 Our hypothesis is that lymphatic endothelial cell junctions transform zipper-like to button-like concurrent with reduction of the cell–cell overlapping areas by TNF-α for fluid clearance under inflammation. However, the confirmation of this hypothesis requires further in vivo study. In contrast to results of immunostaining, Western blot analysis in HDLEC whole cell lysate revealed that the total amounts of VE-cadherin did not alter in TNF-α treated HDLEC in agreement with previous study.48 This might indicate that TNF-α stimulation altered distribution of VE-cadherin to the cell surface and actin cytoskeletal remodeling but not the total amount of protein.
In conclusion, cell junctions of HDLEC are heterogeneous and involve continuous and discontinuous junctions, even at a confluent state. In continuous and discontinuous junctions in HDLEC, the tight junction marker, ZO-1, and the adherens junction marker, VE-cadherin, are co-localized. Under inflammation conditions, the discontinuous junctions without cell–cell overlapping area become dominant, increasing paracellular permeability.
Supplementary Material
Acknowledgments
The authors thank Dr M. Itoh for the T8-754 antibody, and Dr M. Furuse, Dr T. Higashi, and Dr S. Tokuda for helpful discussion. This work was supported by Japan Society for the Promotion of Science (JSPS), KAKENHI, Grant-in-Aid for Young Scientists (B), (Grant Number: 23792340).
Author Disclosure Statement
The authors have no conflicts of interest or financial ties to disclose.
References
- 1.Schulte-Merker S, Sabine A, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol 2011;193:607–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol 2012;180:2561–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev Cell 2009;16:209–221 [DOI] [PubMed] [Google Scholar]
- 4.Dejana E. Endothelial cell–cell junctions: Happy together. Nat Rev Mol Cell Biol 2004;5:261–270 [DOI] [PubMed] [Google Scholar]
- 5.Leach L, Clark P, Lampugnani MG, Arroyo AG, Dejana E, Firth JA. Immunoelectron characterisation of the inter-endothelial junctions of human term placenta. J Cell Sci 1993;104:1073–1081 [DOI] [PubMed] [Google Scholar]
- 6.Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 1999;147:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 1999;147:1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vestweber D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol 2008;28:223–232 [DOI] [PubMed] [Google Scholar]
- 9.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 2008;121:2115–2122 [DOI] [PubMed] [Google Scholar]
- 10.Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res 2009;335:17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baluk P, Fuxe J, Hashizume H, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 2007;204:2349–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmid-Schönbein GW. Microlymphatics and lymph flow. Physiol Rev 1990;70:987–1028 [DOI] [PubMed] [Google Scholar]
- 13.Nguyen VA, Fürhapter C, Obexer P, Stössel H, Romani N, Sepp N. Endothelial cells from cord blood CD133+CD34+ progenitors share phenotypic, functional and gene expression profile similarities with lymphatics. J Cell Mol Med 2009;13:522–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breslin JW, Yuan SY, Wu MH. VEGF-C alters barrier function of cultured lymphatic endothelial cells through a VEGFR-3-dependent mechanism. Lymphat Res Biol 2007;5:105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price GM, Chrobak KM, Tien J. Effect of cyclic AMP on barrier function of human lymphatic microvascular tubes. Microvasc Res 2008;76:46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammer T, Tritsaris K, Hübschmann MV, Gibson J, Nisato RE, Pepper MS, Dissing S. IL-20 activates human lymphatic endothelial cells causing cell signalling and tube formation. Microvasc Res 2009;78:25–32 [DOI] [PubMed] [Google Scholar]
- 17.Safuan S, Storr SJ, Patel PM, Martin SG. A comparative study of adhesion of melanoma and breast cancer cells to blood and lymphatic endothelium. Lymphat Res Biol 2012;10:173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenzie JA, Ridley AJ. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol 2007;213:221–228 [DOI] [PubMed] [Google Scholar]
- 19.Itoh M, Yonemura S, Nagafuchi A, Tsukita S, Tsukita S. A 220-kD undercoat-constitutive protein: Its specific localization at cadherin-based cell–cell adhesion sites. J Cell Biol 1991;115:1449–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huveneers S, Oldenburg J, Spanjaard E, et al. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol 2012;196:641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yonemura S, Itoh M, Nagafuchi A, Tsukita S. Cell-to-cell adherens junction formation and actin filament organization: Similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J Cell Sci 1995;108:127–142 [DOI] [PubMed] [Google Scholar]
- 22.Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol 2000;150:797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrafa E, Caprara V, Di Castro V, Rosanò L, Bagnato A, Spinella F. Endothelin-1 cooperates with hypoxia to induce vascular-like structures through vascular endothelial growth factor-C, -D and -A in lymphatic endothelial cells. Life Sci 2012;91:638–643 [DOI] [PubMed] [Google Scholar]
- 24.Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell 2000;100:209–219 [DOI] [PubMed] [Google Scholar]
- 25.Millán J, Cain RJ, Reglero-Real N, et al. Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol 2010;8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hordijk PL, Anthony E, Mul FP, Rientsma R, Oomen LC, Roos D. Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J Cell Sci 1999;112:1915–1923 [DOI] [PubMed] [Google Scholar]
- 27.Birukova AA, Tian Y, Dubrovskyi O, et al. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J Cell Physiol 2012;227:3405–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukuhara S, Sakurai A, Sano H, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell–cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol 2005;25:136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wittchen ES, Worthylake RA, Kelly P, Casey PJ, Quilliam LA, Burridge K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem 2005;280:11675–11682 [DOI] [PubMed] [Google Scholar]
- 30.Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res 2007;313:2504–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carmeliet P, Lampugnani MG, Moons L, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999;98:147–157 [DOI] [PubMed] [Google Scholar]
- 32.Duncan GS, Andrew DP, Takimoto H, et al. Genetic evidence for functional redundancy of platelet/endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol 1999;162:3022–3030 [PubMed] [Google Scholar]
- 33.Mahooti S, Graesser D, Patil S, Newman P, Duncan G, Mak T, Madri JA. PECAM-1 (CD31) expression modulates bleeding time in vivo. Am J Pathol 2000;157:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graesser D, Solowiej A, Bruckner M, et al. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest 2002;109:383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrithers M, Tandon S, Canosa S, Michaud M, Graesser D, Madri JA. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am J Pathol 2005;166:185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cattelino A, Liebner S, Gallini R, et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J Cell Biol 2003;162:1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukunaga Y, Liu H, Shimizu M, Komiya S, Kawasuji M, Nagafuchi A. Defining the roles of beta-catenin and plakoglobin in cell–cell adhesion: Isolation of beta-catenin/plakoglobin-deficient F9 cells. Cell Struct Funct 2005;30:25–34 [DOI] [PubMed] [Google Scholar]
- 38.Ayalon O, Sabanai H, Lampugnani MG, Dejana E, Geiger B. Spatial and temporal relationships between cadherins and PECAM-1 in cell–cell junctions of human endothelial cells. J Cell Biol 1994;126:247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hordijk PL, Anthony E, Mul FP, Rientsma R, Oomen LC, Roos D. Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J Cell Sci 1999;112:1915–1923 [DOI] [PubMed] [Google Scholar]
- 40.Baluk P, Tammela T, Ator E, et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest 2005;115:247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med 2006;203:2763–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shirasawa M, Sonoda S, Terasaki H, et al. TNF-α disrupts morphologic and functional barrier properties of polarized retinal pigment epithelium. Exp Eye Res 2013;110:59–69 [DOI] [PubMed] [Google Scholar]
- 43.Chaitanya GV, Franks SE, Cromer W, et al. Differential cytokine responses in human and mouse lymphatic endothelial cells to cytokines in vitro. Lymphat Res Biol 2010;8:155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wells SR, Jennings MH, Rome C, Hadjivassiliou V, Papas KA, Alexander JS. Alpha-, gamma- and delta-tocopherols reduce inflammatory angiogenesis in human microvascular endothelial cells. J Nutr Biochem 2010;21:589–597 [DOI] [PubMed] [Google Scholar]
- 45.Polzer K, Baeten D, Soleiman A, Distler J, Gerlag DM, Tak PP, Schett G, Zwerina J. Tumour necrosis factor blockade increases lymphangiogenesis in murine and human arthritic joints. Ann Rheum Dis 2008;67:1610–1616 [DOI] [PubMed] [Google Scholar]
- 46.Polunovsky VA, Wendt CH, Ingbar DH, Peterson MS, Bitterman PB. Induction of endothelial cell apoptosis by TNF alpha: Modulation by inhibitors of protein synthesis. Exp Cell Res 1994;214:584–594 [DOI] [PubMed] [Google Scholar]
- 47.Kajiya K, Kidoya H, Sawane M, Matsumoto-Okazaki Y, Yamanishi H, Furuse M, Takakura N. Promotion of lymphatic integrity by angiopoietin-1/Tie2 signaling during inflammation. Am J Pathol 2012;180:1273–1282 [DOI] [PubMed] [Google Scholar]
- 48.Rival Y, Del Maschio A, Rabiet MJ, Dejana E, Duperray A. Inhibition of platelet endothelial cell adhesion molecule-1 synthesis and leukocyte transmigration in endothelial cells by the combined action of TNF-alpha and IFN-gamma. J Immunol 1996;157:1233–1241 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.