

Abstract
The tradition of animal husbandry in the context of a nomadic lifestyle has been of great significance in the Mongolian society. Both Bactrian camels and horses have been invaluable for the survival and development of human activities in the harsh arid environment of the Mongolian steppe. As camels offer unique and sustainable opportunities for livestock production in marginal agro-ecological zones, we investigated the current genetic diversity of three local Mongolian camel breeds and compared their levels of variation with common native Mongolian camels distributed throughout the country. Based on mitochondrial and nuclear markers, we found levels of genetic diversity in Mongolian populations similar to that reported for Chinese Bactrian camels and for dromedaries. Little differentiation was detected between single breeds, except for a small group originating from the northwestern Mongolian Altai. We found neither high inbreeding levels in the different breeds nor evidence for a population decline. Although the Mongolian camel census size has severely declined over the past 20 years, our analyses suggest that there still exists a stable population with adequate genetic variation for continued sustainable utilization.
Keywords: Bayesian clustering, microsatellites, mitochondrial DNA, phylogeny
Introduction
Mongolia has successfully preserved pasture-supported animal husbandry under the conditions of a traditional nomadic lifestyle. For centuries, Mongolian herders have maintained five types of livestock: Bactrian camels, horses, cattle, sheep and goats. Although the domestication history of the other livestock species is well explored, our understanding of Bactrian camel (Camelus bactrianus) domestication is still incomplete. Recent studies have suggested that domestication took place in the Central and Eastern Asian Steppe around 6000 years ago (Peters & von den Driesch 1997; Trinks et al. 2012; Burger 2012). The relationship between domestic and wild camels (Camelus ferus) is comparable to that of domestic and wild horses (Equus ferus przewalskii). Although closely related, Bayesian reconstructions of the demographic history do not support the Przewalski's horse as direct ancestor of the domestic form (Lau et al. 2009; Warmuth et al. 2012). Similarly, the extant wild two-humped camels in Mongolia and China have been considered as the ancestors of modern domestic Bactrian camels. However, genetic studies have demonstrated high sequence divergence at mitochondrial (Ji et al. 2009; Silbermayr et al. 2010a) and nuclear loci (Jirimutu et al. 2012), excluding the wild camel as the direct ancestor of modern domestic populations.
Camels harbor many specific physiological characteristics that enable the production of beneficial items such as milk, meat and fur despite encountering harsh desert ecological conditions. A majority of the domestic camels in Mongolia, referred to as Mongolian native camels (MNT), are bred as multipurpose animals and do not represent particular morphological or biologic features. Nevertheless, three breeds are currently recognized. The Hos Zogdort (HZ) breed, from Tugrug soum (Gobi-Altai province), is known to have high wool productivity (i.e., annual average of 6.6–8.0 kg of wool per individual). The remaining two breeds are defined in statement A/69 ‘Breed and Line Confirmation’ by the Mongolian Agriculture and Food Ministry (Baldan 2011), as the camels of Galbiin Gobiin Ulaan (GGU) and Haniin Hetsiin Huren (HHH) in Umnugobi province (Fig. 1). Although GGU camels from Hanbogd soum1 are bigger than other Mongolian lines, the HHH breed originating from Mandal-Ovoo soum is very docile and smaller than is the common Bactrian camel (Chuluunbat et al. 2012).
Figure 1.
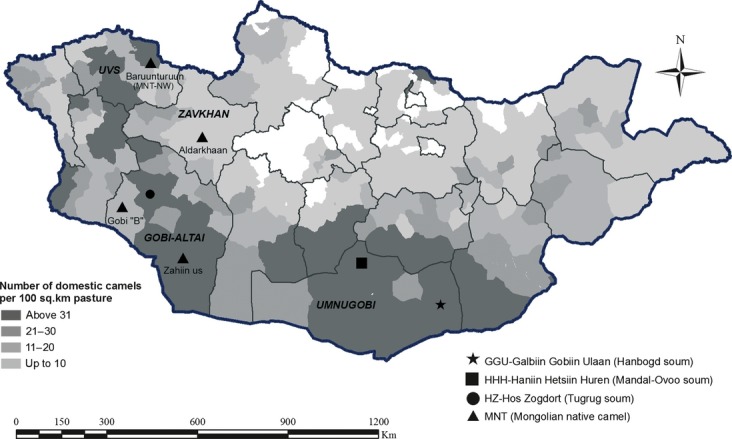
Geographic distribution of the domestic Bactrian camel samples analyzed in the present study. Province names are given in capital letters. The map is color coded according to the Bactrian camel density. The highest density of domestic Bactrian camels (67.6%) is found in the desert steppe area and Gobi Desert region, which constitutes 42% of the Mongolian territory. The remaining Bactrian camel herds are distributed in the northern Great Lakes Depression area (19.5%), in the eastern Mongolian steppe (7.7%) and in the forest-steppe environment (5.2%) (Dorjgotov 2009).
In a previous study focused on the genetic diversity of Chinese Bactrian camels, two Mongolian populations originating close to the Chinese border were included (Jianlin et al. 2004). Additionally, the diversity of Mongolian domestic camel populations was investigated using 31 blood protein loci, of which only two displayed polymorphisms (Koshimoto et al. 1999). The authors reported that the average heterozygosity of Mongolian camels was about 30 times lower than values in horses and sheep and about five times lower than in goats and buffalos, which already seem to have a comparatively lower genetic variability. Considering the low variation in blood protein loci, which potentially underestimate variability (Tapio et al. 2003), we investigated the genetic diversity in Mongolian Bactrian camels using nuclear and mitochondrial markers. We aimed to trace possible differentiation within and between the three local breeds (HHH, HZ, GGU) and common MNT. With the information on population diversity and structure gained from this first comprehensive study on Mongolian camels, we aim to support continuous sustainable and traditional breed management and the preservation of genetic resources.
Materials and methods
Sample collection and DNA extraction
In total, we sampled 206 animals from seven different localities of Mongolia (Fig. 1). The two Bactrian camels lines from Umnogobi province, HHH (n = 39) and GGU (n = 39), in addition to the HZ line (n = 43) from Gobi-Altai province, were of particular interest due to their recognized features. In parallel, we collected MNT samples from four different regions: (i) Aldarkhaan soum, Zavkhan province (Aldarkhaan; n = 26); (ii) Tsogt soum, Gobi-Altai province (Zahiin Us; n = 39); (iii) Bugat soum, Gobi-Altai province (Gobi B; n = 16); and (iv) Baruunturuun soum, Uvs province (MNT-NW; n = 4; Fig. 1). All samples (blood and hair) were collected during routine veterinary procedures and are listed in Table S1.
DNA extraction from EDTA-preserved blood was performed with the QIAmp® DNA kit (Qiagen). Hair samples were digested with a non-commercial lysis buffer (Pfeiffer et al. 2004), and DNA was extracted with the NucleoSpin® Tissue Kit (Macherey-Nagel). Negative controls were performed in every set of extractions and during further amplification steps.
DNA amplification, genotyping and sequencing
We selected a total of 17 microsatellite loci previously shown to successfully amplify alleles at different size ranges (Silbermayr et al. 2010b). The microsatellites were amplified in six multiplex PCRs set according to fragment length and fluorescence labeling (Table S2) following Silbermayr et al. (2010b). Genotyping runs were performed with the MegaBACE 1000, and electropherograms were evaluated with genetic profiler v2.2 (GE Healthcare).
A continuous 803-bp mitochondrial DNA (mtDNA) fragment spanning the end of cytochrome B to the partial control region (CR) (nt15122 nt15924; NC_009628.2) was sequenced in both directions with two overlapping primer pairs (Silbermayr et al. 2010a) on the MegaBACE 1000. The amplicons were aligned to a reference genome (NC_009628.2) using codoncode aligner v3.0.2 (CC Cooperation).
Statistical analyses
Due to the absence of accurate and standardized pedigrees, all genotypes were investigated with coancestry (Wang 2011) to estimate their relatedness coefficient (r) applying the triadic likelihood estimator and 1000 bootstrapping samples for the 95% confidence interval. Closely related individuals (r > 0.5; Wang 2011) were removed for unbiased estimates of neutral genetic parameters and population differentiation inferences.
Parameters of mitochondrial polymorphisms were computed with dnasp 5.1.0.1 (Librado & Rozas 2009) including the number of haplotypes, number of polymorphic sites, mean number of pairwise differences (k) and haplotype diversity (Hd). We calculated the nucleotide diversity π based on the average number of pairwise differences (Nei 1987) and Watterson's θS based on the number of segregating sites (Watterson 1975). Population pairwise distance ΦST, analysis of molecular variance (AMOVA) and a mismatch distribution were estimated using arlequin v3.11 (Excoffier & Lischer 2010). The Kimura-2-parameter substitution model was selected as the best-fit model based on the Akaike information criterion with correction for small sample size (AICc) in jmodeltest 0.1.1 (Posada 2008). Probability values for the null model of sudden expansion were calculated with parametric bootstrap values of 1000 iterations. We used two neutrality tests, Tajima's D and Fu's FS, and assessed their significance with 10 000 permutations. For the inference of population structure at the mitochondrial level, we performed Bayesian clustering for linked loci in baps6 (Corander & Tang 2007). We specified prior upper bound values for the number of clusters (i.e., 3–8) and performed 10 independent runs for each value. The median joining network (MJN) was constructed with network 4.6.1.1 (Bandelt et al. 1999) using default parameters.
The microsatellite polymorphisms within populations, estimated as the total number of alleles (TNA), mean number of alleles per locus (MNA), allelic richness (Ar) standardized for a minimal number of 16 individuals per population, observed (HO) and expected (HE) heterozygosities, as well as nuclear pairwise FST values corrected for multiple testing, were calculated with msanalyzer 4.05 (Dieringer & Schlötterer 2003). Genetic inbreeding coefficients FIS (Weir & Cockerham 1984) were inferred in genetix (Belkhir et al. 1999–2004). A nuclear AMOVA implemented in arlequin v3.11 estimated the partitioning of genetic variation within and among breeds. We inferred population structure based on microsatellites with structure v2.3 (Pritchard et al. 2000) using 50 000 iterations after a burn-in of 10 000 Markov Chain Monte Carlo replications and an admixture model allowing correlated allele frequencies. Four independent simulations for each K (2–8) were performed to identify the most probable clustering solution through examining the modal distribution of Delta K (Evanno et al. 2005). Graphical representations of these statistics were obtained using structure harvester v0.68 (Earl & vonHoldt 2012). The results from multiple runs for each K were concatenated by clumpp (Jacobsson & Rosenberg 2007). To further understand the clustering pattern, we carried out genetic distance-based principal coordinate analysis (PCoA) using genalex v6.5 (Peakall & Smouse 2012). Finally, we inferred excess/deficiency of nuclear heterozygosity to search for signals of population decline/expansion with bottleneck 1.2.0.2 (Cornuet & Luikart 1997) applying 1000 replications. We performed the evaluation using the stepwise mutation (SMM) and two-phase models of microsatellite evolution, with the proportion of SMM and the variance set to 85% and 12% respectively. The significance of the tests was assessed using Wilcoxon sign-rank test (Piry et al. 1999). The mode-shift indicator test was utilized, as stable populations are expected to have larger proportions of alleles at low frequency (Cornuet & Luikart 1997).
Results
We characterized the overall and breed-specific genetic diversity in three local domestic Bactrian camel lines and common Mongolian native camels (Fig. 1). Despite in-depth interviews with the breeders during the sampling procedure, we detected 43 closely related individuals among our sample set. In all estimates of diversity and population divergence, we included only unrelated individuals (r < 0.5; Wang 2011) and considered the camels from the northwestern Mongolian Uvs province (MNT-NW) as a separate population based on our clustering results.
Mitochondrial and nuclear genetic diversity
Within the 83-mtDNA sequence alignment, we detected 10 polymorphic sites (eight transitions and two transversions) recovering five previously described haplotypes (D1–D5; Silbermayr et al. 2010a) and nine new ones (GenBank submission KF640722–KF640727 and KF640729–KF640731). Molecular diversity indices over all Mongolian Bactrian camels are detailed in Table 1. Among the four investigated populations (GGU, HHH, HZ and MNT), Hd and π ranged between 0.600 and 0.020, and from 0.0011 to 0.0032 respectively (Table 1).
Table 1.
Genetic diversity of domestic camel populations determined from mitochondrial and microsatellite data.
| mtDNA (803 bp) | |||||||
|---|---|---|---|---|---|---|---|
| Pop | n | Haplotypes | Var. sites | Hd | π | k | θS |
| HHH | 8 | 6 | 5 | 0.929 (0.084) | 0.0029 (0.002) | 2.393 (1.603) | 1.928 (1.129) |
| HZ | 9 | 6 | 7 | 0.889 (0.091) | 0.0032 (0.003) | 2.566 (1.796) | 2.576 (1.922) |
| GGU | 6 | 3 | 2 | 0.600 (0.215) | 0.0011 (0.001) | 0.867 (0.347) | 0.876 (0.468) |
| MNT | 56 | 9 | 9 | 0.601 (0.067) | 0.0014 (0.002) | 1.112 (0.525) | 1.959 (0.671) |
| MNT-NW | 4 | 1 | 0 | – | – | – | – |
| Total | 83 | 14 | 10 | 0.725 (0.044) | 0.0019 (0.002) | 1.566 (0.869) | 2.004 (0.624) |
| Microsatellite (17 loci) | |||||||
|---|---|---|---|---|---|---|---|
| Pop | n | TNA | MNA | Ar | HO | HE | FIS |
| HHH | 27 | 77 | 4.28 (1.69) | 3.97 | 0.539 (0.239) | 0.547 (0.232) | 0.0156* |
| HZ | 30 | 78 | 4.33 (1.94) | 3.96 | 0.559 (0.244) | 0.544 (0.230) | −0.028* |
| GGU | 35 | 86 | 4.78 (2.55) | 4.02 | 0.495 (0.238) | 0.508 (0.232) | 0.0246* |
| MNT | 54 | 91 | 5.06 (2.46) | 4.05 | 0.523 (0.226) | 0.542 (0.225) | 0.0378* |
| MNT-NW | 4 | 35 | 2.00 (0.82) | n.a. | 0.300 (0.335) | 0.355 (0.270) | 0.0185 |
| Total | 150 | 113 | 7.11 (3.41) | 4.22 | 0.522 (0.224) | 0.546 (0.230) | 0.0436* |
GGU, Galbiin Gobiin Ulaan; HHH, Haniin Hetsiin Huren; HZ, Hos Zogdort; MNT, Mongolian native camels (Aldarkhaan, Zahiin us, Gobi ‘B’); MNT-NW, Western Mongolian native camels; n, number of individuals; Var. sites, variable sites; Hd, Haplotype diversity; π, nucleotide diversity based on the number of pairwise nucleotide differences; k, mean number of pairwise differences; θS, Watterson's theta based on the number of segregating sites; TNA, total number of alleles; MNA, mean number of alleles per locus; Ar, allelic richness per locus calculated for a population based on minimum sample size of 16 diploid individuals; HO/HE, observed/expected heterozygosity; FIS, inbreeding coefficient; n.a., not applicable.
Standard deviations are given in brackets.
P-value < 0.001.
The allelic variation of 17 microsatellite loci over all Mongolian Bactrian camels (n = 150) ranged between three and 12 alleles per locus with a MNA of 7.11. Within the 113 total alleles, we detected 26 private alleles, of which 11 belonged to fthe MNT population. Ar ranged from 3.96 to 4.05 alleles per locus, and heterozygosity levels varied between HO = 0.495–0.539 and HE = 0.508–0.559 respectively. FIS values were significantly different from zero and ranged between 0.0156 and 0.0378 in the local Mongolian populations. A negative FIS = −0.028 was detected in the HZ Bactrian camels from the Gobi Altai (Table 1). Nine of 17 loci contributed to the heterozygote excess in this population (Table S3).
Population structure and differentiation
For the inference of population structure, we performed clustering analyses on the mitochondrial and nuclear datasets independently, without using any prior information about the sample origin. Among the 83 mitochondrial sequences, baps analysis revealed five haplogroups as the best clustering solution (posterior probability = 0.81). With the exception of four samples (MNT-NW) collected in the northwestern Uvs province, which shared a single haplotype, no phylogeographic or breed-specific pattern was observed (Fig. S1). This quasi-panmictic haplotype distribution among the Mongolian camels was also illustrated in the MJN (Fig. 2).
Figure 2.
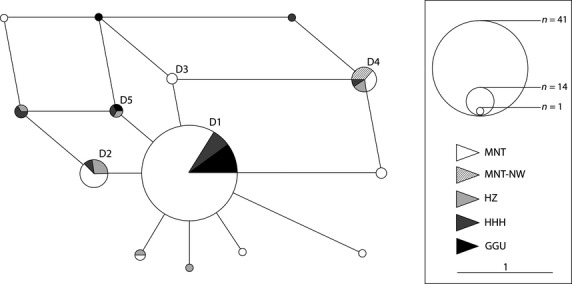
Median-joining network displaying the maximum parsimony relationship between the 14 mitochondrial haplotypes obtained from 83 unrelated domestic camels. D1–D5 refer to mitochondrial haplotypes described in Silbermayr et al. (2010a).
At the nuclear level, Bayesian clustering implemented in structure revealed no population differentiation for a theoretical number of ancestral populations set to K = 2–4. The optimal clustering solution was obtained for K = 5 (Delta K; Fig. S2), which differentiated the MNT-NW individuals from the other populations (Fig. 3, Fig. S3). Increasing the number of populations to K = 6 increased the level of admixture but did not result in geographically or phenotypically meaningful clustering solutions. The alternative clustering method (PCoA) reflected the same distinctiveness of the MNT-NW individuals, while it captured the extensive sharing of diversity among the Mongolian populations (Fig. 4). These clustering results were coherent with the AMOVA, which reported 97% of the nuclear and 73% of the mitochondrial variation to be shared within the defined populations. Nuclear pairwise FST values were significantly different from zero in all but two comparisons and ranged between 0.003 (MNT vs. HHH) and 0.207 (MNT-NW vs. GGU). Mitochondrial ΦST revealed significant differences between the MNT-NW samples and all other populations (0.485–0.833) and between MNT and HHH (0.156; Table 2).
Figure 3.
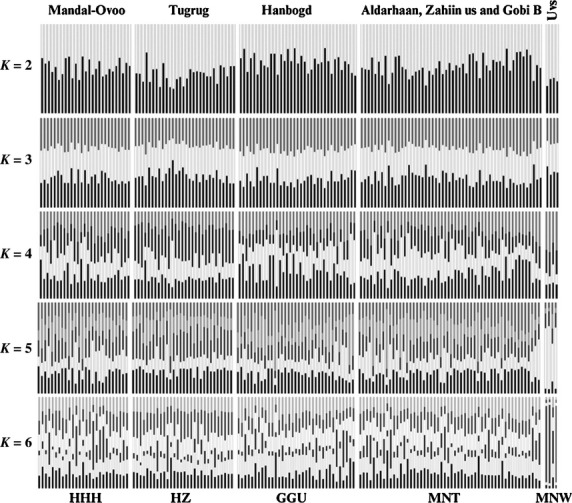
Individual assignment probabilities of 150 Mongolian Bactrian camels to two to six theoretical genetic ancestry groups using structure software. From K = 5 (best clustering solution; Delta K; Fig. S2), northwestern Mongolian individuals (MNT-NW) are slightly differentiated from the other populations.
Figure 4.
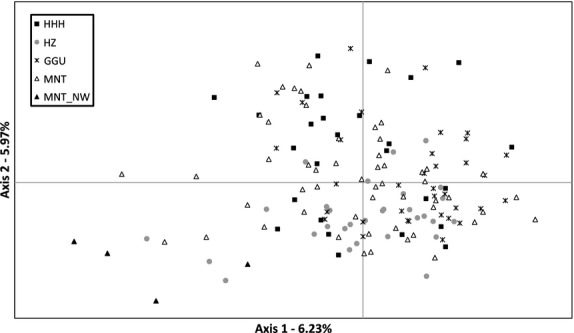
Two-dimensional plot of the principle coordinate analysis performed with genalex showing the extensive sharing of nuclear diversity among three defined breeds (HHH, HZ and GGU) and common native Mongolian camels (MNT). Only the four northwestern individuals (MNT-NW) show little differentiation. The first and second axis explained 6.23% and 5.97% of the variation respectively.
Table 2.
Population pairwise distances based on the 803-bp mtDNA sequences (ФST; below diagonal) and 17 microsatellite loci (FST; above diagonal).
| FST | |||||
|---|---|---|---|---|---|
| HHH | HZ | GGU | MNT | MNT-NW | |
| ΦST | |||||
| HHH | – | 0.014 | 0.020*** | 0.003 | 0.162** |
| HZ | –0.080 | – | 0.050*** | 0.029*** | 0.184*** |
| GGU | 0.024 | 0.036 | – | 0.019*** | 0.207*** |
| MNT | 0.156*** | 0.057 | 0.055 | – | 0.165*** |
| MNT-NW | 0.513 | 0.485 | 0.833*** | 0.682*** | – |
GGU, Galbiin Gobiin Ulaan; HHH, Haniin Hetsiin Huren; HZ, Hos Zogdort; MNT, Mongolian native camels (Aldarkhaan, Zahiin Us, Gobi B); MNT-NW, northwestern Mongolian native camels.
The negative ΦST value in the comparison of HZ/HHH should be interpreted as zero; it reflects the algorithm to estimate this value (Weir & Cockerham 1984) and indicates that more mitochondrial diversity is present within than between these two groups.
*P-value < 0.05, **P-value < 0.01, ***P-value < 0.001.
Population demographic inferences
In the mismatch distribution based on the pairwise differences between individuals, we observed a multimodal distribution (Fig. S4) and a Harpending's raggedness index r = 0.0387 (P > 0.1), which is a statistical measure for the development of population size under the null hypothesis of population expansion. To further evaluate demographic expansion/decline in Mongolian camels, we performed two neutrality tests, which can indicate past demographic events in the absence of selection. Tajima's D (–0.574, P > 0.1) and Fu's FS (–28.005, P < 0.0001) provided negative values, although Tajima's D was not significantly different from zero. We detected a significant number of microsatellite loci with a deficiency of observed vs. expected heterozygosity (P-value < 0.01) under the SMM model implemented in bottleneck software. Finally, the graphical representation of allele frequency distribution presented a L-shape, characteristic of a stable population under mutation-drift equilibrium.
Discussion
Over the past 20 years, Mongolian Bactrian camels have experienced a reduction in their population size from 700 000 to less than half of their previous census size. The effects of such a severe decline might be relevant for future sustainable utilization and conservation of this important livestock species. To evaluate the impact of this recent demographic event on the present genetic variability, we performed a comprehensive survey of mitochondrial and nuclear genetic diversity in three phenotypically different Mongolian domestic breeds in comparison with common Mongolian native camels.
Genetic diversity in the Mongolian camel populations
Among the 14 mitochondrial profiles, we detected nine new haplotypes represented in lower frequencies (Fig. 2). Although the overall mitochondrial diversity (Hd, π) in Mongolian camels was lower than observed in dromedaries (0.744, 0.0036; Al-Mathen F., Charruau P., Peters J., Abdussamad A.M., Raziq A., Faye B., Al-Eknah M., Walzer C., Hanotte O. & Burger P.A., submitted), the nuclear heterozygosities (HO, HE) reached values that were similar to those of Chinese Bactrian camels and slightly higher than those measured in the two previously studied Mongolian populations (Gobi Altai: HO/HE = 0.453/0.526 (n = 17); South Gobi: HO/HE = 0.521/0.545 (n = 39); Jianlin et al. 2004). In comparison with the domestic one-humped species, Mongolian Bactrian camels had similar levels of nuclear variation to Kenyan dromedaries (Mburu et al. 2003) and higher values than the feral Australian camel population (Spencer & Woolnough 2010). This contradicts the extremely low variability described in blood protein loci in Mongolian domestic camels (Koshimoto et al. 1999). Similar discrepancies and underestimation of variation using blood protein loci compared with microsatellites have been described in sheep (Tapio et al. 2003). Among the specific Mongolian camel lines, HZ and HHH from Southern Mongolia reached the highest diversity levels (Table 1). The observed higher total and mean number of alleles per locus detected in the GGU and MNT camels were most likely related to the greater number of individuals sampled from these two groups. Using a standardized population size, the allelic richness did not differ between the respective lines (Table 1).
We evaluated inbreeding in the Mongolian camel populations, which is relevant for breed management and conservation, and found low FIS values within the individual groups (Table 1). The negative value (−2.8%) detected in the HZ line from Gobi Altai indicated heterozygote excess in this camel population. This line also displayed the highest heterozygosity parameters observed among the breeds. These results can be interpreted as possible signs of outbreeding, most likely due to a recent admixture of two (or more) populations that were genetically more distant. A slightly higher FIS value was observed for the overall Mongolian Bactrian camel population; this value may be higher in the case of a non-entirely panmictic population (Silvertown & Charlesworth 2001). In comparison, the inbreeding values of the Mongolian lines were lower than those estimated in Arabian and African one-humped camel populations (4.4–9.2%; Al-Mathen/Charruau et al. submitted) and in Australian dromedaries (13.7–23.2%; Spencer & Woolnough 2010). These results are congruent with the low population structure and the high levels of gene flow observed among Mongolian camel breeds.
Population structure and genetic differentiation
Mongolian camel herders have traditionally selected camels based on morphological and productivity traits for milk, meat and wool (Chuluunbat et al. 2012). Considering this morphological characterization, we investigated the genetic diversity among the described breeds and expected breed-specific population differentiation. However, the clustering solutions of mitochondrial and nuclear DNA showed extensive sharing of diversity and absence of genetic substructure between the geographic populations and breeds (Figs. 4, S1). At the best clustering solutions, we differentiated four individuals from northwestern Mongolia (MNT-NW; Figs. 3 and 4). Although these samples displayed no close relationship as estimated with coancestry, they originated from a single maternal lineage that was also present in three other populations (HHH, HZ and MNT; Fig. 2). Although the MNT-NW camels were sampled in the Uvs province (Fig. 1), they originated from breeders belonging to the ethnic group of the Oirats in the western Mongolian Altai Mountains. The Oirats reached Mongolia only about 100 years ago, coming from northwestern China and Tibet (Chuluunbaatar 2010). Future comparisons with camels from China and Kazakhstan might elucidate the origin of the small, but distinct, differentiation between MNT-NW and other Mongolian camels.
The extensive usage of Bactrian camels as beasts of burden across deserts might explain the high levels of shared genetic variation among the populations (i.e., AMOVA, ΦST and FST estimates) and the weak structure observed with the putatively neutral markers (i.e., baps, PCoA, structure analyses). Low degrees of population differentiation based on neutral markers have been reported in several marine organisms inhabiting high-connectivity environments (DeWoody & Avise 2000; Grosberg & Cunningham 2001). In the case of the well-managed Old World camelids, high gene flow has artificially been induced and anticipated over centuries of trans-national trading. In this study, we observed the effects of this genetic homogenization in Mongolian Bactrian camels through the lack of distinct population differentiation. The time of recent anthropogenic selection into breeds might have been too short for genetic drift to act in the isolated populations and to leave significant signals in neutral markers. Recent candidate-gene-based and genome-wide approaches to detect selection and population differentiation presented evidence for stronger divergence in potentially adaptive genes and distinct population subdivision in highly migratory species (Corander et al. 2013; Defaveri et al. 2013). Therefore, targeting genes that have been under natural or anthropogenic selection might reveal (adaptive) divergence and population differentiation in specific regions in the Bactrian camel genome.
Population demographic inferences
The star-shape distribution of haplotypes in the MJN centered on high-frequency haplotypes (Fig. 2) indicates population expansion, possibly a result of an initial population reduction typical during domestication. Although the observed distribution of the sequence mismatches was multimodal, which corresponds to a stable population (Rogers & Harpending 1992), the modeled curve showed a smooth distribution characteristic of expanding populations (Fig. S3). Harpending's r, which takes larger values for multimodal distributions commonly found in stationary populations, was small but not significant. Therefore, we could not reject the null hypothesis of population expansion under the sudden-expansion model (Schneider & Excoffier 1999). Both neutrality tests (Tajima's D and Fu's FS) produced negative results, which could be interpreted as signals of population expansion in the absence of selection. Although only one neutrality test (Fu's FS) was significant, this test has been shown to be especially sensitive to population expansion (Fu 1997). These demographic signals may have captured a past domestication expansion that reached a maximum during the usage of Bactrian camels for regular transport in a nomadic lifestyle and along trading routes until they were replaced by motorized vehicles. The expansion signal at the mtDNA level might prevail over the signal of the rapid and (too) recent population decline that has occurred in Mongolian Bactrian camels during the past 20 years (3–4 generations considering a generation time of 6 years). However, based on nuclear microsatellites, a key test to detect a recent bottleneck in a population is to determine whether an excess of observed relative to expected heterozygosity calculated from the allelic diversity exists. During a reduction in the effective population size, low-frequency alleles (<0.01) are lost with a higher probability, whereas heterozygosity is reduced at a constant rate (Allendorf & Luikart 2007). The bottleneck results did not reveal significant patterns of heterozygosity excess, and the mode-shift test displayed an allelic frequency distribution characteristic of a non-declining population. Therefore, we cannot corroborate the hypothesis of a recent genetic bottleneck in the Mongolian population. On the contrary, we detected significant heterozygosity deficiency indicating population expansion. One reason why we failed to detect a bottleneck might be related to the fact that a recent event will more likely be traced in the case of an isolated population that does not experience incorporation of new migrants (Cornuet & Luikart 1997). As a rule of thumb, one migrant per generation will counteract the effects of isolation and genetic drift (Mills & Allendorf 1996). Considering the history of Bactrian camels along trading routes between Asia and Europe, we would not describe the Mongolian camel population as isolated but, rather, would expect continuous gene flow with neighboring regions. We cannot exclude, however, that a strong post-domestication expansion might have obscured the signal of the recent reduction in population size.
Conclusions
Among the domesticated Old World camelids (Camelini), Mongolian Bactrian camels harbor a genetic diversity comparable to that of Chinese Bactrian camels and dromedaries. After removing closely related individuals, we detected neither high inbreeding in the different Mongolian breeds nor evidence for a bottleneck. Our analyses rather suggested a stable or expanding population. However, the actual number of Mongolian Bactrian camels has severely declined over the past 20 years. If the negative trend continues, it might lead to a reduced genetic variability in Mongolian camels that could affect production traits as well as the potential for adaptation. Our results highlight the importance of preserving the current variation in the Mongolian camel populations as a highly valuable, desert livestock species. Based on neutral markers, we see little population structure in Mongolian Bactrian camels. This leads us to the conclusion that the phenotypic differentiation observed in the Mongolian breeds might be due mainly to recent anthropogenic selection, which will change allele frequencies in selected genes rather than in neutral markers. Future studies using candidate-gene or genome-wide approaches will be useful to detect loci under selection for breed differentiation and to target potential effects of the recent population decline.
Acknowledgments
We express our sincere thanks to the Mongolian Academy of Sciences (MAS) for granting official permission for Mongolian domestic Bactrian camel research. We are most grateful to Ts. Janchiv (head: Institute of Biology, MAS), D. Enkhnasan, D. Altanchimeg, G. Tsogtjargal and all breeders providing samples and information. P. Burger is recipient of an APART fellowship, Austrian Academy of Science and acknowledges funding from the Austrian Science Foundations FWF P24706 and the Eurasia-Pacific Uninet (EPU-31/2011).
Footnotes
Soum: second level administrative subdivision of Mongolia, equivalent to the district. The 21 provinces of Mongolia are divided into 329 soums.
Supporting information
Additional supporting information may be found in the online version of this article.
Figure S1 Bayesian analysis of population structure (baps) of 83 mitochondrial sequences (803 bp).
Figure S2 Distribution Delta K of Mongolian domestic camel.
Figure S3 Individual assignment probabilities of 150 Mongolian Bactrian camels to 2–6 theoretical genetic ancestry groups using structure software.
Figure S4 Mismatch distribution based on the pairwise differences between individuals.
Table S1 Details on the samples used in this study.
Table S2 Seventeen microsatellite markers applied in the Bactrian camel (Camelus bactrianus).
Table S3 Inbreeding coefficients FIS per microsatellite locus.
References
- 1.Allendorf FW, Luikart G. Conservation and the Genetics of Population. Oxford: Blackwell Publishing Ltd; 2007. p. 642. [Google Scholar]
- 2.Baldan T. The Studying of Domestic Camel in the Last 50 Years. Ulaanbataar: Institute of Animal Husbandry; 2011. p. 127. (in Mongolian language) [Google Scholar]
- 3.Bandelt H, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 4.Belkir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. France: Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier; 1999. –2004) [Google Scholar]
- 5.Burger P. Genetic traces of domestication in Old World camelids. In: Knoll EM, Burger PA, editors. Camels in Asia and North Africa – Interdisciplinary Perspectives on their Past and Present Significance. Vienna: Austrian Academy of Sciences Press; 2012. pp. 18–24. [Google Scholar]
- 6.Chuluunbaatar O. The cultural and ethnic identity of the Oirat peoples. In: Lehner E, Harrer A, Sint H, editors. Along the Great Wall: Architecture and Identity in China and Mongolia. Vienna: IVA-ICRA; 2010. pp. 165–72. [Google Scholar]
- 7.Chuluunbat B, Ulziisaihan T, Tumennasan K. Population genetic study of Mongolian two humped camel (Camelus bactrianus. Proceedings of Institute of Biology. 2012;28:17–20. [Google Scholar]
- 8.Corander J, Tang J. Bayesian analysis of population structure based on linked molecular information. Mathematical Biosciences. 2007;205:19–31. doi: 10.1016/j.mbs.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Corander J, Majander KK, Lu Cheng L, Merilä J. High degree of cryptic population differentiation in the Baltic Sea herring Clupea harengus. Molecular Ecology. 2013;22:2931–40. doi: 10.1111/mec.12174. [DOI] [PubMed] [Google Scholar]
- 10.Cornuet JM, Luikart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1997;144:2001–14. doi: 10.1093/genetics/144.4.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Defaveri J, Shikano T, Shimada Y, Merilä J. High degree of genetic differentiation in marine three-spined sticklebacks (Gasterosteus aculeatus. Molecular Ecology. 2013;22:4811–28. doi: 10.1111/mec.12430. [DOI] [PubMed] [Google Scholar]
- 12.DeWoody JA, Avise JC. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. Journal of Fish Biology. 2000;56:461–73. [Google Scholar]
- 13.Dieringer D, Schlötterer C. microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes. 2003;3:167–9. [Google Scholar]
- 14.Dorjgotov D. National Atlas of Mongolia. Ulaanbataar: Institute of Geology; 2009. p. 199. [Google Scholar]
- 15.Earl D, vonHoldt B. structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources. 2012;4:359–61. [Google Scholar]
- 16.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology Notes. 2005;14:2611–20. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 17.Excoffier L, Lischer H. arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10:564–7. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 18.Fu YX. Statistical test of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grosberg R, Cunningham CW. Genetic structure in the sea: from populations to communities. In: Bertness MD, Gaines SD, Hay ME, editors. Marine Community Ecology. Sunderland: Sinauer Associates; 2001. pp. 61–84. [Google Scholar]
- 20.Jacobsson M, Rosenberg NA. clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–6. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 21.Ji R, Cui P, Ding F, Geng J, Gao H, Zhang H, Yu J, Hu S, Meng H. Monophyletic origin of domestic Bactrian camel (Camelus bactrianus) and its evolutionary relationship with the extant wild camel (Camelus bactrianus ferus. Animal Genetics. 2009;40:377–82. doi: 10.1111/j.1365-2052.2008.01848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jianlin H, Ochieng JW, LKhagva B, Hanotte O. Genetic diversity and relationship of domestic Bactrian camels (Camelus bactrianus) in China and Mongolia. Journal of Camel Practice and Research. 2004;11:97–9. [Google Scholar]
- 23.Jirimutu, Bactrian Camel Genome Consortium Genome sequences of wild and domestic bactrian camels. Nature Communications. 2012;3:1202. doi: 10.1038/ncomms2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koshimoto C, Amano T, Nozawa K, Tanabe Y, Munkhtoyaa B, Tumennasan H, Zhanchiv T. Blood protein/enzyme polymorphisms in the two humped camel (Camelus bactrianus) of Mongolia. Report of the Society for Researches on Native Livestock. 1999;17:95–102. [Google Scholar]
- 25.Lau AN, Peng L, Goto H, Chemnick L, Ryder OA, Makova KD. Horse domestication and conservation genetics of Przewalski's horse inferred from sex chromosomal and autosomal sequences. Molecular Biology and Evolution. 2009;26:199–208. doi: 10.1093/molbev/msn239. [DOI] [PubMed] [Google Scholar]
- 26.Librado P, Rozas J. dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 27.Mburu DN, Ochieng JW, Kuria SG, Jianlin H, Kaufmann B, Rege JEO, Hanotte O. Genetic diversity and relationships of indigenous Kenyan camel (Camelus dromedarius) populations: implications for their classification. Animal Genetics. 2003;34:26–32. doi: 10.1046/j.1365-2052.2003.00937.x. [DOI] [PubMed] [Google Scholar]
- 28.Mills LS, Allendorf FW. The one-migrant-per-generation rule in conservation and management. Conservation Biology. 1996;10:1509–18. [Google Scholar]
- 29.Nei M. Molecular Evolutionary Genetics. New York: Columbia University Press; 1987. p. 512. [Google Scholar]
- 30.Peakall R, Smouse PE. genalex 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics. 2012;28:2537–9. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters J, von den Driesch A. The two-humped camel (Camelus bactrianus): new light on its distribution, management and medical treatment in the past. Journal of Zoology. 1997;242:651–79. [Google Scholar]
- 32.Pfeiffer I, Volkel I, Taubert H, Brenig B. Forensic DNA-typing of dog hair: DNA-extraction and PCR amplification. Forensic Science International. 2004;141:149–51. doi: 10.1016/j.forsciint.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Piry S, Luikart G, Cornuet J-M. bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. Journal of Heredity. 1999;90:502–3. [Google Scholar]
- 34.Posada D. jModelTest: Phylogenetic model averaging. Molecular Biology and Evolution. 2008;25:1253–6. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 35.Pritchard J, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution. 1992;9:552–69. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 37.Schneider S, Excoffier L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999;152:1079–89. doi: 10.1093/genetics/152.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silbermayr K, Orozco-terWengel P, Charruau P, Enkhbileg D, Walzer C, Vogl C, Schwarzenberger F, Kaczensky P, Burger PA. High mitochondrial differentiation levels between wild and domestic Bactrian camels: a basis for rapid detection of maternal hybridization. Animal Genetics. 2010a;41:315–8. doi: 10.1111/j.1365-2052.2009.01993.x. [DOI] [PubMed] [Google Scholar]
- 39.Silbermayr K, Tero N, Charruau P, Enkhbileg D, Walzer C, Burger PA. Isolation and characterization of nine new microsatellite loci in the domestic Bactrian camel (Camelus bactrianus) and amplification in the wild Bactrian camel (C. ferus. Molecular Ecology Resources. 2010b;10:1106–8. [Google Scholar]
- 40.Silvertown JW, Charlesworth B. Introduction to Plant Population Biology. 4th edition. Oxford: Wiley-Blackwell; 2001. p. 65. [Google Scholar]
- 41.Spencer PBS, Woolnough AP. Assessment and genetic characterisation of Australian camels using microsatellite polymorphisms. Livestock Science. 2010;129:241–5. [Google Scholar]
- 42.Tapio M, Miceikiené I, Vilkki J, Kantanen J. Comparison of microsatellite and blood protein diversity in sheep: inconsistencies in fragmented breeds. Molecular Ecology. 2003;12:2045–56. doi: 10.1046/j.1365-294x.2003.01893.x. [DOI] [PubMed] [Google Scholar]
- 43.Trinks A, Burger PA, Beneke N, Burger J. Simulations of populations ancestry of the two-humped camel (Camelus bactrianus. In: Knoll EM, Burger PA, editors. Camels in Asia and North Africa. Interdisciplinary Perspectives on their Significance in Past and Present. Vienna: Austrian Academy of Sciences Press; 2012. pp. 79–86. [Google Scholar]
- 44.Wang J. coancestry: a program for simulating, estimating and analyzing relatedness and inbreeding coefficients. Molecular Ecology Resources. 2011;11:141–5. doi: 10.1111/j.1755-0998.2010.02885.x. [DOI] [PubMed] [Google Scholar]
- 45.Warmuth V, Eriksson A, Bower MA, et al. Reconstructing the origin and spread of horse domestication in the Eurasian steppe. Proceedings of the National Academy of Sciences of the United States of America. 2012;22:8202–6. doi: 10.1073/pnas.1111122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watterson G. On the number of segregating sites in genetical models without recombination. Theoretical Population Biology. 1975;7:256–76. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 47.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Bayesian analysis of population structure (baps) of 83 mitochondrial sequences (803 bp).
Figure S2 Distribution Delta K of Mongolian domestic camel.
Figure S3 Individual assignment probabilities of 150 Mongolian Bactrian camels to 2–6 theoretical genetic ancestry groups using structure software.
Figure S4 Mismatch distribution based on the pairwise differences between individuals.
Table S1 Details on the samples used in this study.
Table S2 Seventeen microsatellite markers applied in the Bactrian camel (Camelus bactrianus).
Table S3 Inbreeding coefficients FIS per microsatellite locus.