

Abstract
Helicobacter pylori incites a futile inflammatory response, which is the key feature of its immunopathogenesis. This leads to the ability of this bacterial pathogen to survive in the stomach and cause peptic ulcers and gastric cancer. Myeloid cells recruited to the gastric mucosa during Helicobacter pylori infection have been directly implicated in the modulation of host defense against the bacterium and gastric inflammation. Heme oxygenase-1 (HO-1) is an inducible enzyme that exhibits anti-inflammatory functions. Our aim was to analyze the induction and role of HO-1 in macrophages during H. pylori infection. We now show that phosphorylation of the H. pylori virulence factor cytotoxin associated gene A (CagA) in macrophages results in expression of hmox-1, the gene encoding HO-1, through p38/nuclear factor (erythroid-derived 2)-like 2 signaling. Blocking phagocytosis prevented CagA phosphorylation and HO-1 induction. The expression of HO-1 was also increased in gastric mononuclear cells of human patients and macrophages of mice infected with cagA+ H. pylori strains. Genetic ablation of hmox-1 in H. pylori-infected mice increased histologic gastritis, which was associated with enhanced M1/Th1/Th17 responses, decreased Mreg response, and reduced H. pylori colonization. Gastric macrophages of H. pylori-infected mice and macrophages infected in vitro with this bacterium showed an M1/Mreg mixed polarization type; deletion of hmox-1 or inhibition of HO-1 in macrophages caused an increased M1 and a decreased of Mreg phenotype. These data highlight a mechanism by which H. pylori impairs the immune response and favors its own survival via activation of macrophage HO-1.
Introduction
Helicobacter pylori infects half of the world's population and is the causative agent of chronic gastritis, peptic ulcer disease and gastric mucosa-associated lymphoid tissue lymphoma. Long-term infection is a major risk factor for the development of gastric cancer, the second leading cause of cancer deaths worldwide. H. pylori expresses several virulence factors that impact disease outcome. Most of the H. pylori strains that provoke neoplastic transformation possess the cytotoxin-associated gene (cag) pathogenicity island (1), which carries genes encoding a type 4 secretion system (T4SS) and the virulence factor CagA (2, 3). When injected into the cytoplasm of gastric epithelial cells (3), CagA is sequentially phosphorylated on tyrosine residues by c-Src and Abl kinases (4) and then causes signaling events in host cells (5-7).
Besides this interaction with gastric epithelial cells, H. pylori has an impact on the recruitment and differentiation of lymphoid cells in the gastric mucosa. Thus, H. pylori infection results in a mixed Th1/Th17-dominant T cell response, which contributes to the establishment of chronic gastritis (8, 9). It has been also demonstrated that the H. pylori-induced Treg response plays a role in failure of specific immunity, thus favoring the persistence of the bacterium in its ecological niche (10). Moreover, H. pylori interacts with myeloid cells either directly, when bacteria cross the epithelial barrier and reach the lamina propria (11), or indirectly, through the release of bacterial products (12).
Macrophages play an essential role in host defense against bacterial infection and in the regulation of inflammatory processes, including during H. pylori infection (13). In response to various signals from the extracellular milieu, macrophages can be polarized into different populations of activated cells exhibiting different phenotype, receptor, and cytokine secretion patterns (14). Classically activated macrophages, also called M1 macrophages, interact with Th1 lymphocytes and exhibit microbicidal activity by producing oxygen radicals and NO, the latter through enhanced expression of inducible NO synthase (iNOS). In contrast, IL-4-stimulated wound-healing macrophages (M2 cells) contribute to the production of the extracellular matrix and exhibit indirect regulatory effects on the immune response. Regulatory macrophages (Mreg, also called type II-activated macrophages) synthesize high levels of IL-10 that limits inflammation, but predisposes the host to infections (15). It has been shown that gastric macrophages show features of the M1 profile during H. pylori infection (16). Nonetheless, we have found that gastric macrophages from H. pylori-infected mice exhibit activation of the arginase/ornithine decarboxylase metabolic pathway, a functional feature of M2 macrophages (17, 18), and an increase of M2 markers has been evidenced in the gastric mucosa from infected patients (19). Moreover, studies have associated macrophage production of IL-10, the typical Mreg cytokine, with infection by H. pylori (16, 19). Together, these data suggest that macrophage polarization during H. pylori infection is not a canonical process and results in a phenotypically mixed population of cells.
The direct effect of H. pylori on the molecular/cellular events that orchestrate macrophage polarization remains unknown. In the present work, we show that H. pylori induces macrophage hmox-1, the gene encoding heme oxygenase-1 (HO-1), a potent anti-inflammatory and antioxidant enzyme (20). This occurs by signaling events requiring CagA phosphorylation and the activation of p38 and nuclear factor (erythroid-derived 2)-like 2 (NRF-2). The activity of HO-1 in H. pylori-infected macrophages results in a switch of polarization toward a reduction of the M1 population and an increase of the Mreg profile, leading to a failure of innate and adaptive immune responses.
Materials and Methods
Reagents
The HO-1 inhibitor chromium mesoporphyrin (CrMP) was obtained from Frontier Scientific. The AP-1 inhibitor SR11302 (10 μM) was purchased from Santa Cruz Biotechnology. The following pharmacological compounds were obtained from Calbiochem: the NF-κB inhibitor Bay 11-7082 ((E)3-[(4-methylphenyl)sulfonyl]-2-propenenitrile; 5 μM); the ERK1/2 inhibitor ERKi (3-(2-aminoethyl)-5-((4-ethoxyphenyl)methylene)-2,4-thiazolidinedione, HCl; 20 μM); the JNK inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one, 1,9-pyrazoloanthrone; 1 μM); the p38 inhibitor SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole; 2 μM); the PI3K inhibitor LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one; 10 μM); the c-Src inhibitor PP1 (4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo-d-3,4-pyrimidine); and cytochalasin D (10 μM), an inhibitor of actin polymerization.
Human tissues
Biopsies from gastric tissues were obtained from human subjects in Colombia as described (21), under protocols approved by the ethics committees of the local hospitals and of the Universidad del Valle in Cali, Colombia, as well as the Institutional Review Board at Vanderbilt University. The cagA status of H. pylori was determined from these tissues by PCR analysis performed on isolated colonies (21).
Bacteria, animals and infections
We used the cagA+ H. pylori strains 60190, 7.13, PMSS1, and G27. The ureA, cagE, cagA, vacA, and flaA isogenic mutants constructed in the strain 60190 (22, 23), and the strain G27 lacking the CagA phosphorylation domains (cagAEPISA; 24) were also used.
C57BL/6×FVB hmox-1+/− mice were bred to generate wild-type (WT) and hmox-1−/− mice, as described (25, 26); hmox-1+/− breeder mice were provided by Anupam Agarwal (University of Alabama, Birmingham, AL). The genotypes were verified by PCR using primer sets for hmox-1 and neo (Supplemental Table 1). Animals were used under protocol M/05/176 approved by the IACUC at Vanderbilt University. Mice were infected intragastrically 3 times, every two days, with 109 H. pylori PMSS1. Animals were sacrificed after two months. Colonization was assessed by qPCR using H. pylori ureA gene and mouse 18S rRNA primers (Supplemental Table 1) as described (18).
Purification of gastric macrophages
Macrophages were isolated from mouse stomach exactly as described (17, 27).
Cells, infections, and transfection
The murine macrophage cell line RAW 264.7 was maintained in DMEM containing 10% FBS, HEPES and sodium pyruvate. Peritoneal cells from WT or hmox-1−/− mice were collected after intraperitoneal injection of PBS. Cells were counted, plated and macrophages were purified by washing away nonadherent cells after 1 h of incubation. RAW 264.7 cells or peritoneal macrophages were stimulated with H. pylori at a multiplicity of infection of 100. All pharmacological inhibitors of signaling pathways were added 30 min prior to activation.
To determine the levels of adhesion and phagocytosis of H. pylori, RAW 264.7 cells were washed thoroughly five times with PBS after infection, incubated or not for one hour with 200 μg/ml gentamicin, and lysed in 0.1 % saponin for 30 min at 37°C. The number of bacteria in each lysate was determined by counting the CFUs after plating serial dilutions on blood agar plates.
RAW 264.7 cells in Opti-MEM I Reduced Serum Media (Invitrogen) were transfected using Lipofectamine 2000 with 100 nM ON-TARGETplus siRNAs (Dharmacon) directed against hmox-1, nrf-2 or lmnA, or with 100 nM SignalSilence siRNAs (Cell Signaling) directed against murine p38 or erk1. After 6 h, cells were washed, maintained 36 h in serum-containing antibiotic-free medium, and then stimulated.
Immunostaining
Immunohistochemistry was performed on human gastric tissues as described (18, 23) using a rabbit polyclonal anti-human/mouse HO-1 Ab (1:500; StressGen). Slides were reviewed and scored by a gastrointestinal pathologist (M.B.P.) who was blinded to the clinical status of the subjects. The percentage of mononuclear cells staining positively for HO-1 was determined in each patient by counting the cells with moderate or strong intensity staining on antral biopsies. Immunofluorescence for HO-1, iNOS, and F4/80 was performed on murine gastric tissues (18) using the Abs described in Supplemental Table 2.
Luminex assay
Gastric tissues were lysed in CelLytic™ MT Reagent (Sigma) containing the Protease Inhibitor Cocktail (Set III, Calbiochem) and protein concentrations were determined using the BCA Protein Assay (Pierce). Samples were assayed using a magnetic bead-based protein detection assay for IL-17 using a Millipore FlexMap 3D Luminex machine.
Flow cytometry
Immune cells were isolated from the total glandular stomach by enzymatic digestion (27). Cells were stained for HO-1 and for F4/80 using the Abs described in Supplemental Table 2. Stained cells were analyzed with an LSRII flow cytometer (BD Biosciences) and FlowJo software (Tree Star, Inc.).
Analysis of mRNA levels
RNA purification, reverse transcription, and real-time PCR were performed as described (23) using the primers listed in Supplemental Table 1.
Western Blot analysis
RAW 264.7 cells were lysed using RIPA buffer or NE-PER Nuclear Protein Extraction Kit (Pierce) containing the Protease Inhibitor Cocktail (Set III, Calbiochem) and the Phosphatase Inhibitor Cocktail (Set I, Calbiochem). Protein concentrations were determined using the BCA Protein Assay (Pierce). Western blotting was performed using 10 μg of protein per lane. Primary and secondary Abs are listed in Supplemental Table 2. Densitometric analysis of Western blots was performed with ImageJ 1.45s software (rsbweb.nih.gov/ij/).
Statistics
All the data shown represent the mean ± SEM. Student's t test or ANOVA with the Newman-Keuls test were used to determine significant differences between two groups or to analyze significant differences among multiple test groups, respectively. In the case of the staining for HO-1 in human subjects, nonparametric testing was conducted with the Kruskal-Wallis test followed by Dunn's Multiple Comparisons test.
Results
H. pylori stimulates hmox-1 expression in macrophages
There was a significant increase in hmox-1 mRNA in macrophages infected with H. pylori strains 7.13, 60190, or PMSS1 compared to uninfected cells (Figs. 1A). However, the level of hmox-1 mRNA was 5.6 ± 0.7-fold and 4.3 ± 0.9-fold more elevated in macrophages infected with H. pylori 60190 and PMSS1, respectively, than in those stimulated with the strain 7.13 (Fig. 1A). We also demonstrated that hmox-1 mRNA expression was upregulated in peritoneal macrophages isolated from C57BL/6 mice and infected ex vivo with H. pylori 60190 (Fig. 1A). HO-1 protein expression was also rapidly induced in RAW 264.7 cells infected with H. pylori 60190, peaking 6 h post-inoculation (Fig. 1B). Interestingly, we found that H. pylori-induced hmox-1 mRNA expression was significantly inhibited when the bacteria were separated from the macrophages using a 0.22 μm filter support (Fig. 1C). Further, we observed that hmox-1 mRNA expression (Fig. 1D) and the phagocytosis of H. pylori by macrophages (Fig. 1E) were both reduced in infected macrophages treated with cytochalasin D that prevents phagocytosis of H. pylori (28). Lastly, we found that H. pylori 7.13, which induced hmox-1 relatively poorly, was significantly less phagocytized by RAW 264.7 cells than the strains 60190 or PMSS1 (Fig. 1E). It should be noted that there was complete killing of H. pylori when the macrophages cocultured with bacteria in the presence of cytochalasin D were treated with gentamicin (Fig. 1E), validating that these bacteria were extracellular. These results suggest that H. pylori phagocytosis is required to induce HO-1 in macrophages.
Figure 1.
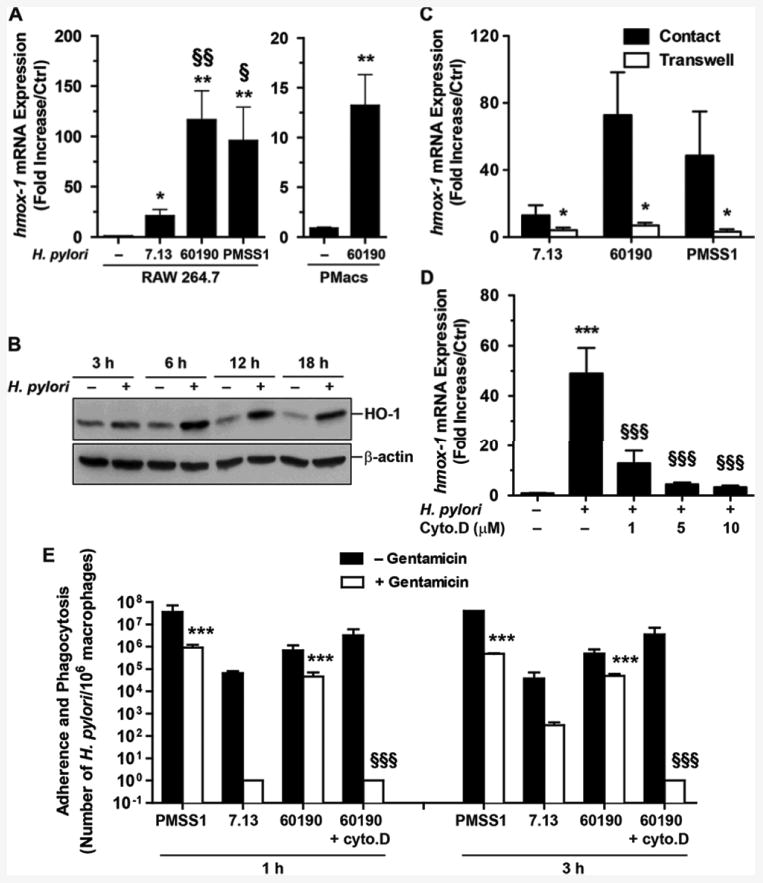
Effect of H. pylori on HO-1 induction in macrophages. A, hmox-1 RNA expression in RAW 264.7 cells and in murine peritoneal macrophages (PMacs) infected for 6 h with H. pylori. *P < 0.05, **P < 0.01 vs. Ctrl; §P < 0.05, §§P < 0.01 vs. cells infected with strain 7.13; n = 5 for RAW 264.7 cells and n = 3 for PMacs. B, Levels of HO-1 in macrophages infected with H. pylori 60190. Data representative of 4 independent experiments. C, Induction of hmox-1 by H. pylori in contact with the cells or separated from macrophages by Transwell filter supports. *P < 0.05 vs. contact. D, Effect of cytochalasin D (Cyto.D) on hmox-1 transcript levels in RAW 264.7 cells. ***P < 0.001 vs. uninfected cells; §§§P < 0.001 vs. cells infected with the strain 60190; n = 3. E, Determination of H. pylori adherence plus phagocytosis (– Gentamicin) and phagocytosis (+ Gentamicin) by macrophages. Gentamicin added to H. pylori without macrophages killed 100% of the bacteria (data not shown). ***P < 0.001 compared to the number of H. pylori 7.13 bacteria phagocytized by RAW 264.7 cells (+ Gentamicin); §§§P < 0.001 vs. the level of phagocytosis of H. pylori 60190; n = 3.
H. pylori-induced HO-1 in macrophages requires CagA phosphorylation
We then assessed which bacterial factor was implicated in hmox-1 expression. There was a significant reduction of hmox-1 mRNA levels in RAW 264.7 cells infected with H. pylori cagA− compared to macrophages infected with the WT strain or with the flaA, cagE, ureA, or vacA mutants (Fig. 2A). This difference between the cagA and cagE mutants suggests that CagA, but not the T4SS, is involved in hmox-1 expression. We then assessed the effect of phospho-CagA (p-CagA) in HO-1 induction. We first observed that CagA was rapidly phosphorylated in infected macrophages (Fig. 2B); importantly, the phosphorylation of CagA was also observed when macrophages were infected with a H. pylori strain with deletion of cagE, thus lacking a functional T4SS; this demonstrates that CagA is phosphorylated in macrophages independently of the T4SS. Moreover, we found that the levels of CagA and p-CagA were greater in macrophages infected with the strains 60190 or PMSS1 than with strain 7.13 (Fig. 2C), which correlated with the level of phagocytosis depicted in Fig. 1E. Further, the levels of intracellular p-CagA and CagA were reduced when macrophages infected with the HO-1-inducing H. pylori strain 60190 were pretreated with cytochalasin D (Fig. 2D), proving that phagocytosis is an essential step for CagA phosphorylation in macrophages. Moreover, the reduction in phosphorylation of CagA when RAW 264.7 cells infected with strain 60190 were pre-treated with the c-Src inhibitor PP1 (Fig. 2E) correlated with a marked attenuation in the expression of hmox-1 (Fig. 2F). Lastly, the hmox-1 gene was significantly less expressed in macrophages stimulated with a cagAEPISA mutant strain than with WT H. pylori (Fig. 2G), demonstrating the involvement of p-CagA in inducible transcription of hmox-1.
Figure 2.
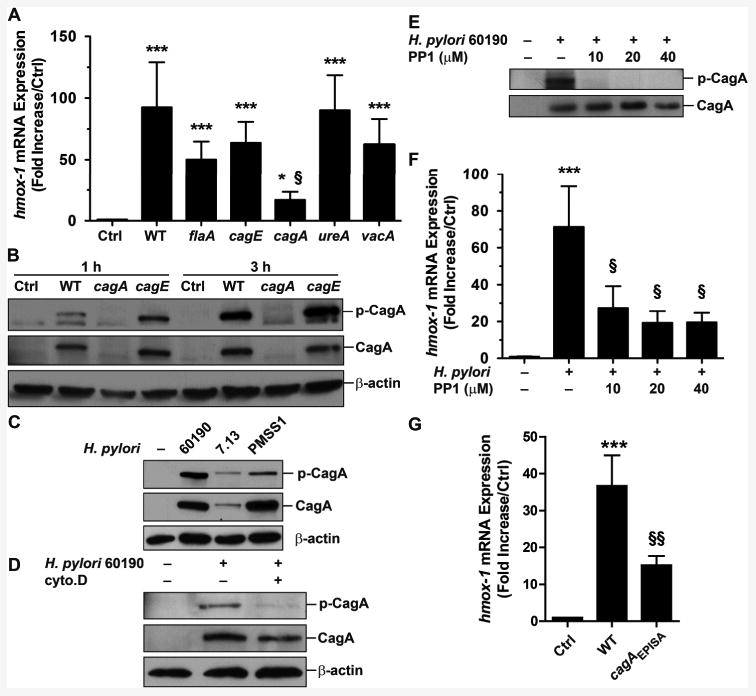
Effect of H. pylori virulence factors on hmox-1 expression. A, RAW 264.7 cells were infected for 6 h with WT H. pylori 60190 or with various isogenic mutants. The expression of hmox-1 was analyzed by real-time PCR. *P < 0.05, ***P < 0.001 vs. Ctrl; §P < 0.05 vs. cells infected with WT; n = 5. B, Analysis of CagA phosphorylation in RAW 264.7 cells infected with H. pylori 60190 or with the cagA or cagE mutant strains. Data representative of 4 independent experiments for each. C, CagA phosphorylation in cells infected with 60190, 7.13, or PMSS1 for 3 h; data are representative of 3 experiments. D, RAW 264.7 cells pretreated with cytochalasin D (Cyto.D) were infected 2 h with H. pylori 60190; after a 1 h gentamicin treatment, CagA delivery and phosphorylation was analyzed. Data representative of 3 independent experiments. E and F, Effect of increasing concentrations of the c-Src inhibitor PP1 on CagA phosphorylation (E) and on hmox-1 expression (F) in macrophages. ***P < 0.001 vs. Ctrl; §P < 0.05 vs. H. pylori-infected macrophages; n = 3. G, hmox-1 RNA expression in RAW 264.7 cells infected for 6 h with H. pylori G27 or the cagAEPISA mutant. ***P < 0.001 vs. Ctrl; §§P < 0.05 vs. macrophages infected with the WT strain; n = 5.
Induction of HO-1 by H. pylori is mediated by p38 and NRF-2
As shown in Fig. 3A, the specific inhibition of p38 by SB203580 resulted in a significant reduction of H. pylori-induced hmox-1 mRNA expression, whereas inhibitors of ERK1/2, JNK, PI3K, NF-κB, or AP-1 had no effect. None of these pharmacologic inhibitors had a significant effect on hmox-1 expression in uninfected cells (data not shown). The data with the p38 inhibitor was confirmed by the use of siRNA directed against p38 (Fig. 3B), which significantly inhibited hmox-1 mRNA expression in H. pylori-stimulated macrophages (Fig. 3C); in contrast, the erk1 siRNA (Fig. 3B) had no effect on hmox-1 induction (Fig. 3C). Then, because we found that HO-1 induction was mediated by p-CagA and by p38, we determined whether p38 activation was dependent on CagA phosphorylation. Fig. 3D depicts that the phosphorylation of p38 on Thr180/Tyr182 was decreased in macrophages i) pre-treated with PP1 and infected with H. pylori 60190 or ii) infected with the cagA mutant strain, when compared to RAW 264.7 cells infected with H. pylori 60190. Together, these results show that p-CagA signals in macrophages to activate p38. In accordance with the level of phagocytosis (Fig. 1E) and of CagA phosphorylation (Fig. 2C) with the various H. pylori strains, we found that p38 was less activated in macrophages infected with H. pylori 7.13 than with the strain 60190 (Fig. 3E). It has been reported that NRF-2 is a transcription factor activated by p38 that may transactivate the hmox-1 gene (29); consistent with this, we found that blocking of NRF-2 expression using siRNA (Fig. 3F) resulted in a significant reduction of H. pylori-induced hmox-1 mRNA expression (Fig. 3G).
Figure 3.
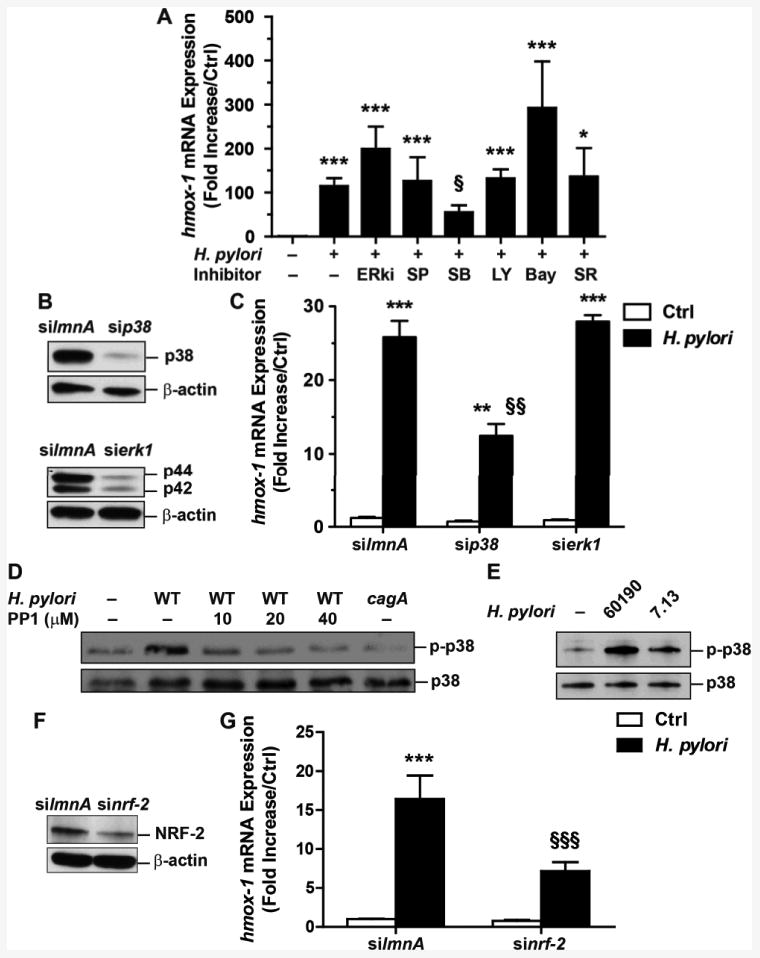
Molecular regulation of hmox-1 transcription in macrophages. A, hmox-1 mRNA expression in RAW 264.7 cells pre-treated with ERKi, SP600125 (SP), SB203580 (SB), LY294002 (LY), Bay11-7082 (Bay), or SR11302 (SR) and infected with H. pylori for 6 h. ***P < 0.001 vs. Ctrl; §P < 0.05 vs. infected cells; n = 5. B, Western blots showing the effect of knockdown of p38 and p42/p44 in RAW 264.7 cells transfected with lmnA, p38, or erk1 siRNAs. C, Levels of hmox-1 mRNA in macrophages transfected with siRNAs directed against lmnA, p38, or erk1 and then treated with H. pylori for 6 h. ***P < 0.001, **P < 0.01 vs. Ctrl; §§P < 0.01 vs. cells transfected with lmnA or erk1 siRNA and infected with H. pylori; n = 5. D, Levels of p-p38 and p38 in macrophages pretreated with PP1 and infected with H. pylori or with the cagA mutant. Representative data of 3 independent experiments. E, p38 phosphorylation in RAW 264.7 cells infected with the strains 60190 or 7.13. F, Effect of nrf-2 siRNA on knockdown of NRF-2 in RAW 264.7 cells. G, Levels of hmox-1 mRNA in macrophages transfected with siRNAs directed against lmnA or nrf-2 and then treated with H. pylori for 6 h. ***P < 0.001 vs. Ctrl; §§§P < 0.001 vs. cells transfected with lmnA and infected with H. pylori; n = 6.
HO-1 is induced in gastric macrophages during H. pylori infection
To demonstrate the in vivo relevance of our findings, we evaluated the presence of HO-1 in mononuclear cells of gastric tissues of infected patients in which the cagA status of the infecting H. pylori strains was known (21). Tissues from subjects infected with cagA+ H. pylori strains exhibited more staining in mononuclear cells than tissues from controls or patients infected with cagA– strains (Figs. 4A and 4B); in particular, strong staining of cells with the appearance of tissue macrophages was detected. Moreover, we observed that HO-1 levels were increased in C57BL/6 mice infected for 2 months with H. pylori PMSS1 that retains a functional T4SS in vivo (30), when compared to uninfected mice (Fig. 5A and Supplemental Fig. 1), and that HO-1 staining co-localized to cells that were positive for the macrophage marker F4/80 (Fig. 5A and Supplemental Fig. 1). To confirm this observation, we isolated gastric immune cells and analyzed F4/80 and HO-1 expression by flow cytometry. A representative flow cytometric dot plot (Fig. 5B) and analysis performed from multiple animals (Fig. 5C) demonstrate a significantly increased percentage of F4/80+/HO-1+ cells in infected mice compared to control animals. Further, the expression levels of HO-1 in gastric macrophages were also enhanced in the isolated gastric macrophages from H. pylori-infected mice (Figs. 5D and 5E).
Figure 4.
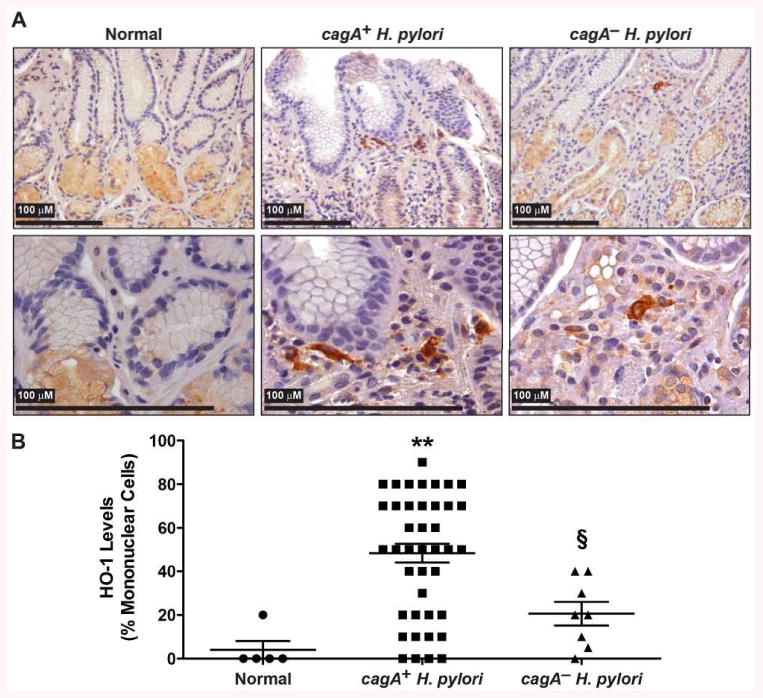
Expression of HO-1 in patients infected with H. pylori. A, Representative HO-1 immunoperoxidase staining in gastric tissues. B, Quantification of staining score for HO-1 in mononuclear cells. **P < 0.01 vs. uninfected patients; §P < 0.05 vs. individuals infected with cagA+ H. pylori. Each symbol is a different subject.
Figure 5.
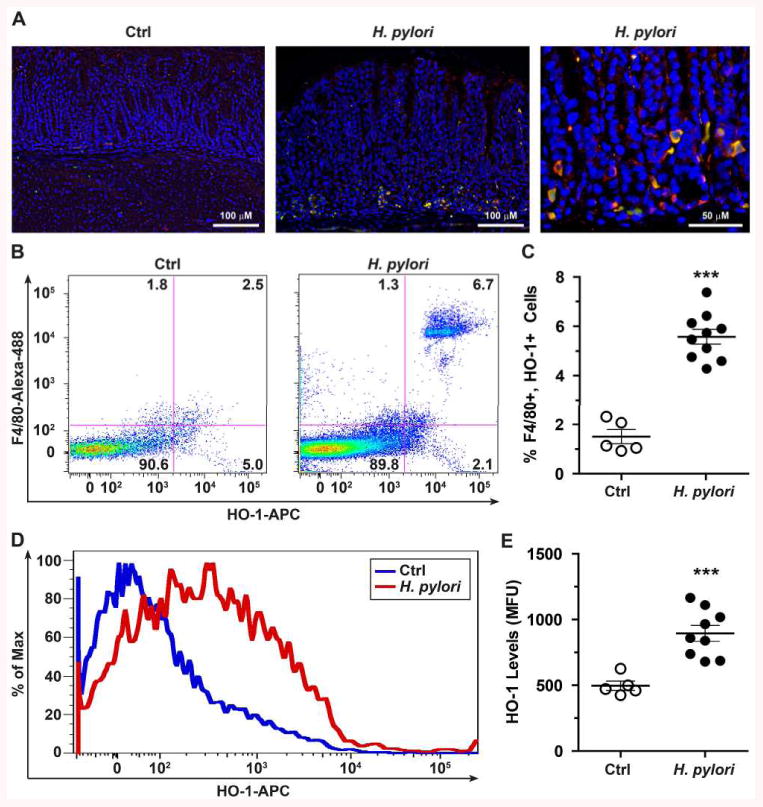
Expression of HO-1 in gastric macrophages during H. pylori infection. A, Immunofluorescence performed in the gastric tissue of C57BL/6 mice infected or not with H. pylori PMSS1 for 2 months. The macrophage marker F4/80, HO-1, and nuclei were detected with TRITC (red), DyLight 488 (green), and DAPI (blue), respectively; merged images are shown, with cells double-positive for F4/80 and HO-1 depicted by yellow color. B-E, Gastric cells were isolated from mice and analyzed by flow cytometry for the expression of F4/80 and HO-1. Representative dot plots with percent of cells in each quadrant (B) and flow cytometric analysis of HO-1 levels in mean fluorescence units (D). The summary data are presented in (C) and (E). ***P < 0.001 vs Ctrl; each symbol represents a different mouse.
Genetic ablation of HO-1 exacerbates gastritis and restores immunity to H. pylori
To further investigate the role of macrophage HO-1 in the pathophysiology of H. pylori infection, we infected WT and hmox-1-deficient mice for 2 months with strain PMSS1. There was a significant increase in gastric inflammation in infected hmox-1−/− mice compared to WT animals, as demonstrated by histologic gastritis scores (Fig. 6A) and representative histologic sections (Fig. 6B). We also found that the mRNA expression of the genes encoding the M1 markers iNOS, TNF-α and IL-12p40 was increased, and conversely, the mRNA level of the prototype Mreg cytokine IL-10 was decreased, in gastric macrophages isolated from hmox-1−/− mice, when compared to those from WT animals (Fig. 6C); in accordance with this, iNOS protein immunolocalizing to gastric macrophages was more induced in the gastric tissue of infected hmox-1−/− mice than WT animals (Fig. 6D). In addition, there were more transcripts of the genes encoding IFN-γ and IL-17 (Fig. 6E), the prototype cytokines of Th1 and Th17 responses, and more IL-17 protein (Fig. 6F) in gastric tissues from infected hmox-1−/− mice compared to infected WT animals. Consistent with the increased M1, Th1, and Th17 immune response in the hmox-1−/− mice, gastric colonization by H. pylori was significantly reduced with hmox-1 deletion (Fig. 6G). These data establish that HO-1 downregulates gastric inflammation and favors H. pylori survival.
Figure 6.
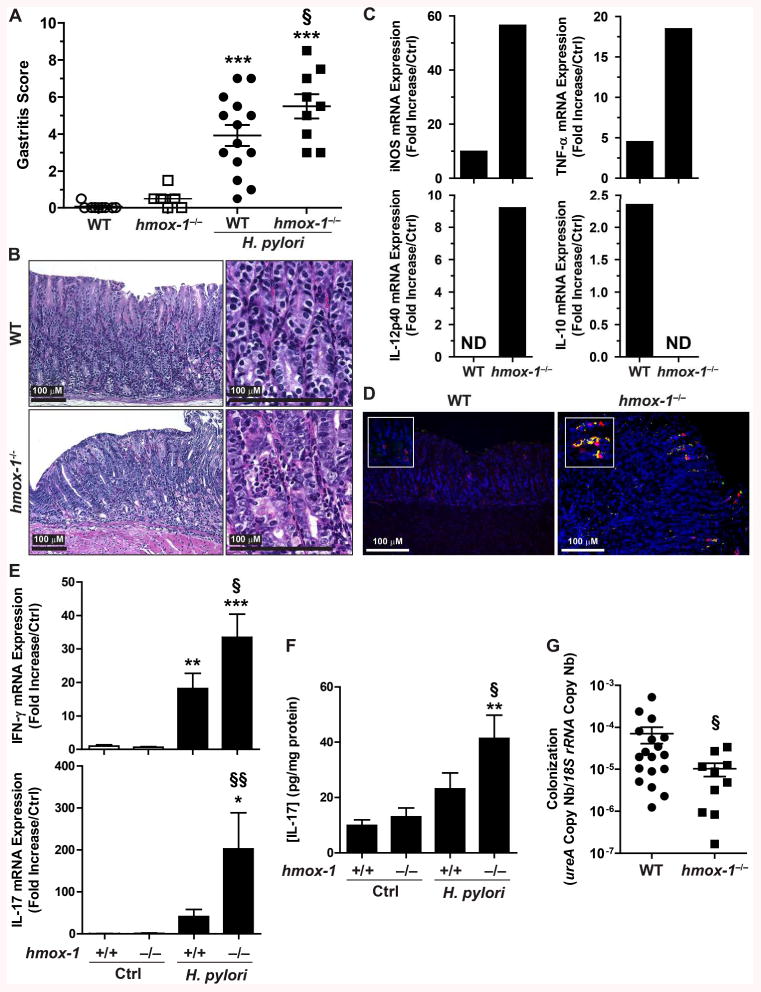
Effect of hmox-1 deletion on the outcome of H. pylori infection. WT and hmox-1−/− mice were infected with H. pylori PMSS1 for 2 months. A and B, Levels of gastritis. C, Expression of iNOS, TNF-α, IL-12p40, and IL-10 genes in gastric macrophages. Macrophages were purified from the gastric tissues of 3 WT mice, 5 H. pylori-infected WT mice, 3 hmox-1−/− mice, and 5 hmox-1−/− mice infected with H. pylori. The RNA from the gastric macrophages from each mouse was extracted and pooled in each group of mice before analysis by RT-qPCR. Values are expressed as fold increase compared to uninfected mice; ND, no PCR product detected. D, iNOS expression. Immunofluorescence for the macrophage marker F4/80 (red), iNOS (green), and nuclei (blue) in the gastric tissue of H. pylori-infected mice. Merged images are shown, with the cells double-positive for iNOS and F4/80 evidenced by yellow color. E, Expression levels of IFN-γ and IL-17 mRNAs in gastric tissues. F, Concentration of IL-17 in the gastric tissues. G, Colonization of the stomach by H. pylori. For A, D, and E, *P < 0.05, **P < 0.01, ***P < 0.001 vs. uninfected animals; §P < 0.05, §§P < 0.01 vs. infected WT mice.
H. pylori-induced HO-1 regulates macrophage polarization
Because our studies indicated that HO-1 induction in gastric macrophages during H. pylori infection is associated with decreased iNOS and M1 cytokine expression and increased IL-10 expression (Fig. 6C) in WT mice, we reasoned that HO-1 may directly affect macrophage polarization. To test this hypothesis, we infected resident peritoneal macrophages from WT and hmox-1−/− mice with H. pylori for 24 h ex vivo, and analyzed mRNA expression of polarization markers. The genes encoding the M1 markers iNOS, TNF-α, IL-12p40, and IL-1β, and the Mreg markers IL-10, LIGHT, and CCL1 were significantly induced by H. pylori in WT macrophages (Fig. 7A and Supplemental Fig. 2); among the eight M2 marker genes tested, only CCL17 was significantly induced during the infection of WT macrophages (Fig. 7A and Supplemental Fig. 2). These results suggest that H. pylori-infected macrophages exhibit a predominantly mixed M1/Mreg phenotype. Remarkably, the expression levels of iNOS, TNF-α, IL-12p40, and CXCL10 (M1 populations) were significantly increased in infected macrophages from hmox-1-deficient mice when compared to WT macrophages (Fig. 7A and Supplemental Fig. 2). Inversely, the M2 (CCL17) and Mreg (IL-10, LIGHT, and CCL1) genes were less expressed in infected hmox-1−/− macrophages than in WT cells (Fig. 7A and Supplemental Fig. 2). In accordance with these data, we found that significantly more NO and less IL-10 were released by infected macrophages from hmox-1−/− mice than from WT mice (Fig. 7B).
Figure 7.
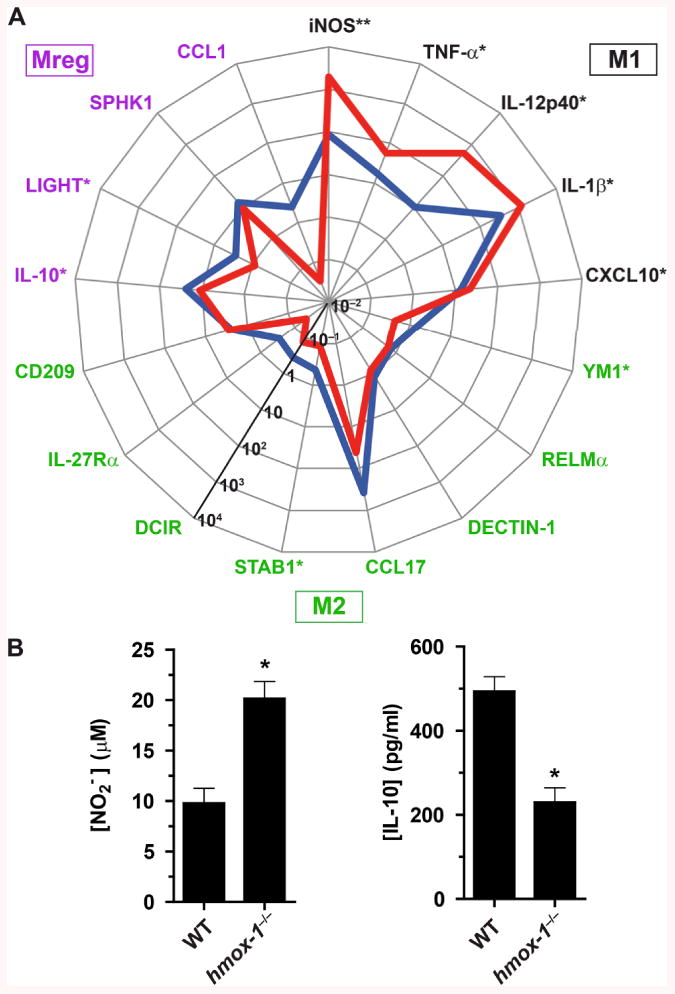
Macrophage polarization in response to H. pylori. A, The mRNA levels of the genes encoding markers of the M1, M2, and Mreg populations were analyzed in peritoneal macrophages from WT (blue line) or hmox-1−/− (red line) mice infected with H. pylori 60190 for 24 h; n = 6 mice for each genotype. For each gene, asterisks denote significant differences between WT and hmox-1−/− mice (*P < 0.05, **P < 0.01). B, Concentrations of NO2− and IL-10 in the supernatant of peritoneal macrophages from WT and hmox-1−/− mice infected for 24 h with H. pylori. *P < 0.05 vs. WT; n = 3-6 mice.
To further investigate the role of HO-1 on the modulation of the expression of the genes encoding M1 and Mreg markers, we used siRNA directed against hmox-1 (Fig. 8A) or the HO-1 inhibitor CrMP to block the expression and the activity of HO-1 in RAW 264.7 cells, respectively. We observed that knockdown or pharmacological inhibition of HO-1 resulted in increased expression of iNOS and in a concomitant decrease in expression of IL-10 in H. pylori-infected macrophages (Figs. 8B and 8C). Collectively these data support the contention that macrophage HO-1 downregulates M1 polarization and favors an Mreg phenotype during H. pylori infection.
Figure 8.
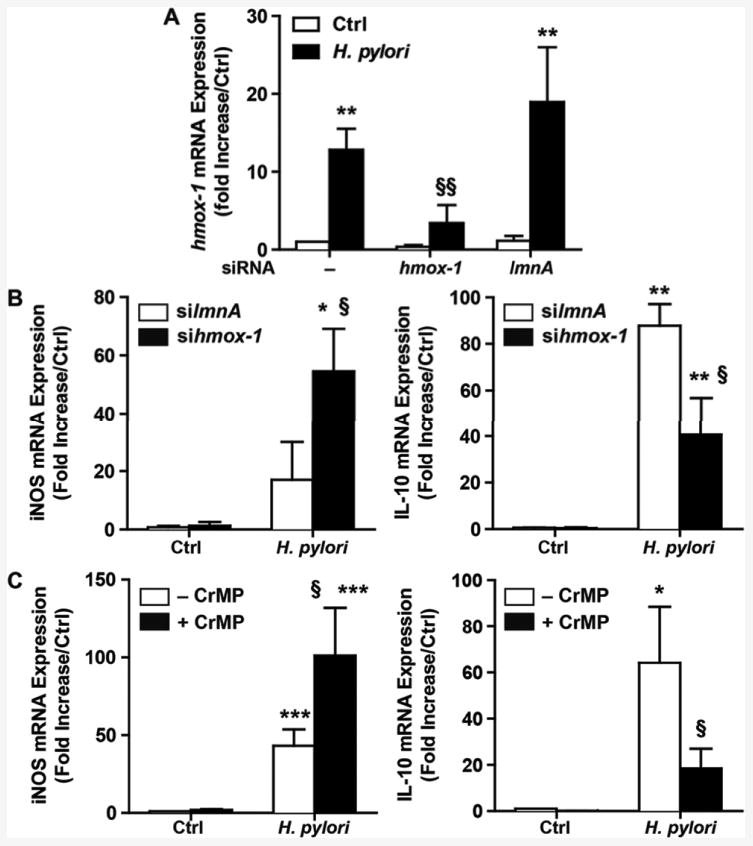
Regulation of macrophage activation by HO-1. A, The expression of hmox-1 was analyzed in RAW 264.7 cells that were transfected or not with siRNA against hmox-1 or lmnA before infection with H. pylori. **P < 0.01 vs. uninfected macrophages; §§P < 0.01 vs. cells not transfected or transfected with silmnA and infected with H. pylori. B and C, Levels of iNOS and IL-10 mRNA expression in RAW 264.7 transfected with siRNA against lmnA or hmox-1 (B) or treated with CrMP (C), and infected with H. pylori for 24 h. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Ctrl; §P < 0.05 vs. cells infected with H. pylori and transfected with lmnA (B) or not treated with CrMP (C); n = 3.
Discussion
Both innate and adaptive immunity play a cardinal role in controlling bacterial burden of H. pylori within the gastric mucosa (9, 18, 31). Nonetheless, the bacterium has elaborated numerous strategies to prevent the efficiency of the host immune response to survive in its ecological niche (32). In this context, we have identified a specific process by which H. pylori downregulates the inflammatory response of macrophages. The induction of HO-1 by H. pylori in murine macrophages through a p-CagA/p38/NRF-2-dependent pathway favors the polarization of macrophages towards an Mreg phenotype. Our finding has direct significance in vivo, since we have also demonstrated that HO-1 is induced in gastric macrophages of H. pylori-infected C57BL/6 mice. Lastly, this work also establishes that H. pylori-induced macrophage HO-1 restricts gastritis and favors colonization. In the same way, we have previously shown that the experimental induction of HO-1 in the gastric tissue by a treatment with hemin before H. pylori infection decreases the level of acute gastric inflammation (23).
The induction of HO-1 in macrophages is mostly known as a cellular response to oxidative or nitrosative stress (33). However, bacterial endotoxins (34) or invasive pathogens, such as Mycobacterium tuberculosis (35) or Leishmania mexicana (36), can also induce HO-1. The present work shows for the first time that H. pylori stimulates hmox-1 expression in macrophages. It has been reported that these cells can be activated by numerous factors released by H. pylori, such as urease (12), Hsp60 (37), or LPS (38). Others have shown that contact between macrophages and H. pylori is required to stimulate the production of IL-18 by the human macrophage cell line THP-1 (39) and that phagocytosis contributes to maximal activation of dendritic cells (28). Accordingly, we found that separating H. pylori from macrophages or the inhibition of phagocytosis resulted in a failure of hmox-1 expression. Further, our experiments have established that CagA reaches the cytoplasm of macrophages after phagocytosis independently of the T4SS, is phosphorylated by c-Src, and induces HO-1 in macrophages. The phosphorylation of CagA in the murine macrophage cell line J774 has been reported (40). However, a cleaved form of CagA was evidenced in J774 cells infected for 4 h and 6 h (40), while we found intact CagA protein after a 1 h or 3 h infection. The difference in infection time may explain this difference. Interestingly, we found that the H. pylori strain 7.13 is less phagocytized by macrophages than the strain PMSS1 and 60190; in accordance with this, the protein CagA from the strain 7.13 is less phosphorylated and this results in less induction of hmox-1. Because we found that the phosphorylation of CagA in macrophages is not dependent on the presence of a T4SS, it should be noted that the ability of various strains of H. pylori to express and inject CagA in gastric epithelial cells is not relevant to what occurs in mononuclear cells.
While CagA has been implicated in cellular events leading to macrophage apoptosis (41), we have now discovered that p-CagA also signals in macrophages to stimulate the inducible transcription of hmox-1 through the p38-NRF-2 pathway. The implication of this transduction pathway in hmox-1 expression has been reported in macrophages stimulated with IL-10 (29), α-lipoic acid (42), or cobalt protoporphyrin (43); further, the genetic ablation of NRF-2 completely suppressed hmox-1 transcription in peritoneal macrophages stimulated with diesel exhaust particles (44). Our results are consistent with the fact that the kinase p38 is rapidly activated in gastric epithelial cells by a molecular mechanism involving CagA (45, 46), and in monocytes/macrophages infected with H. pylori (47) or stimulated with purified H. pylori products including VacA or HP0175, a peptidyl prolyl cis-, trans-isomerase (48, 49). The ability of these other H. pylori components to activate p38 may explain why in our experiments the complete inhibition of CagA phosphorylation did not entirely suppress p38 phosphorylation and hmox-1 expression.
Although the polarization of macrophages is usually initiated by cytokines and bacterial endotoxins, mediators of the innate immune response may also regulate the differentiation of myeloid cells (50, 51). Here we demonstrate that H. pylori-induced HO-1 is a regulator of macrophage polarization by tipping the M1/Mreg balance in favor of an Mreg phenotype. In support of the contention that HO-1 orchestrates the Mreg switching, it has been reported that hmox-1 is one of the genes significantly upregulated in bone marrow-derived macrophages polarized into Mregs when compared to an M1 population (15) and that HO-1 is induced by M-CSF in IL-10-producing macrophages (52). Additionally, the transfer of a functional hmox-1 cDNA using adenoviral delivery has been shown to enhance IL-10 production from alveolar macrophages that attenuates LPS-induced acute lung injury in mice (53) .
Various immunological mechanisms, such as impaired NO production (18, 54) and recruitment of regulatory T cells (10), may explain the persistence of H. pylori within the gastric mucosa. The Mreg population is known to dampen the immune response, which results in the decrease of inflammation (55) and/or in the progression of infectious diseases (56, 57). Moreover, these macrophages are efficient antigen-presenting cells inducing T-cell responses that are dominated by the production of anti-inflammatory cytokines (58). Accordingly, we found that the genetic deletion of hmox-1 leads to increased gastritis and decreased colonization in H. pylori-infected mice. Moreover, HO-1 products have been shown to regulate the expression of bacterial virulence factors, such as the dormancy regulon of M. tuberculosis (35). HO-1 might thus have a direct effect on H. pylori growth/virulence, and this deserves further investigation.
This work reveals another mechanism by which the H. pylori virulence factor CagA contributes to H. pylori pathogenesis, by causing signaling in macrophages that induces HO-1. This directly shapes the inflammatory response and favors the immune evasion of this pathogen. Conversely, we have shown that H. pylori inhibits HO-1 in gastric epithelial cells in vitro, as well as in the stomach of mice or humans infected with cagA+ H. pylori strains (23); we have also demonstrated that HO-1 inhibits H. pylori-induced c-Src activation and consequently CagA phosphorylation in gastric epithelial cells (59). Hence, the H. pylori-induced downregulation of HO-1 in epithelial cells can be a mechanism by which this pathogen facilitates phosphorylation of CagA and p-CagA-dependent neoplastic transformation. Therefore, the activation of HO-1 in macrophages and the inhibition of HO-1 in gastric epithelial cells are cellular mechanisms that both favor H. pylori persistence and pathogenesis. In this context, we propose that a specific crosstalk exists between H. pylori and host HO-1. This cell-dependent dichotomous regulation of HO-1 expression orchestrated by CagA represents an example of a successful adaptation of a pathogenic bacterium in its ecological niche.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants R01DK053620 and R01AT004821 (to K.T.W.), P01CA116087 (to R.M.P and K.T.W.), UL1RR024975 (Vanderbilt CTSA, Pilot Project to K.T.W.), P01CA028842 (to P.C. and K.T.W.), the Vanderbilt Digestive Disease Research Center Grant (P30DK058404), the Vanderbilt Cancer Center Support Grant (P30CA068485) and by Merit Review Grant 1I01BX001453 from the Office of Medical Research, Department of Veterans Affairs (to K.T.W.). A.P.G. is supported in part by the Philippe Foundation.
Abbreviations used in this article
- cag
cytotoxin-associated gene
- CrMP
chromium mesoporphyrin
- HO-1
heme oxygenase-1
- iNOS
inducible NO synthase
- NRF-2
nuclear factor (erythroid-derived 2)-like 2
- T4SS
type IV secretion system
- WT
wild-type
References
- 1.Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, Schiavon S, Guariso G, Ceroti M, Nitti D, Rugge M, Plebani M, Atherton JC. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135:91–99. doi: 10.1053/j.gastro.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 2.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 4.Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–1566. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segal ED, Lange C, Covacci A, Tompkins LS, Falkow S. Induction of host signal transduction pathways by Helicobacter pylori. Proc Natl Acad Sci USA. 1997;94:7595–7599. doi: 10.1073/pnas.94.14.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backert S, Moese S, Selbach M, Brinkmann V, Meyer TF. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol Microbiol. 2001;42:631–644. doi: 10.1046/j.1365-2958.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 7.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, Tanaka S, Azuma T, Hatakeyama M. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–17216. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 8.Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, Luthra GK, Brooks EG, Graham DY, Reyes VE, Ernst PB. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology. 1998;114:482–492. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 9.Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, Wu C, Mao XH, Jia KR, Wang FJ, Guo H, Flavell RA, Zhao Z, Liu KY, Xiao B, Guo Y, Zhang WJ, Zhou WY, Guo G, Zou QM. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol. 2010;184:5121–5129. doi: 10.4049/jimmunol.0901115. [DOI] [PubMed] [Google Scholar]
- 10.Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito T, Kobayashi D, Uchida K, Takemura T, Nagaoka S, Kobayashi I, Yokoyama T, Ishige I, Ishige Y, Ishida N, Furukawa A, Muraoka H, Ikeda S, Sekine M, Ando N, Suzuki Y, Yamada T, Suzuki T, Eishi Y. Helicobacter pylori invades the gastric mucosa and translocates to the gastric lymph nodes. Lab Invest. 2008;88:664–681. doi: 10.1038/labinvest.2008.33. [DOI] [PubMed] [Google Scholar]
- 12.Gobert AP, Mersey BD, Cheng Y, Blumberg DR, Newton JC, Wilson KT. Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol. 2002;168:6002–6006. doi: 10.4049/jimmunol.168.12.6002. [DOI] [PubMed] [Google Scholar]
- 13.Kaparakis M, Walduck AK, Price JD, Pedersen JS, van Rooijen N, Pearse MJ, Wijburg OL, Strugnell RA. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect Immun. 2008;76:2235–2239. doi: 10.1128/IAI.01481-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quiding-Jarbrink M, Raghavan S, Sundquist M. Enhanced M1 macrophage polarization in human Helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS One. 2010;5:e15018. doi: 10.1371/journal.pone.0015018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, Gobert AP, Chaturvedi R, Wilson KT. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol. 2011;186:3632–3641. doi: 10.4049/jimmunol.1003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, de Sablet T, Piazuelo MB, Sarvaria AR, Cheng Y, Closs EI, Casero RA, Jr, Gobert AP, Wilson KT. Polyamines impair immunity to Helicobacter pylori by inhibiting L-arginine uptake required for nitric oxide production. Gastroenterology. 2010;139:1686–1698. 1698 e1681–1686. doi: 10.1053/j.gastro.2010.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fehlings M, Drobbe L, Moos V, Renner Viveros P, Hagen J, Beigier-Bompadre M, Pang E, Belogolova E, Churin Y, Schneider T, Meyer TF, Aebischer T, Ignatius R. Comparative analysis of the interaction of Helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infect Immun. 2012;80:2724–2734. doi: 10.1128/IAI.00381-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 21.de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, Bravo LE, Sicinschi LA, Delgado AG, Mera RM, Israel DA, Romero-Gallo J, Peek RM, Jr, Cover TL, Correa P, Wilson KT. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut. 2011;60:1189–1195. doi: 10.1136/gut.2010.234468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peek RM, Jr, Blaser MJ, Mays DJ, Forsyth MH, Cover TL, Song SY, Krishna U, Pietenpol JA. Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Res. 1999;59:6124–6131. [PubMed] [Google Scholar]
- 23.Gobert AP, Asim M, Piazuelo MB, Verriere T, Scull BP, de Sablet T, Glumac A, Lewis ND, Correa P, Peek RM, Jr, Chaturvedi R, Wilson KT. Disruption of nitric oxide signaling by Helicobacter pylori results in enhanced inflammation by inhibition of heme oxygenase-1. J Immunol. 2011;187:5370–5379. doi: 10.4049/jimmunol.1102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA. 1997;94:10919–10924. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol. 2000;278:F726–F736. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- 27.Chaturvedi R, Asim M, Lewis ND, Algood HM, Cover TL, Kim PY, Wilson KT. L-arginine availability regulates inducible nitric oxide synthase-dependent host defense against Helicobacter pylori. Infect Immun. 2007;75:4305–4315. doi: 10.1128/IAI.00578-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kranzer K, Sollner L, Aigner M, Lehn N, Deml L, Rehli M, Schneider-Brachert W. Impact of Helicobacter pylori virulence factors and compounds on activation and maturation of human dendritic cells. Infect Immun. 2005;73:4180–4189. doi: 10.1128/IAI.73.7.4180-4189.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nature Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 30.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Müller A. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology. 2011;140:199–209. doi: 10.1053/j.gastro.2010.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rad R, Brenner L, Krug A, Voland P, Mages J, Lang R, Schwendy S, Reindl W, Dossumbekova A, Ballhorn W, Wagner H, Schmid RM, Bauer S, Prinz C. Toll-like receptor-dependent activation of antigen-presenting cells affects adaptive immunity to Helicobacter pylori. Gastroenterology. 2007;133:150–163 e153. doi: 10.1053/j.gastro.2007.04.071. [DOI] [PubMed] [Google Scholar]
- 32.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 33.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 34.Camhi SL, Alam J, Otterbein L, Sylvester SL, Choi AM. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am J Respir Cell Mol Biol. 1995;13:387–398. doi: 10.1165/ajrcmb.13.4.7546768. [DOI] [PubMed] [Google Scholar]
- 35.Shiloh MU, Manzanillo P, Cox JS. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe. 2008;3:323–330. doi: 10.1016/j.chom.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pham NK, Mouriz J, Kima PE. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect Immun. 2005;73:8322–8333. doi: 10.1128/IAI.73.12.8322-8333.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gobert AP, Bambou JC, Werts C, Balloy V, Chignard M, Moran AP, Ferrero RL. Helicobacter pylori heat shock protein 60 mediates interleukin-6 production by macrophages via a toll-like receptor (TLR)-2-, TLR-4-, and myeloid differentiation factor 88-independent mechanism. J Biol Chem. 2004;279:245–250. doi: 10.1074/jbc.M307858200. [DOI] [PubMed] [Google Scholar]
- 38.Perez-Perez GI, Shepherd VL, Morrow JD, Blaser MJ. Activation of human THP-1 cells and rat bone marrow-derived macrophages by Helicobacter pylori lipopolysaccharide. Infect Immun. 1995;63:1183–1187. doi: 10.1128/iai.63.4.1183-1187.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamauchi K, Choi IJ, Lu H, Ogiwara H, Graham DY, Yamaoka Y. Regulation of IL-18 in Helicobacter pylori infection. J Immunol. 2008;180:1207–1216. doi: 10.4049/jimmunol.180.2.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Odenbreit S, Gebert B, Puls J, Fischer W, Haas R. Interaction of Helicobacter pylori with professional phagocytes: role of the cag pathogenicity island and translocation, phosphorylation and processing of CagA. Cell Microbiol. 2001;3:21–31. doi: 10.1046/j.1462-5822.2001.00088.x. [DOI] [PubMed] [Google Scholar]
- 41.Menaker RJ, Ceponis PJ, Jones NL. Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Infect Immun. 2004;72:2889–2898. doi: 10.1128/IAI.72.5.2889-2898.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogborne RM, Rushworth SA, O'Connell MA. Alpha-lipoic acid-induced heme oxygenase-1 expression is mediated by nuclear factor erythroid 2-related factor 2 and p38 mitogen-activated protein kinase in human monocytic cells. Arterioscler Thromb Vasc Biol. 2005;25:2100–2105. doi: 10.1161/01.ATV.0000183745.37161.6e. [DOI] [PubMed] [Google Scholar]
- 43.Paiva CN, Feijo DF, Dutra FF, Carneiro VC, Freitas GB, Alves LS, Mesquita J, Fortes GB, Figueiredo RT, Souza HS, Fantappie MR, Lannes-Vieira J, Bozza MT. Oxidative stress fuels Trypanosoma cruzi infection in mice. J Clin Invest. 2012;122:2531–2542. doi: 10.1172/JCI58525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 45.Keates S, Keates AC, Warny M, Peek RM, Jr, Murray PG, Kelly CP. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol. 1999;163:5552–5559. [PubMed] [Google Scholar]
- 46.Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol. 2009;183:8099–8109. doi: 10.4049/jimmunol.0900664. [DOI] [PubMed] [Google Scholar]
- 47.Bhattacharyya A, Pathak S, Datta S, Chattopadhyay S, Basu J, Kundu M. Mitogen-activated protein kinases and nuclear factor-kappaB regulate Helicobacter pylori-mediated interleukin-8 release from macrophages. Biochem J. 2002;368:121–129. doi: 10.1042/BJ20020555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pathak SK, Basu S, Bhattacharyya A, Pathak S, Banerjee A, Basu J, Kundu M. TLR4-dependent NF-kappaB activation and mitogen- and stress-activated protein kinase 1-triggered phosphorylation events are central to Helicobacter pylori peptidyl prolyl cis-, trans-isomerase (HP0175)-mediated induction of IL-6 release from macrophages. J Immunol. 2006;177:7950–7958. doi: 10.4049/jimmunol.177.11.7950. [DOI] [PubMed] [Google Scholar]
- 49.Hisatsune J, Nakayama M, Isomoto H, Kurazono H, Mukaida N, Mukhopadhyay AK, Azuma T, Yamaoka Y, Sap J, Yamasaki E, Yahiro K, Moss J, Hirayama T. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-kappaB activation. J Immunol. 2008;180:5017–5027. doi: 10.4049/jimmunol.180.7.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark K, MacKenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, Peggie M, Plater L, Pedrioli PG, McIver E, Gray NS, Arthur JS, Cohen P. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc Natl Acad Sci USA. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van den Bossche J, Lamers WH, Koehler ES, Geuns JM, Alhonen L, Uimari A, Pirnes-Karhu S, Van Overmeire E, Morias Y, Brys L, Vereecke L, De Baetselier P, Van Ginderachter JA. Pivotal Advance: Arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J Leukoc Biol. 2012;91:685–699. doi: 10.1189/jlb.0911453. [DOI] [PubMed] [Google Scholar]
- 52.Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiol. 2010;215:788–795. doi: 10.1016/j.imbio.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 53.Inoue S, Suzuki M, Nagashima Y, Suzuki S, Hashiba T, Tsuburai T, Ikehara K, Matsuse T, Ishigatsubo Y. Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum Gene Ther. 2001;12:967–979. doi: 10.1089/104303401750195926. [DOI] [PubMed] [Google Scholar]
- 54.Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, Mobley HL, Wilson KT. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci USA. 2001;98:13844–13849. doi: 10.1073/pnas.241443798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tierney JB, Kharkrang M, La Flamme AC. Type II-activated macrophages suppress the development of experimental autoimmune encephalomyelitis. Immunol Cell Biol. 2009;87:235–240. doi: 10.1038/icb.2008.99. [DOI] [PubMed] [Google Scholar]
- 56.Bleharski JR, Li H, Meinken C, Graeber TG, Ochoa MT, Yamamura M, Burdick A, Sarno EN, Wagner M, Rollinghoff M, Rea TH, Colonna M, Stenger S, Bloom BR, Eisenberg D, Modlin RL. Use of genetic profiling in leprosy to discriminate clinical forms of the disease. Science. 2003;301:1527–1530. doi: 10.1126/science.1087785. [DOI] [PubMed] [Google Scholar]
- 57.Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J Exp Med. 2005;201:747–754. doi: 10.1084/jem.20041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol. 2002;72:101–106. [PubMed] [Google Scholar]
- 59.Gobert AP, Verriere T, de Sablet T, Peek RM, Jr, Chaturvedi R, Wilson KT. Haem oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells. Cell Microbiol. 2013;15:145–156. doi: 10.1111/cmi.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.