

Abstract
Mycobacterium indicus pranii (MIP) is an atypical mycobacterial species possessing strong immunomodulatory properties. It is a potent vaccine candidate against tuberculosis, promotes Th1 immune response and protects mice from tumours. In previous studies, we demonstrated higher protective efficacy of MIP against experimental tuberculosis as compared with bacillus Calmette–Guérin (BCG). Since macrophages play an important role in the pathology of mycobacterial diseases and cancer, in the present study, we evaluated the MIP in live and killed form for macrophage activation potential, compared it with BCG and investigated the underlying mechanisms. High levels of tumour necrosis factor-α, interleukin-12p40 (IL-12p40), IL-6 and nitric oxide were produced by MIP-stimulated macrophages as compared with BCG-stimulated macrophages. Prominent up-regulation of co-stimulatory molecules CD40, CD80 and CD86 was also observed in response to MIP. Loss of response in MyD88-deficient macrophages showed that both MIP and BCG activate the macrophages in a MyD88-dependent manner. MyD88 signalling pathway culminates in nuclear factor-κB/activator protein-1 (NF-κB/AP-1) activation and higher activation of NF-κB/AP-1 was observed in response to MIP. With the help of pharmacological inhibitors and Toll-like receptor (TLR) -deficient macrophages, we observed the role of TLR2, TLR4 and intracellular TLRs in MIP-mediated macrophage activation. Stimulation of HEK293 cells expressing TLR2 in homodimeric or heterodimeric form showed that MIP has a distinctly higher level of TLR2 agonist activity compared with BCG. Further experiments suggested that TLR2 ligands are well exposed in MIP whereas they are obscured in BCG. Our findings establish the higher macrophage activation potential of MIP compared with BCG and delineate the underlying mechanism.
Keywords: bacterial, cytokines, Toll-like receptors
Introduction
Mycobacterium indicus pranii (MIP), earlier known as Mycobacterium w, was selected from a panel of mycobacteria for its ability to evoke a cell-mediated immune response against Mycobacterium leprae.1,2 Its use as an adjunct to standard chemotherapy against multibacillary leprosy resulted in significantly enhanced bacillary clearance and shortened recovery time. Protective effects against leprosy and tuberculosis were also observed in the household contacts of leprosy patients.3–5 MIP was further evaluated against Mycobacterium tuberculosis and provided protection against experimental tuberculosis in both BCG-responder as well as non-responder strains of mice.6 Consistent with the beneficial effects shown against tuberculosis and leprosy, MIP shares antigenic determinants with M. leprae and M. tuberculosis.7 Underscoring its uniqueness, recent studies have placed MIP between slow-growing and fast-growing species.8 Further studies suggested that MIP possesses strong immunomodulatory properties and enhances the immune response when used as an adjuvant along with a recombinant anti-human chorionic gonadotrophin vaccine.9 Heat-killed MIP is approved for human use and is available commercially.
Macrophages are major phagocytic cells strategically located throughout the body to clear invading pathogens and cellular debris. Classically activated macrophages produce pro-inflammatory cytokines, reactive nitrogen species, antimicrobial peptides and offer protection against a variety of bacteria, protozoa and viruses.10 Pathogenic bacteria such as M. tuberculosis, however, have evolved the mechanisms to evade the microbicidal activities of macrophages. Mycobacterium tuberculosis inhibits the phagolysosomal fusion and uses the phagosomal compartments as niches for its growth and multiplication. Similarly, the Leishmania parasite exploits the macrophages for its growth and multiplication. Interestingly, MIP-activated macrophages have been shown to contain the Leishmania parasite in an inducible nitric oxide synthase-dependent manner.11 Classically activated macrophages can also lyse tumour cells and activate CD8+ T cells to offer protection from tumour.12,13 However, macrophages possess a high degree of plasticity and their normal function is altered in neoplastic diseases. Growing tumours promote M2 macrophages and take advantage of their tissue-repairing and homeostatic properties. Tumour-associated macrophages produce high levels of vascular endothelial growth factor, interleukin-8 (IL-8) and IL-10, which support tumour growth.14 Accordingly, high infiltration of tumour-associated macrophages is associated with poor prognosis of malignant disease.
In previous studies, we observed that live MIP provides significantly strong protection against tuberculosis in mice and guinea pigs compared with killed MIP and bacillus Calmette–Guérin (BCG). Although MIP is approved for human use in heat-killed form, it is a non-pathogenic mycobacterium and is cleared from the host within 4–6 weeks. We therefore used MIP in both live and killed forms in our studies. Analysis of the immune response revealed that MIP promotes the Th1 response and leads to interferon-γ production by lung-infiltrating immune cells.15,16 The whole genome sequence of MIP has been published recently and consistent with our findings in silico analysis demonstrated a much higher fraction of putative antigenic proteins in MIP compared with BCG.17 Given the importance of Th1 immune response in protection against cancer, we also evaluated MIP for cancer immunotherapy. Significant protection from tumour and activation of an anti-tumour immune response were observed in MIP-treated mice.18 Similar findings have been reported by others and the role of interferon-γ has been demonstrated in the MIP-mediated anti-tumour response.19 Since macrophages play a central role in the pathology of mycobacterial diseases and cancer, in the present study, we evaluated MIP in both live and heat-killed form for macrophage activation potential and examined the underlying mechanism. As BCG is a widely used vaccine against tuberculosis and is also effective against superficial bladder cancer,20 we included BCG in this study and compared it with MIP. Our results showed higher activation of macrophage by live MIP compared with killed MIP and BCG. Both MIP and BCG activated the macrophages in MyD88 and nuclear factor-κB/activator protein-1 (NF-κB/AP1) -dependent manner. With the help of pharmacological inhibitors and Toll-like receptor (TLR) -deficient macrophages, we noted the role of TLR2, TLR4 and intracellular TLRs in macrophage activation by MIP. Using HEK293 cells expressing TLR2, we further observed that MIP and BCG have differential TLR2 engagement properties. Our findings demonstrate higher macrophage activation potential of MIP and implicate the role of TLRs in differential activation of macrophages by MIP and BCG.
Materials and methods
Ethics statement
All animal experiments described in this manuscript were approved by the institutional animal ethics committee of the National Institute of Immunology, New Delhi and performed in accordance with the guidelines from the same: IAEC approval no. 205/08/11.
Mice
Six- to eight-week-old inbred C57BL/6, IL10−/− and MyD88−/− mice were obtained from the animal facility of the National Institute of Immunology, New Delhi. TLR2−/− and TLR4−/− mice were a kind gift from Dr Ruslan Medzhitov (Yale University School of Medicine, New Haven, CT) to Dr Gobardhan Das and were maintained at the International Centre for Genetic Engineering and Biotechnology, New Delhi.
Mycobacteria
Glycerol stocks of MIP and BCG Danish 1331 were thawed and cultured in 7H9 medium containing 0·05% Tween-80 and 0·05% glycerol, and supplemented with 10% albumin-dextrose-catalase. MIP was cultured in a shaker incubator (100 r.p.m.) and BCG was grown as a standing culture, with intermittent manual shaking, at 36°. Log-phase cultures were harvested by centrifugation at 2000 g and washed in PBS containing 3% fetal bovine serum (PBS-3). Bacterial aggregates were removed from BCG suspension by additional centrifugation of suspension at 50 g for 10 min. Purity of cultures was tested by plating MIP and BCG cultures on nutrient agar plates. Counts of MIP and BCG were determined on the basis of optical density. Heat-killed MIP was prepared by autoclaving bacterial suspension at 15 psi for 15 min.
Isolation of peritoneal macrophages
One millilitre aged thioglycollate broth was given intraperitoneally per mouse and peritoneal exudate cells were harvested after 96 hr from sacrificed mice. Macrophages were purified on the basis of plastic adherence. Peritoneal exudate cells were plated in cell culture flasks in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution (RPMI-10). After 2 hr, non-adherent cells were removed by three or four washes with PBS and macrophages were harvested by incubating the cells with non-enzymatic cell dissociation solution followed by scraping.
Stimulation of RAW 264.7 macrophages, peritoneal macrophages and HEK 293 cells
RAW 264.7 macrophages were maintained in RPMI-10 medium. For stimulation, 3·0 × 105 cells were plated per well in 96-well flat-bottom plates in duplicate. Heat-killed MIP (MIP-K), live MIP (MIP-L) and BCG were added to the culture at the indicated multiplicities of stimulation (MOS) and culture supernatants were collected after 24 hr. For kinetics studies, MIP-K, MIP-L and BCG were used at MOS of 10 and culture supernatants were collected after 12, 24 and 36 hr. For peritoneal macrophage stimulation, 1·5 × 106 cells were plated per well of 24-well plates and stimulated with MIP-K, MIP-L or BCG at the indicated MOS in the presence of 20 U/ml recombinant murine interferon-γ. Culture supernatants were collected after 24 hr and stored at −20°. 293tlr2, 293tlr2/1, and 293tlr2/6 cells were purchased from Invivogen (San Diego, CA) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (DMEM-10), 1% penicillin-streptomycin solution, 10·0 μg/ml blasticidin S and 100 μg/ml Normocin. For stimulation, 1·0 × 105 cells were plated per well in 96-well flat-bottom plates and MIP-K, MIP-L or BCG was added in native or sonicated form at the MOS of 50. Culture supernatants were collected at 48 hr and analysed for IL-8.
Measurement of cytokines and nitric oxide
Cytokine level in the culture supernatants was determined according to the instruction-manual provided with the ELISA kits purchased from BD Biosciences (San Diego, CA) or e-Bioscience (San Diego, CA). Nitric oxide (NO) level was determined with the help of Griess reagents system and optical density was measured at 540 nm.
Flow cytometry
RAW 264.7 cells were cultured in 24-well plates (1.5x106 cells/well) and stimulated with MIP-K, MIP-L and BCG at MOS of 10 in the presence of 20 U/ml interferon-γ. Cells were harvested after 24 hr and incubated with phycoerythrin-conjugated anti-mouse CD40, CD80, and CD86 (purchased from BD Biosciences or e-Bioscience) and washed with ice-cold PBS. Stained cells were run on BD LSR Flow Cytometer and data were analysed with Cyflogic 1.2.1 software (CyFlo Ltd, Turku, Finland).
Analysis of NF-κB/AP-1 activation
For analysis of NF-κB/AP-1 activation, 1·0 × 105 RAW-Blue cells (purchased from Invivogen) were cultured per well of 96-well plates in DMEM-10 supplemented with 200 μg/ml Zeocin and 100 μg/ml Normocin and stimulated with MIP or BCG and the indicated MOS. Culture supernatants were collected after 24 hr and secreted embryonic alkaline phosphatase (SEAP) level was determined with QUANTI-Blue substrate (Invivogen), according to the instructions provide by the supplier.
Inhibition of NF-κB/AP-1 and TLR signalling
RAW 264.7 cells were cultured in 96-well plates (3×105 cells/well) in RPMI-10 medium and incubated with curcumin, Cli095, OxPAPC or chloroquine, for inhibition of NF-κB/AP-1, TLR4, TLR2 and TLR4, and intracellular TLRs, respectively, at indicated concentrations. After 1 hr, MIP and BCG were added to the culture. Culture supernatants were collected after 24 hr and analysed for tumour necrosis factor-α (TNF-α) level.
Sonication of bacteria
The MIP-K, MIP-L and BCG suspended in DMEM-10 were sonicated for 10 cycles (1 min on, 1 min off) at 60% power using a Sonoplus sonicator (Bandelin Electronics, Berlin, Germany).
Statistical analysis
All statistical analyses were performed with the help of GraphPad Prism 5.0 software (GraphPad, San Diego, CA). Data were analysed by one-way analysis of variance with Tukey's multiple comparison test applied post analysis. A P value of < 0·05 was considered significant.
Results
MIP-stimulated macrophages produced higher levels of pro-inflammatory cytokines compared with BCG-stimulated macrophages
Peritoneal macrophages were stimulated with MIP-K, MIP-L or BCG at increasing MOS and culture supernatants were analysed for pro-inflammatory cytokines. Significantly high levels of TNF-α, IL-12p40 and IL-6 were produced by MIP-L-stimulated macrophages compared with MIP-K-stimulated and BCG-stimulated macrophages (Fig. 1a–c). Moderate levels of TNF-α, IL-12p40 and IL-6 were produced in response to MIP-K. Both MIP-K and MIP-L induced significantly high levels of these cytokines compared with BCG. Macrophage killing in response to MIP and BCG, as determined on the basis of propidium iodide staining, was observed to be comparable with unstimulated macrophages (data not shown). It indicated that MIP and BCG do not show significant cytotoxicity against macrophages at MOS of ≤ 20. Further, MIP-K-stimulated and MIP-L-stimulated macrophages produced high levels of NO compared with BCG-stimulated macrophages but the difference was not significant (Fig. 1d). Reactive nitrogen species are important effector molecules produced by activated macrophages to kill intracellular pathogens and tumour cells. MIP-K, MIP-L and BCG led to dose-dependent increases in the production of pro-inflammatory cytokines and NO by macrophages.
Figure 1.
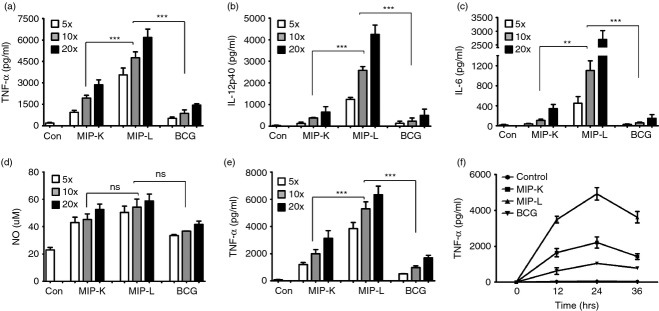
Production of pro-inflammatory cytokines and nitric oxide (NO) by heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG) -stimulated macrophages. Peritoneal macrophages were stimulated with MIP-K, MIP-L and BCG and culture supernatants were analysed by ELISA. Higher levels of tumour necrosis factor-α (TNF-α), interleukin-12p40 (IL-12p40) and IL-6 production was observed in response to MIP-L. MIP-K led to moderate level of these cytokines (a–c). High level of NO was also observed in MIP-L and MIP-K-stimulated macrophages compared with BCG, but difference was not significant. (d). RAW 264.7 macrophages were also stimulated with MIP-K, MIP-L and BCG, and a high level of TNF-α was produced in response to MIP-L. A moderate level of TNF-α was produced by MIP-K-stimulated RAW 264.7 macrophages (e). Time-kinetics of RAW 264.7 macrophage response to MIP-K, MIP-L and BCC was studied and at all time-points, higher levels of TNF-α production were observed in response to MIP-L followed by MIP-K (f). Data shown are mean ± SEM of three independent experiments. **P < 0·01, ***P < 0·001 and ns, not significant.
RAW 264.7 macrophages were also evaluated for their response to MIP and BCG. RAW 264.7 is an adherent murine monocyte/macrophage cell line derived from Abelson murine leukaemia virus-induced tumour.21 RAW 264.7 cells produced TNF-α in a dose-dependent manner when stimulated with MIP-K, MIP-L and BCG. Level of TNF-α produced in response to MIP-L was significantly high compared with that produced in response to MIP-K and BCG (Fig. 1e). As BCG inhibits phagolysosomal fusion to some extent, we wondered if BCG could induce a higher response at a later time-point. But time-kinetics of TNF-α production by RAW 264.7 macrophages in response to MIP and BCG showed that MIP-L induces a higher level of TNF-α at all time-points (Fig. 1f). These data showed that MIP leads to significantly high levels of pro-inflammatory cytokine production by primary macrophage as well as RAW 264.7 macrophage cell line as compared with BCG.
MIP-stimulated macrophages showed higher up-regulation of co-stimulatory molecules compared with BCG-stimulated macrophages
Antigen-presenting cells up-regulate the expression of co-stimulatory molecules upon activation. These co-stimulatory molecules interact with their cognate receptors and lead to activation of the adaptive immune response.22 We asked whether macrophages stimulated with MIP-K, MIP-L or BCG up-regulates the expression of CD40, CD80 and CD86. To examine this, RAW 264.7 macrophages were stimulated with MIP-K, MIP-L and BCG for 24 hr, stained with fluorochrome-conjugated antibodies, and analysed by flow cytometry. Substantially high expression of CD40, CD80 and CD86 was observed in the macrophages stimulated with MIP-L (Fig. 2). Maximum up-regulation was observed in the expression of CD40 among all three molecules analysed. Moderate expression of co-stimulatory molecules was observed in MIP-K-stimulated macrophages. Cytokine and co-stimulatory markers data established the high activation of macrophages by MIP compared with BCG.
Figure 2.

Expression of activation markers by macrophages stimulated with heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG). RAW 264.7 macrophages were stimulated with MIP-K, MIP-L and BCG at a multiplicity of stimulation of 10. Cells were stained with phycoerythrin-conjugated anti-mouse CD40, CD80 and CD86 monoclonal antibodies and analysed by flow cytometry. MIP-L was leading to higher up-regulation of co-stimulatory molecules. Moderate up-regulation was observed in response to MIP-K. Representative histograms are shown. Composite Mean Fluorescence Intensity (= % positive cells × mean fluorescence intensity of positive cells) is also shown as a bar graph. Data shown are mean ± SEM of two independent experiments. *P < 0·05.
Differential macrophage activation in response to MIP and BCG is not mediated by IL-10
Interleukin-10 is an immune-suppressive cytokine with pleiotropic effects including down-regulation of co-stimulatory molecules and decreased production of pro-inflammatory cytokines by antigen-presenting cells. Pathogenic mycobacteria and BCG are known to induce IL-10 production by dendritic cells. Interleukin-10 acts in an autocrine manner to impair dendritic cell activation and antigen-presenting functions.23,24 We examined the putative role of IL-10 in the differential response of macrophages to MIP and BCG. But we did not observe a significant increase in IL-10 production by MIP-K-, MIP-L- and BCG-stimulated macrophages compared with unstimulated macrophages (data not shown). This indicated that IL-10 has no role in the differential activation of macrophages by MIP and BCG. It was further confirmed with the help of IL-10-deficient macrophages. MIP-K, MIP-L and BCG led to similar responses from wild-type and IL-10-deficient macrophages, as indicated by TNF-α and IL-12p40 production (Fig. 3a,b). This ruled out involvement of IL-10 differential activation of macrophages by MIP and BCG.
Figure 3.
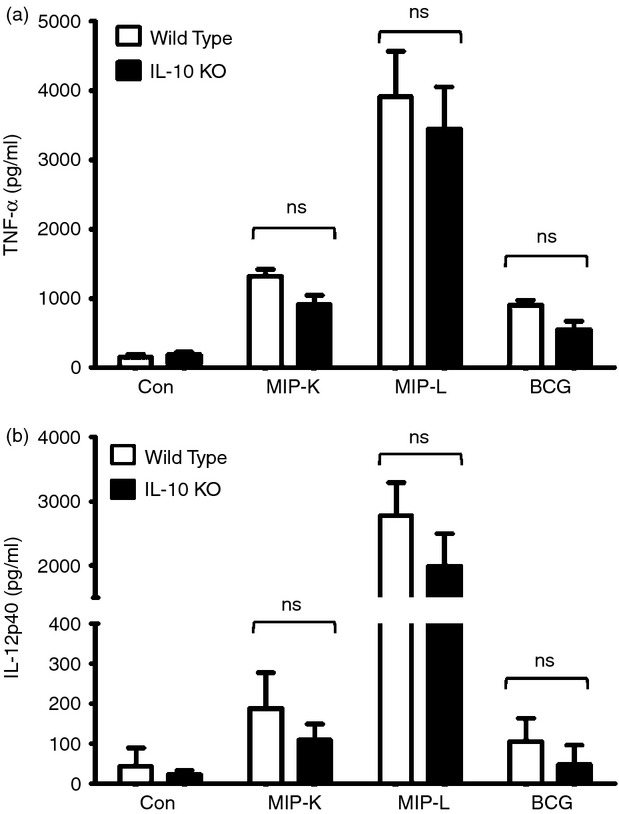
Response of interleukin-10 deficient (IL-10−/−) and wild-type macrophages to heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG). Peritoneal macrophages from IL-10−/− and wild-type mice were stimulated with MIP-K, MIP-L and BCG at a multiplicity of stimulation of 10. A similar response in terms of tumour necrosis factor-α (TNF-α) and IL-12-p40 production was observed in IL-10−/− and wild-type macrophages. Data shown are mean ± SEM of three independent experiments. ns, not significant.
Both MIP and BCG activated the macrophages through MyD88- and NF-κB/AP-1-dependent pathway
Toll-like receptors are evolutionarily conserved pattern recognition receptors playing major roles in the recognition of microbes and their products by immune cells. MyD88 adapter protein is associated with cytoplasmic tail of TLRs and passes information inside cells.25,26 To examine the role of MyD88 in macrophage activation, wild-type and MyD88-deficient macrophages were stimulated with MIP-K, MIP-L and BCG and culture supernatants were analysed for pro-inflammatory cytokines. Loss of response to MIP and BCG as indicated by drastically reduced TNF-α and IL-12p40 production, was observed in MyD88-deficient macrophages (Fig. 4a,b). This showed that both MIP and BCG activate the macrophages in a MyD88-dependent manner.
Figure 4.
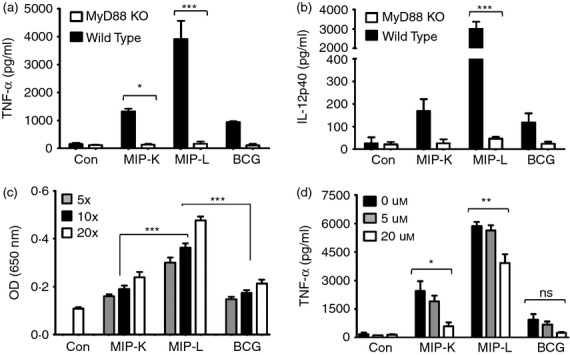
Role of MyD88 and nuclear factor-κB/activator protein 1 ((NF-κB/AP-1) in heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG) mediated activation of macrophages. Peritoneal macrophages from MyD88−/− and wild-type mice were stimulated with MIP-K, MIP-L and BCG. Response to MIP and BCG was lost in MyD88−/− macrophages, as shown by drastically reduced levels of tumour necrosis factor-α (TNF-α) and interleukin-12 p40 (IL-12p40) in culture supernatants (a,b). Role of NF-κB/AP-1 in macrophage activation by MIP or BCG was determined by stimulating RAW-Blue cells, which express NF-κB/AP-1 inducible secreted alkaline phosphatase (SEAP). Higher levels of SEAP activity were observed in response to MIP-L (c). The role of NF-κB/AP-1 in MIP-mediated macrophage activation was determined with the help of pharmacological inhibitor. RAW 264.7 macrophages were incubated with curcumin at the indicated concentration for 30 min and then stimulated with MIP-K, MIP-L and BCG. Dose-dependent decrease in TNF-α production in response to MIP-K, MIP-L and BCG was observed in the presence of curcumin (d). Data shown are mean ± SEM of three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 and ns, not significant.
Signalling through MyD88 culminates in the translocation of NF-κB and AP-1 into nucleus and subsequent transcription of inducible genes including those encoding for pro-inflammatory cytokines.26 With the help of NF-κB/AP-1-inducible SEAP-expressing Raw-Blue cells, we noted significantly high NF-κB/AP-1 activation in response to MIP-L (Fig. 4c). Dose-dependent increase in SEAP level was induced by both MIP and BCG. The role of NF-κB/AP-1 in MIP- and BCG-mediated macrophage activation was further confirmed by stimulating RAW 264.7 macrophages in the presence of curcumin – an inhibitor of NF-κB/AP-1.27 Reduced production of TNF-α in response to MIP and BCG was observed in the presence of curcumin (Fig. 4d). These results showed that both MIP and BCG activate the macrophage through an MyD88- and NF-κB/AP-1-dependent pathway.
TLR2, TLR4 and intracellular TLRs have distinct roles in MIP-mediated macrophage activation
Loss of response in MyD88-deficient macrophages indicated the major role of TLRs in macrophage activation by MIP. Contribution of different TLRs in MIP-mediated macrophage activation was therefore further examined. RAW 264.7 macrophages were stimulated with MIP and BCG in the presence of pharmacological inhibitors of TLR4 (Cli095), TLR2 and TLR4 (OxPAPC) and intracellular TLRs (chloroquine)28 and TNF-α level was examined in the culture supernatants. Diminished TNF-α production by MIP-K-, MIP-L- and BCG-stimulated macrophages was noted in the presence of Cli095 (Fig. 5a). This indicated the role of TLR4 in MIP- and BCG-mediated macrophage activation. Decreased TNF-α production in response to MIP-L was also observed in the presence of chloroquine, indicating the role of intracellular TLRs in macrophage activation by MIP-L (Fig. 5b). However, a dramatic reduction in the TNF-α production by MIP-K-, MIP-L- and BCG-stimulated macrophage was observed in the presence of OxPAPC, which indicated the major role of TLR2 in MIP-mediated and BCG-mediated macrophage activation (Fig. 5c).
Figure 5.
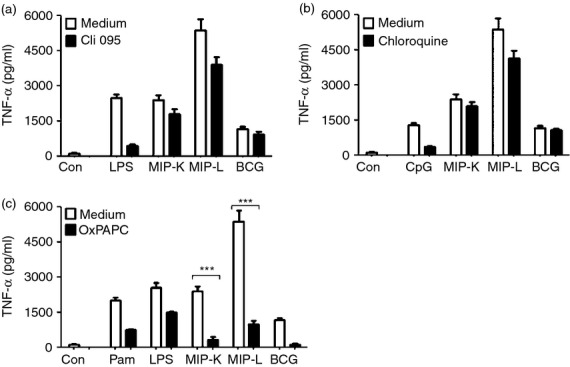
Role of Toll-like receptors (TLRs) in macrophage activation by heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG). RAW 264.7 macrophages were incubated with 1·5 μg/ml Cli095 (TLR4 inhibitor), 80 μg/ml OxPAPC (TLR2 and TLR4 inhibitor) or 20 μm chloroquine (intracellular TLR inhibitor) for 60 min. MIP-K, MIP-L and BCG were subsequently added at multiplicities of infection of 10 and culture supernatants were analysed for tumour necrosis factor-α (TNF-α) after 24 hr. Diminished levels of TNF-α production by MIP-K, MIP-L and BCG stimulated macropahges were observed in the presence of Cli095 (a). Decreased TNF-α production by MIP-L-stimulated macrophages was also observed in the presence of chloroquine (b). Dramatic reduction in TNF-α production by MIP-K, MIP-L and BCG-stimulated macrophages was observed in the presence of OxPAPC (c). Pam-Pam3CSK4, LPS-lipopolysachharide, CpG-CpG DNA were used as positive controls. Data shown are mean ± SEM of three independent experiments. ***P < 0·001 and ns, not significant.
The role of TLRs in macrophage activation by MIP was further confirmed with the help of macrophages isolated from TLR2−/− and TLR4−/− mice. Consistent with the results obtained with pharmacological inhibitors, decreased production of TNF-α and IL-12p40 in response to MIP and BCG was observed in TLR4-deficient macrophages. A dramatic reduction in TNF-α and IL-12p40 production in response to MIP and BCG stimulation, however, was observed in TLR2-deficient macrophages (Fig. 6a,b). These results confirmed the major role of TLR2 in macrophage activation by MIP and BCG.
Figure 6.
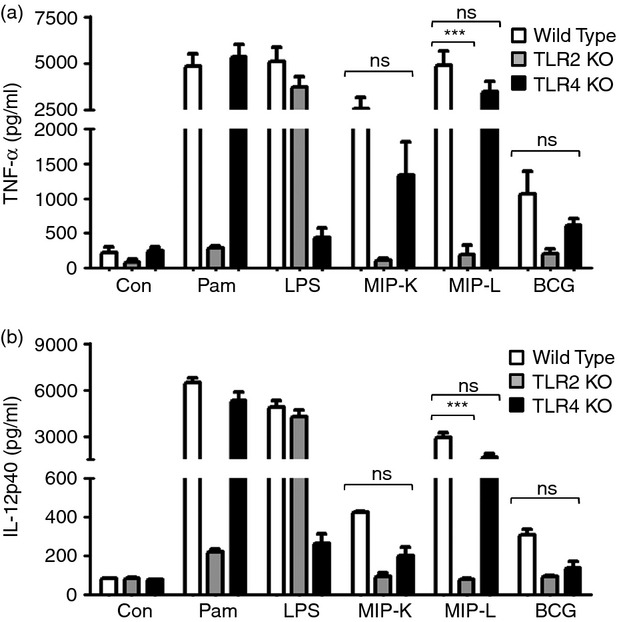
Response of Toll-like receptor (TLR) -deficient macrophages to heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG). Peritoneal macrophages from TLR2−/−, TLR4−/− and wild-type mice were stimulated with MIP-K, MIP-L and BCG at multiplicities of infection of 10. Culture supernatants were analysed for tumour necrosis factor-α (TNF-α) and interleukin-12 p40 (IL-12p40) by ELISA. Diminished production of TNF-α and IL-12p40 in response to MIP-K, MIP-L and BCG was observed in TLR4−/− macrophages. Dramatic reduction in the TNF-α and IL-12p40 production in response to MIP-K, MIP-L and BCG was observed in TLR2−/− macrophages. Pam (Pam3CSK4, 10 ng/ml) and LPS (Lipopolysaccharide, 10 ng/ml) were used as positive controls. Data shown are mean ± SEM of two independent experiments. ***P < 0·001 and ns, not significant.
MIP-K, MIP-L and BCG have differing TLR2 agonist property
Toll-like receptor 2 can recognize a wide array of microbial molecules, which include peptidoglycans, lipoteichoic acid, lipoarabinomannan, zymosan and lipoproteins. The remarkable ability of TLR2 to function as a homodimer or as heterodimer with TLR1 and TLR6 is responsible for the recognition of a wide spectrum of microbial molecules by TLR2.29 Since the above studies showed the major role of TLR2 in macrophage activation by MIP and BCG, we further delineated the TLR2 engagement by MIP and BCG. A well-established HEK293-TLR system was used to examine this.30 HEK293 cells expressing murine TLR2/2 homodimer (293tlr2/2 cells), TLR2/1 heterodimer (293tlr2/1 cells) and TLR2/6 heterodimer (293tlr2/6 cells) were stimulated with MIP-K, MIP-L or BCG in native or sonicated form and culture supernatants were analysed for IL-8. The sonicated form was used to expose the potential TLR ligands, which might be hidden or inaccessible in the native form of mycobacteria.
Interleukin-8 production by 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells in response to native MIP-L indicated that native MIP-L engages all three forms of TLR2, namely TLR2/2, TLR2/1 and TLR2/6 (Fig. 7a,b,c, respectively). In comparison to 293tlr2/2 cells and 293tlr2/6 cells, the 293tlr2/1 cells showed a high response to native MIP-L, indicating a higher level of TLR2/1 ligands in native MIP-L. Further, native MIP-L induced significantly higher levels of IL-8 by 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells in comparison to native MIP-K and BCG, suggesting a higher level of TLR2 ligands in native MIP-L. Comparable levels of IL-8 were produced in response to native MIP-L and sonicated MIP-L, indicating that TLR2 ligands are well exposed in the native MIP-L and sonication does not help to further increase the availability of TLR2 ligands. Native MIP-K showed decreased engagement of TLR2/1, TLR2/6 and TLR2/2 compared with MIP-L. Surprisingly, BCG, when used in native form, showed minimal TLR2 agonist properties. Comparable levels of IL-8 were produced by unstimulated and native BCG-stimulated 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells. Interestingly, however, when used in sonicated form, BCG engaged all three form of TLR2. This showed that TLR2 ligands in native BCG are not well exposed and sonication increases the availability of ligands leading to substantial IL-8 production. Increased IL-8 production was also observed with sonicated MIP-K as compared with its native MIP-K, suggesting that level of exposed TLR2 ligands decreases due to autoclaving. Taken together, these results demonstrate the different TLR2/2, TLR2/1, TLR2/6 agonist property of MIP-L, MIP-K and BCG. In native form, MIP-L has a higher level of TLR2 agonist activity followed by MIP-K. BCG in native form is inefficient in engaging TLR2 expressed in HEK 293 cells.
Figure 7.
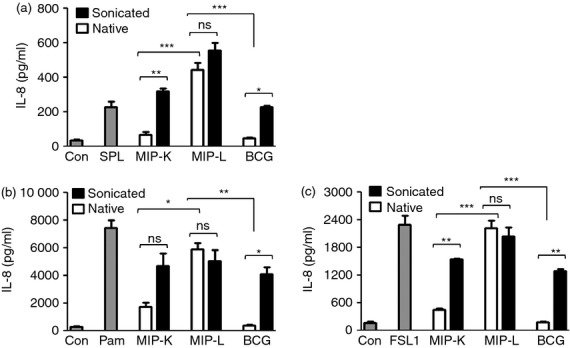
Delineation of Toll-like receptor (TLR2) signalling by heat-killed Mycobacterium indicus pranii (MIP-K), live Mycobacterium indicus pranii (MIP-L) and bacillus Calmette–Guérin (BCG). HEK293 cells expressing TLR2/2, TLR2/1 or TLR2/6 (a, b and c, respectively) were stimulated with MIP-K, MIP-L and BCG in native or sonicated form at a multiplicity of infection of 50. Culture supernatants were collected after 48 hr and analysed for interleukin-8 (IL-8) by ELISA. Higher levels of IL-8 production from 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells were observed in response to native and sonicated MIP-L. Maximum level of IL-8 was produced by 293tlr2/1 cells. Native MIP-K led to moderate levels of IL-8 from 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells. Native BCG did not lead to IL-8 production from either of 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells, indicating that TLR2 ligands are not well exposed in native BCG. Substantial levels of IL-8 were produced by 293tlr2/2, 293tlr2/1 and 293tlr2/6 cells in response to sonicated BCG. SPL-Streptococcus pneumoniae lysate (TLR2/2 ligand, MOS = 50), Pam-Pam3CSK4 (TLR2/1 ligand, 0·2 μg/ml), FSL1 (TLR2/6 ligand, 1 μg/ml) were used as positive controls. Data shown are mean ± SEM of three independent experiments. *P < 0·05, **P < 0·01, ***P < 0·001 and ns, not significant.
Discussion
Initially evaluated as an immune-therapeutic against leprosy, MIP has generated a lot of interest because of its strong immunomodulatory activity and the promising results obtained against tuberculosis,9,31. In our previous studies, where MIP was evaluated as a vaccine candidate against tuberculosis, we found significantly higher protection in MIP-treated animals compared with those treated with BCG.15 Strong Th1 response induced by MIP prompted us to evaluate it for cancer immunotherapy. Encouraging results including lower tumour burden, higher immune cell infiltration in tumour mass, and higher T-cell and NK-cell cytotoxicity against tumour cells were observed in MIP-treated mice.18 Macrophages are important components of the mononuclear phagocytic system, strategically located throughout the body to ingest and process foreign materials and to recruit other immune cells to the site of insult. M1 macrophages produce pro-inflammatory cytokines and reactive nitrogen intermediates, and offer protection against infectious diseases and neoplastic conditions. M2 macrophages on the other hand have regenerative and tissue-repairing functions and have been implicated in a variety of diseases.32 Our results show that a significantly higher level of pro-inflammatory cytokines and nitric oxide were produced from macrophages stimulated with MIP-L. MIP-K also showed a higher response compared with BCG. Higher activation of macrophages by MIP can explain the beneficial effects of MIP seen against leprosy and tuberculosis. Activated macrophages can potentially destroy the intracellular pathogens and can process and present the antigens to T cells leading to activation of the adaptive immune response.
In addition to production of cytokines and presentation of processed antigens, expression of co-stimulatory molecules is indispensible for activation of the adaptive immune response.33 Co-stimulatory molecules, namely CD40, CD80 and CD86, bind with their receptors and provide the necessary signal for activation of adaptive immune response. Our results show that MIP-L leads to the highest up-regulation of CD40, CD80 and CD86 by macrophages, followed by MIP-K. BCG induced lower expression of these markers. In the absence of an optimal cytokine milieu and co-stimulatory molecules, activation of adaptive responses would be impaired and this may partly explain the poor efficacy of BCG vaccine.
Toll-like receptors play an important role in the recognition of lipoarabinomannans and various other microbial molecules.34 Engagement of TLRs has a wide range of effects, which include production of pro-inflammatory cytokines and up-regulation of co-stimulatory molecules, enhanced antigen presentation by antigen-presenting cells and differentiation of monocytes into macrophages and dendritic cells.35–37 MyD88 is an adapter molecule located downstream to TLRs (except TLR3) and is involved in the intracellular passage of the information.25 In the absence of MyD88 adapter protein, both innate and adaptive immune responses are impaired. Our results demonstrate the crucial role of MyD88 in macrophage activation by MIP or BCG. Using the TLR signalling inhibitors, we established the role of different TLRs in the activation of macrophage by MIP. TLR2 had a prominent role in macrophage activation by MIP or BCG, even though TLR4 and intracellular TLRs were also important in macrophage activation. Response of TLR knockout macrophages to MIP and BCG stimulation further confirmed the results obtained with TLR signalling inhibitor and established the major role of TLR2 in macrophage activation by MIP and BCG.
Toll-like receptor 2 can recognize a variety of microbial molecules, including peptidoglycans, lipoteichoic acid, lipoarabinomannan, zymosan and lipoprotein. This remarkable ability of TLR2 to recognize different types of molecules is owing to its cooperation with other TLRs. Accordingly, TLR2 may function as homodimer (TLR2/2) or as heterodimer with TLR1 (TLR2/1) or TLR6 (TLR2/6) and it increases the spectrum of potential TLR2 ligands.29 Given that TLR2 has a major role in activation of macrophage by MIP and BCG, we further delineated the TLR2 signalling by MIP and BCG. HEK293-expressing murine TLR2 alone or in combination with TLR1 or TLR6 were used for this. In concurrence with superior macrophage activation, MIP-L show higher levels of TLR2/2, TLR2/1 and TLR2/6 agonistic activity compared with MIP-K and BCG. Decreased ability of MIP-K to engage TLR2 and to subsequently activate the macrophages can be explained because heat treatment will denature the potent TLR ligands. Interestingly, despite showing TLR2 engagement in macrophages, BCG did not engage either of TLR2/2, TLR2/1 or TLR2/6 in HEK 293 cells. We speculated that TLR2 agonists are not in exposed form in the BCG which resulted in to lack of response in TLR2-expressing HEK 293 cells. To address this, we used MIP and BCG in sonicated form. Interestingly, when used in sonicated form, BCG led to a substantial response from TLR2-expressing HEK cells. Hence, even though BCG has potent TLR2 ligands, they are not in exposed form and cannot engage respective TLRs efficiently. Hiding of putative TLR ligands can be an important strategy of pathogenic mycobacteria to evade immune-recognition. BCG also inhibits phagolysosomal fusion, which would further prevent degradation of bacterial component and subsequent release of putative ligands including the ones which stimulate intracellular TLRs. Clearly, MIP-L has distinctly higher levels of TLR2 ligands, which seems to be well exposed. BCG also has putative TLR2 ligands but these ligands are not in exposed form and therefore cannot engage the TLR2 efficiently. Taken together, our findings establish the superior activation of macrophages by MIP-L followed by MIP-K compared with BCG and implicates the role of TLRs in differential response of macrophages to MIP and BCG.
Acknowledgments
PK and RT performed the experiments; PK, GD and SB designed the study; PK and SB prepared the manuscript. This work was supported by the core research grant from the National Institute of Immunology, New Delhi, India.
Disclosures
The authors declare no commercial or financial conflict of interest.
References
- 1.Saxena VK, Singh US, Singh AK. Bacteriological study of a rapidly growing strain of Mycobacterium. Lepr India. 1978;50:588–96. [PubMed] [Google Scholar]
- 2.Deo MG. Anti-leprosy vaccines – field trials and future prospects. Indian J Lepr. 1984;56:764–75. [PubMed] [Google Scholar]
- 3.Talwar GP, Zaheer SA, Mukherjee R, et al. Immunotherapeutic effects of a vaccine based on a saprophytic cultivable mycobacterium, Mycobacterium w in multibacillary leprosy patients. Vaccine. 1990;8:121–9. doi: 10.1016/0264-410x(90)90134-8. [DOI] [PubMed] [Google Scholar]
- 4.Katoch K, Katoch VM, Natrajan M, et al. Treatment of bacilliferous BL/LL cases with combined chemotherapy and immunotherapy. Int J Lepr Other Mycobact Dis. 1995;63:202–12. [PubMed] [Google Scholar]
- 5.Sharma P, Misra RS, Kar HK, Mukherjee A, Poricha D, Kaur H, Mukherjee R, Rani R. Mycobacterium w vaccine, a useful adjuvant to multidrug therapy in multibacillary leprosy: a report on hospital based immunotherapeutic clinical trials with a follow-up of 1–7 years after treatment. Lepr Rev. 2000;71:179–92. doi: 10.5935/0305-7518.20000020. [DOI] [PubMed] [Google Scholar]
- 6.Singh IG, Mukherjee R, Talwar GP. Resistance to intravenous inoculation of Mycobacterium tuberculosis H37Rv in mice of different inbred strains following immunization with a leprosy vaccine based on Mycobacterium w. Vaccine. 1991;9:10–4. doi: 10.1016/0264-410x(91)90309-t. [DOI] [PubMed] [Google Scholar]
- 7.Yadava A, Mukherjee R. An immunodominant 30-kDa antigen of a candidate anti-leprosy vaccine, Mycobacterium w, shares T and B cell determinants with M. leprae and M. tuberculosis. Med Microbiol Immunol. 1993;182:243–53. doi: 10.1007/BF00579623. [DOI] [PubMed] [Google Scholar]
- 8.Saini V, Raghuvanshi S, Talwar GP, Ahmed N, Khurana JP, Hasnain SE, Tyagi AK. Polyphasic taxonomic analysis establishes Mycobacterium indicus pranii as a distinct species. PLoS ONE. 2009;4:e6263. doi: 10.1371/journal.pone.0006263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purswani S, Talwar GP, Vohra R, Pal R, Panda AK, Lohiya NK, Gupta JC. Mycobacterium indicus pranii is a potent immunomodulator for a recombinant vaccine against human chorionic gonadotropin. J Reprod Immunol. 2011;91:24–30. doi: 10.1016/j.jri.2011.06.099. [DOI] [PubMed] [Google Scholar]
- 10.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adhikari A, Majumder S, Banerjee S, Gupta G, Bhattacharya P, Majumdar SB, Saha B, Majumdar S. Mycobacterium indicus pranii (Mw)-mediated protection against visceral leishmaniasis: involvement of TLR4 signalling. J Antimicrob Chemother. 2012;67:2892–902. doi: 10.1093/jac/dks315. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi H, Shimada Y, Sawachi K, et al. Lipopolysaccharide-activated alveolar macrophages having cytotoxicity toward lung tumor cells through cell-to-cell binding-dependent mechanism. Anticancer Res. 2010;30:3159–65. [PubMed] [Google Scholar]
- 13.Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110:11103–8. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–22. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 15.Gupta A, Geetha N, Mani J, Upadhyay P, Katoch VM, Natrajan M, Katoch VM, Bhaskar S. Immunogenicity and protective efficacy of “Mycobacterium w” against Mycobacterium tuberculosis in mice immunized with live versus heat-killed M. w by the aerosol or parenteral route. Infect Immun. 2009;77:223–31. doi: 10.1128/IAI.00526-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta A, Ahmad FJ, Ahmad F, Gupta UD, Natarajan M, Katoch VM, Bhaskar S. Protective efficacy of Mycobacterium indicus pranii against tuberculosis and underlying local lung immune responses in guinea pig model. Vaccine. 2012;30:6198–209. doi: 10.1016/j.vaccine.2012.07.061. [DOI] [PubMed] [Google Scholar]
- 17.Saini V, Raghuvanshi S, Khurana JP, Ahmed N, Hasnain SE, Tyagi AK. Massive gene acquisitions in Mycobacterium indicus pranii provide a perspective on mycobacterial evolution. Nucleic Acids Res. 2012;40:10832–50. doi: 10.1093/nar/gks793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmad F, Mani J, Kumar P, Haridas S, Upadhyay P, Bhaskar S. Activation of anti-tumor immune response and reduction of regulatory T cells with Mycobacterium indicus pranii (MIP) therapy in tumor bearing mice. PLoS ONE. 2011;6:e25424. doi: 10.1371/journal.pone.0025424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rakshit S, Ponnusamy M, Papanna S, Saha B, Ahmed A, Nandi D. Immunotherapeutic efficacy of Mycobacterium indicus pranii in eliciting anti-tumor T cell responses: critical roles of IFNγ. Int J Cancer. 2012;130:865–75. doi: 10.1002/ijc.26099. [DOI] [PubMed] [Google Scholar]
- 20.Alexandroff AB, Jackson AM, O'Donnell MA, James K. BCG immunotherapy of bladder cancer: 20 years on. Lancet. 1999;353:1689–94. doi: 10.1016/S0140-6736(98)07422-4. [DOI] [PubMed] [Google Scholar]
- 21.Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–7. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 23.Demangel C, Bertolino P, Britton WJ. Autocrine IL-10 impairs dendritic cell (DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. Eur J Immunol. 2002;32:994–1002. doi: 10.1002/1521-4141(200204)32:4<994::AID-IMMU994>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 24.Geijtenbeek TB, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, Van Kooyk Y. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 26.Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–82. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- 27.Han SS, Keum YS, Seo HJ, Surh YJ. Curcumin suppresses activation of NF-κB and AP-1 induced by phorbol ester in cultured human promyelocytic leukemia cells. J Biochem Mol Biol. 2002;35:337–42. doi: 10.5483/bmbrep.2002.35.3.337. [DOI] [PubMed] [Google Scholar]
- 28.Forgione N, Tropepe V. Toll-like signaling and the cytokine IL-6 regulate histone deacetylase dependent neuronal survival. PLoS ONE. 2012;7:e41033. doi: 10.1371/journal.pone.0041033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hajjar AM, O'Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–9. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 30.Huang LY, Dumontelle JL, Zolodz M, Deora A, Mozier NM, Golding B. Use of toll-like receptor assays to detect and identify microbial contaminants in biological products. J Clin Microbiol. 2009;47:3427–34. doi: 10.1128/JCM.00373-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faujdar J, Gupta P, Natrajan M, Das R, Chauhan DS, Katoch VM, Gupta UD. Mycobacterium indicus pranii as stand-alone or adjunct immunotherapeutic in treatment of experimental animal tuberculosis. Indian J Med Res. 2011;134:696–703. doi: 10.4103/0971-5916.90999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32:463–88. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 33.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–18. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 35.Datta SK, Raz E. Induction of antigen cross-presentation by Toll-like receptors. Springer Semin Immunopathol. 2005;26:247–55. doi: 10.1007/s00281-004-0174-2. [DOI] [PubMed] [Google Scholar]
- 36.Krutzik SR, Tan B, Li H, et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med. 2005;11:653–60. doi: 10.1038/nm1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]