

Abstract
Occupational asthma (OA) is characterized by allergic airway inflammation and hyperresponsiveness, leading to progressive airway remodeling and a concomitant decline in lung function. The management of OA remains suboptimal in clinical practice. Thus, establishing effective therapies might overcome the natural history of the disease. We evaluated the ability of human adipose-tissue-derived mesenchymal stem cells (hASCs), either unmodified or engineered to secrete the IL-33 decoy receptor sST2, to attenuate the inflammatory and respiratory symptoms in a previously validated mouse model of OA to ammonium persulfate (AP). Twenty-four hours after a dermal AP sensitization and intranasal challenge regimen, the animals received intravenously 1×106 cells (either hASCs or hASCs overexpressing sST2) or saline and were analyzed at 1, 3, and 6 days after treatment. The infused hASCs induced an anti-inflammatory and restorative program upon reaching the AP-injured, asthmatic lungs, leading to early reduction of neutrophilic inflammation and total IgE production, preserved alveolar architecture with nearly absent lymphoplasmacytic infiltrates, negligible smooth muscle hyperplasia/hypertrophy in the peribronchiolar areas, and baseline airway hyperreactivity (AHR) to methacholine. Local sST2 overexpression barely increased the substantial efficacy displayed by unmodified hASCs. Thus, hASCs may represent a viable multiaction therapeutic capable to adequately respond to the AP-injured lung environment by resolving inflammation, tissue remodeling, and bronchial hyperresponsiveness typical of OA.
Introduction
In industrialized countries occupational asthma (OA) is the leading cause of respiratory diseases from occupational origin, and it can affect up to 30 per 100,000 inhabitants [1]. This condition accounts for 10% of people with asthma, causing severe respiratory pathology in young individuals and, therefore, involves a major socioeconomic burden. Persulfate salts used in hairdressing and cosmetics are well-known etiological agents involved in OA. These chemicals may be responsible for up to 5.8% of all cases of OA [2], representing the second, third, or fourth leading cause of OA according to different reports [2,3]. The mechanism by which persulfate salts cause OA is not well known, although based on clinical studies most authors support an immunologic mechanism [2,4].
Nowadays, management of OA is largely preventive and symptomatic, aimed avoiding or reducing work exposure to the causative agent and optimizing the standard medical treatment with bronchodilators and inhaled glucocorticoids [5]. Nevertheless, pharmacological treatment has proven of limited utility [6]. Regarding OA caused by persulfate salts, it has been observed in a small number of patients that their condition seems to improve if they avoid exposure, although asthma symptoms and bronchial hyperresponsiveness may persist [7]. Therefore, a more causal intervention being able to attenuate the immune-inflammatory process while reversing remodeling in the asthmatic airways should be highly desirable to avoid the progressive decline of lung function in these patients.
Due to their regenerative potential and recently characterized immune modulatory and anti-inflammatory properties, mesenchymal stem cells (MSCs) have been proposed as novel therapeutic agents in a wide variety of immune-inflammatory processes leading to tissue injury, including asthma [8,9]. Indeed, MSCs can be easily isolated from several tissues using minimally invasive procedures. Remarkably, fat tissue has proven a rich source of adipose-tissue-derived MSCs (termed ASCs) [10]. Shortly after being administered systemically, MSCs are primarily localized into the lung due to their size (20 μm in diameter), larger than the width of the lung microcapillaries. Additionally, enhanced lung retention is achieved in a proinflammatory microenvironment [11]. Finally, MSCs can be gene enhanced for local production of therapeutically relevant transgenic paracrine factors [12]. Consequently, several studies have recently reported preventive and/or therapeutic benefit of unmodified or genetically engineered MSC transplantation in several acute and chronic airway pathologies [13–15].
Our group has recently shown, in an OA murine model, that inhalation of persulfate salts induces an asthmatic response characterized by neutrophilic inflammation, raised serum IgE levels, increased T and B lymphocyte presence and activity in auricular and cervical draining lymph nodes, and airway hyperresponsiveness [16]. In the present study we have evaluated the therapeutic potential of human adipose-tissue-derived mesenchymal stem cells (hASCs), either unmodified or genetically engineered to overexpress sST2 [17]—a natural decoy receptor to IL-33 [18], key alarmin in innate eosinophilic airway inflammation—resolving the underlying immune-inflammatory process and the chronic airway hyperreactivity (AHR) inherent in the validated ammonium persulfate [(NH4)2S2O8; AP]–induced OA model.
Materials and Methods
Animals
Male BALB/c mice (∼20 g, 6-week old) were obtained from Harlan. The mice were housed in filter-top cages in a conventional animal house with 12 h dark/light cycles and received lightly acidified water and pelleted food (Trouw Nutrition) ad libitum. All procedures involving mice were reviewed and approved by the Ethical Committee of Animal Experimentation from the Vall d'Hebron Research Institute (Barcelona, Spain) (CEEA study No. 61/09).
Isolation and characterization of ASCs
Discarded human adipose tissue from healthy adult women undergoing elective lipoaspiration was used as the source of hASCs, under the Institutional Ethics Committee approval as described elsewhere [17,19]. The raw lipoaspirate (200 mL) was washed extensively in phosphate-buffered saline (PBS) and incubated in PBS containing 0.075% type II collagenase (Life Technologies) at 37°C for 1 h to digest the extracellular matrix. The protease activity was neutralized with Dulbecco's modified Eagle's medium (DMEM; Life Technologies) plus 10% fetal bovine serum (FBS). The resulting suspension was filtered through a 70-μm nylon strainer; centrifuged; resuspended in α-MEM (Life Technologies) containing 15% FBS, 1% penicillin/streptomycin, 1% l-glutamine, and 2.5 μg/mL amphotericin (all from Life Technologies); and seeded overnight in culture flasks (Corning, Inc.) at 37°C in a 5% CO2 atmosphere. Next day, the cell monolayer was washed once more with PBS to remove nonadherent cells and cellular debris and replaced with fresh medium. The cells were expanded until 80% confluence and amplified through periodic passaging (split ratio 1:2).
Flow cytometry to assess the expression levels of different surface markers was performed on an FACScan cytometer using CellQuest software (BD Biosciences). The hASCs were stained with fluorescein-isothiocyanate (FITC) or phycoerythrin (PE)–conjugated antibodies against CD44, CD29 (Immunostep), CD90 (eBioscience), CD14, HLA-DR, and CD34 (BD Biosciences) and the corresponding fluorescent isotype-matched negative control antibodies (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/scd).
Adipocytic differentiation was induced by seeding hASCs for 21 days in Iscove's modified Dulbecco's medium (IMDM; Life Technologies) supplemented with 0.5 mM isobutyl methyl xantine, 1 μM hydrocortisone, and 0.1 mM indomethacin, and visualized by Oil Red-O staining. Osteocytic differentiation was induced by culturing hASCs in IMDM containing 0.1 μM dexametasone, 10 mM β-glycerol phosphate, and 0.2 mM ascorbic acid for 21 days, and visualized after Alizarin Red staining. All hASC differentiation reagents were from Sigma-Aldrich (Supplementary Fig. S1B).
Mouse adipose-tissue-derived mesenchymal stem cells (mASCs) were obtained from the inguinal fat pads of female BALB/c mice, and isolated and characterized analogously to hASCs.
Bioluminescence imaging
Mice were administered, via the lateral tail vein, 1×106 hASCs transduced with a bicistronic lentiviral vector (LV-T2A) encoding the firefly luciferase (fLuc) and the EGFP under the control of a cytomegalovirus promoter [17]. To track fLuc-expressing hASCs, mice were intraperitoneally injected with D-luciferin (150 mg/kg) (Promega) dissolved in PBS and imaged at different times (ranging from 10 min to 5 days) in an IVIS® Spectrum Imaging System (Caliper Life Sciences). Anesthesia was performed in an induction chamber with 3% isoflurane in 100% oxygen at a flow rate of 1.25 L/min and maintained in the IVIS system with a 2.5% of the above mixture at 0.5 L/min. Bioluminescent signaling on the lungs was measured as integrated photons/second. Imaging data were analyzed using the Living Image 4.1 (Caliper Life Sciences).
sST2 cloning, lentiviral vector production, and hASC transduction
The synthesis and production of VSV-G-pseudotyped, high-titer lentiviral particles from the bicistronic expression vector pWPT-sST2FLAG2AEGFP (pWPT-sST2-EGFP) and its corresponding control pWPT-EGFP have been previously described [17]. pWPT-sST2-EGFP encodes, in a 5′ to 3′ orientation, the C-terminus FLAG-tagged murine sST2 cDNA, the picornaviral 2A sequence for cotranslational cleavage, and the reporter EGFP cDNA. pWPT-EGFP encodes only the EGFP cDNA. Both lentiviral vectors are under control of the strong constitutive human EF-1alpha promoter (Supplementary Fig. S2).
hASCs from passage 2–3 were transduced in suspension with the previous lentiviral vector particles at several multiplicities of infection (MOI), ranging from 1 to 40. The culture medium was replaced after 16 h to remove lentiviral particles. At 48 h after transduction, the EGFP expression was analyzed by both flow cytometry and fluorescence microscopy. The recombinant mouse sST2 protein expression was assessed by a specific mouse T1/ST2 sandwich enzyme-linked immunosorbent assay (ELISA; mdbioproducts) according to the manufacturer's instructions, and by western blot analysis using an anti-FLAG antibody [17]. The functional activity of the secreted sST2-FLAG decoy was evaluated by assessing its ability to interfere with proinflammatory IL-33-T1/ST2 signaling in P815 mouse mastocytoma cells [17].
The hASCs administered to the OA mouse model were previously transduced with the appropriate lentiviral vector particles at an optimized MOI of 20. Both pWPT-sST2-EGFP-transduced hASCs (hASC-sST2) and pWPT-EGFP-transduced hASCs (hASC) reached an average of 80%±5% transduction efficiency. Moreover, hASC-sST2 attained a persistent level of secreted sST2 of 35±15 ng/mL [17].
Mouse model of chemical-induced asthma
Schematic approach followed for the hASC-based treatment of experimental OA (Fig. 1) according to the previously published model [16]. On days 0 and 7, the mice received dermal applications of 5% AP or vehicle (DMSO) on the dorsum of both ears (20 μL). On day 14, they received, under light anesthesia with isoflurane (Forene; Abbott Laboratories), an intranasal instillation (40 μL) of 1% AP (challenge) or vehicle (saline; PBS). Experimental groups are as follows: DMSO/SAL–PBS and DMSO/AP–PBS (negative control, nonasthmatic untreated mice); DMSO/SAL–hASC, DMSO/SAL–hASC-sST2, DMSO/AP–hASC, and DMSO/AP–hASC-sST2 (evaluation of potential side-effects of xenogeneic hASC administration in nonasthmatic mice); AP/AP–PBS (positive control, asthmatic untreated mice); and AP/AP–hASC and AP/AP–hASC-sST2 (evaluation of the therapeutic effects of hASC administration in asthmatic mice). The first abbreviation identifies the agent used for the dermal application on days 0 and 7 (sensitization), the second abbreviation identifies the agent administered via intranasal instillation on day 14 (challenge), and the third abbreviation identifies the treatment on day 15 [1×106 cells (hASC or hASC-sST2 in PBS), or PBS alone (250 μL) via tail vein injection].
FIG. 1.

Experimental design and intervention in the OA BALB/c mouse model. The initial sensitization step involves dermal application of 5% AP or vehicle (DMSO) on the dorsum of both ears (20 μL) on days 0 and 7. On day 14, the animals receive an intranasal instillation (40 μL) of 1% AP (challenge) or vehicle (saline). On day 15, the therapeutic approach consists of an intravenous injection of engineered mesenchymal stem cells (1×106 cells; 250 μL) overexpressing either EGFP (hASC) or both sST2 and EGFP (hASC-sST2), or control vehicle (saline). Mice are then sacrificed at days 16, 18, and 21 to assess therapeutic efficacy. AP, ammonium persulfate; DMSO, dimethylsulfoxide; hASCs, human adipose-tissue-derived mesenchymal stem cells; OA, occupational asthma.
Blood retrieval, bronchoalveolar lavage, and cell count
After the methacholine tests, the mice were deeply anesthetized by an intraperitoneal injection of pentobarbital (Nembutal, Sanofi Santé Animale, CEVA). Blood was sampled from the retro-orbital plexus; the lungs were lavaged, in situ, three times with 0.7 mL of sterile saline; and the recovered fluid was pooled. Cells were counted and the bronchoalveolar lavage fluid (BALF) was centrifuged (1000 g, 10 min). The supernatant was frozen (−80°C) until further analyses. For differential cell counts, 250 μL of the resuspended cells (100,000 cells/mL) were spun (300 g, 6 min; Cytospin 3, Shandon, TechGen) onto microscope slides, air-dried, and stained (Diff-Quik method; Medical Diagnostics). For each sample, 500 cells were counted for the number of macrophages, eosinophils, neutrophils, and lymphocytes.
Immunoassays
IL-33 was measured in undiluted sera by standard ELISA techniques, according to the manufacturer's instructions (Biosource). The OptEIA Mouse IgE set from Pharmingen (BD Biosciences) was used to evaluate total serum IgE from diluted serum (1:5 dilution). Measurements were performed according to the manufacturer's instructions. Further, the concentrations of inflammatory cytokines IL-6, IL-13, IFN-γ, and IL-2 were estimated in undiluted BALF supernatants through a luminex-based immunoassay (Millipore).
Flow cytometry analysis
The relative number of BALF mouse lymphocyte subpopulations was assessed by flow cytometry using specific antibodies against CD3, CD4, and CD8 (Life Technologies).
Histopathology and immunohistochemistry
The trachea from an independent set of DMSO/SAL–PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2 animals undergoing neither AHR analysis nor BAL was cannulated at day 21 after the initial AP sensitization step, and the lungs were fixed by inflation to total lung capacity with 1 mL of 4% paraformaldehyde. Following overnight fixation, the lung tissue was dehydrated with an ethanol gradient and embedded in paraffin, cut into 3-μm-thick sections, and stained with hematoxylin and eosin. Images were taken with an Olympus BX41 microscope (Olympus Life Science Research GmbH) using 40× and 20× objectives. To determine morphology and inflammatory infiltrate, images were evaluated by a pathologist who was blinded to the identity of the slides.
For immunohistochemistry, sliced 5-μm sections were mounted on poly-L-lysine-coated glass slides, deparaffinized, and rehydrated. Immunogenic retrieval was performed incubating the slides in 10 mM citrate buffer (pH 6.0) for 20 min in a microwave oven. Following several PBS-T (PBS plus 0.1% Triton X-100) washes, tissue sections were blocked by 2-h incubation in 5% goat serum diluted in PBS-T at room temperature. Further, PBS-T-washed sections were incubated overnight at 4°C in a humid chamber with the appropriate primary antibodies (all diluted 1:100 in PBS-T). Namely, the reporter EGFP was immunolocalized with a biotinylated goat polyclonal anti-GFP (ab6658; Abcam), and the FLAG tag was identified with a rabbit DYKDDDDK Tag antibody (Cell Signaling Technology). Next day, for FLAG tag detection, sections were additionally incubated with a biotin-conjugated goat anti-rabbit IgG antibody (Invitrogen) diluted 1:1000 in PBS for 1 h at room temperature. The endogenous peroxidase was blocked with 3% H2O2 for 10 min in the dark. Finally, the sections were incubated with a biotinylated horseradish peroxidase-avidin complex (ImmunoPure® ABC Peroxidase Staining Kit; Pierce), stained with the peroxidase substrate diaminobenzidine tetrahydrochloride (DAB; Pierce), and counterstained with hematoxylin. Specificity control slides were analogously stained using nonimmune rabbit or nonimmune goat IgG (1:100 dilution; Abcam) as primary antibodies.
RNA isolation and analysis
Mouse lung tissue was immediately frozen in liquid nitrogen after euthanasia. Total lung RNA was isolated using the RNeasy Mini kit (Qiagen). After isolation, RNA samples were DNase treated for 30 min at 37°C to remove contaminating DNA (PE Applied Biosystems). Gene expression analysis from hASC-treated OA (AP/AP) and control (DMSO/SAL) mouse lung RNA was performed through reverse transcription using the High Capacity cDNA Reverse Transcription Kit followed by real-time quantitative TaqMan polymerase chain reaction (TaqMan RT-qPCR; PE Applied Biosystems). The predesigned, human gene-specific primers and probe sets for each human gene analyzed (ICAM1; Hs00164932_m1), PTGS2 (COX-2; Hs00153133_m1), and (TGFB1; Hs00998133_m1), and for the endogenous housekeeping human gene used for normalization (GUSB; Hs99999908_m1) were obtained from Assays-on-Demand™ Gene Expression Products (PE Applied Biosystems), assayed in triplicate, and analyzed in a spectrofluorimetric thermal cycler (ABI PRISM 7300 Sequence Detector; PE Applied Biosystems). Data quantification was carried out through the comparative Ct method.
Airway physiology
To assess the asthmatic profile of all mice not undergoing histological analysis, reactivity to methacholine was evaluated at days 16 and 21 after the initial AP sensitization step. AHR was measured using a forced oscillation technique with the Flexivent system (FlexiVent, SCIREQ). Airway resistance (R) and elastance (E) were quantified using a “snapshot” protocol, as previously described [16]. For each mouse, R and E were plotted against methacholine concentration (from 0 to 10 mg/mL) and the area under the curve (AUC) was calculated.
Statistical analysis
Data are presented as mean values±SDs and were analyzed using one-way analysis of variance (ANOVA) followed by a Dunnett post hoc test (Graphpad Prism 5.00; Graphpad Software, Inc.). A level of P<0.05 (two tailed) was considered statistically significant.
Results
Intravenously transplanted hASCs localize into the lung and undergo anti-inflammatory activation within the AP-induced asthmatic airways
Noninvasive bioluminescent tracking of intravenously infused fLuc-expressing hASCs into control mice revealed their exclusive although transient homing into the lungs [exponential decay; biological half-life (t1/2) ∼1.5 days] (Supplementary Fig. S3).
Moreover, the subacute proinflammatory milieu of the AP-sensitized and -challenged (AP/AP) mouse lungs, analogously to that recently reported for lipopolysaccharide-induced acutely injured mouse lungs (t1/2 ∼3.5 days) [17], stimulated retention and activation of circulating hASCs (Fig. 2). Indeed, AP-induced asthmatic lung-localized hASCs showed (1) early and sustained transcriptional upregulation of ICAM-1 (20- to 60-fold induction), which has been found critical for their T cell adhesive and immune modulatory capacities [20]; and (2) late upregulation of the transcript encoding for the inducible enzyme COX-2 (sixfold induction), whose product prostaglandin E2 increases IL-10 secretion from macrophages to reduce the inflammatory response [21]. Conversely, the Th17-inducing cytokine TGF-β was upregulated neither at day 16 nor at day 18 after the initial AP sensitization step.
FIG. 2.

Intrapulmonary homing and activation status of hASCs in AP-induced OA mice. Anti-inflammatory profile of infused hASCs in the murine lung. The relative mRNA expression of the gene coding for the adhesion molecule human ICAM-1, and the hASC immune modulatory genes COX-2 and TGF-β in hASC-treated murine lung tissue was determined by TaqMan RT-qPCR at days 16 and 18 after the initial AP sensitization step. Experimental groups are DMSO/SAL–hASC and AP/AP–hASC. Data are represented as fold induction of the indicated gene from the AP/AP–hASC group relative to the same gene from the DMSO/SAL–hASC group (n=3–4), ***P<0.001 compared with DMSO/SAL–hASC.
Involvement of proinflammatory IL-33 in the OA mouse model
At the protein level, both hASCs (overexpressing EGFP) and hASC-sST2 (overexpressing EGFP and FLAG-sST2) were immunolocalized within the lung parenchyma in AP-sensitized and -challenged (AP/AP) mice (Fig. 3A).
FIG. 3.
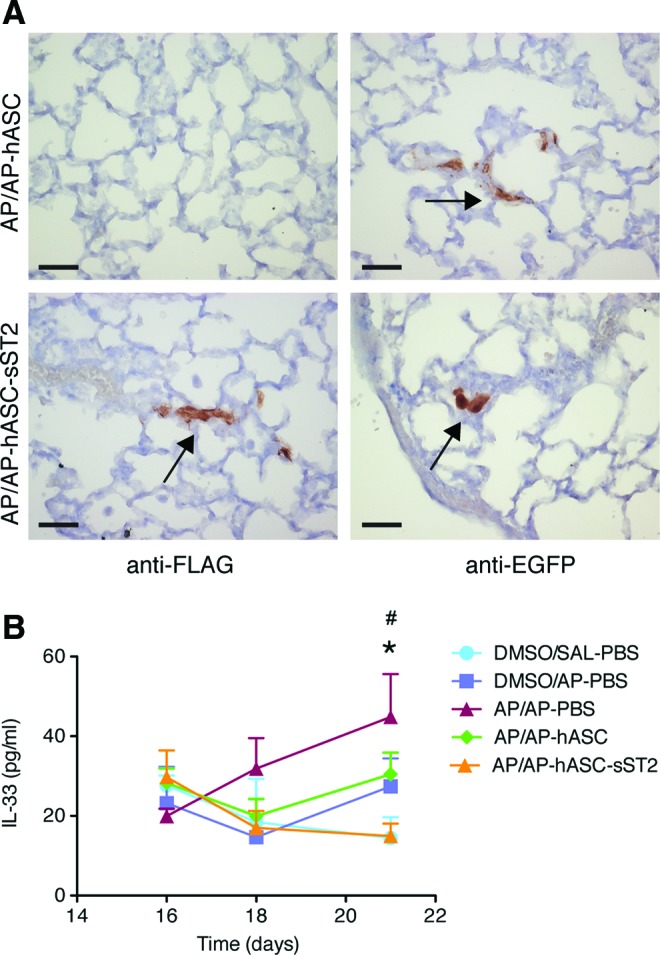
Proinflammatory IL-33 involvement in the OA mouse model. (A) Immunohistochemical localization of pWPT-EGFP- and pWPT-sST2-EGFP-transduced hASCs overexpressing EGFP or both murine sST2-FLAG and EGFP, respectively, in the OA model. Representative lung section images of AP-challenged, hASC-treated mice (AP/AP–hASC) (upper panels), and AP-challenged, hASC-sST2-treated mice (AP/AP–hASC-sST2) (lower panels) day 16 after the initial AP sensitization step. Arrows indicate the brown label detection of sST2-FLAG-overexpressing hASCs (lower left panel), and of EGFP-overexpressing hASCs (right panels). Scale bar=10 μm. (B) Serum IL-33 dynamics in the OA mouse model. Serum IL-33 levels were quantified by ELISA in both treated and nontreated mice at 16, 18, and 21 days after the initial AP sensitization step. Experimental groups are DMSO/SAL–PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2. Values are means±SDs (n=4–7 mice per group). *P<0.05, AP/AP–PBS group compared with both DMSO/SAL–PBS and AP/AP–hASC-sST2 groups; #P<0.05, AP/AP–hASC group compared with both DMSO/SAL–PBS and AP/AP–hASC-sST2 groups. SAL, saline; PBS, phosphate-buffered saline; AP, ammonium persulfate; hASC, hASCs overexpressing EGFP; hASC-sST2, hASCs overexpressing sST2 and EGFP. Color images available online at www.liebertpub.com/scd
The alarmin IL-33, released by damaged epithelia, acts on cells of both the adaptive and innate immune systems and has a central role driving inflammation in allergic airway disease [18]. Accordingly, the serum IL-33 levels increased progressively after AP sensitization and challenge in asthmatic untreated mice (AP/AP–PBS) (Fig. 3B). In contrast, only the asthmatic hASC-treated mice overexpressing the IL-33 decoy receptor sST2 (AP/AP–hASC-sST2), but not the asthmatic hASC-treated mice (AP/AP–hASC), maintained the baseline IL-33 level corresponding to control, unchallenged, and untreated mice (DMSO/SAL–PBS). Thus, at day 21 after the initial AP sensitization step, the IL-33 values from hASC-sST2-treated (AP/AP–hASC-sST2) mice reached statistical significance when compared with those from both untreated (AP/AP–PBS) and hASC-treated (AP/AP–hASC) asthmatic animals.
hASCs are able to inhibit airway neutrophilia and IgE production in the AP-induced OA mice
The total cell count in the BALFs of OA mice did not experience a significant variation with time after AP sensitization and challenge (Fig. 4A). In contrast, the percentage of proinflammatory neutrophils was found increased early after AP challenge in the sensitized mice (AP/AP–PBS), although gradually decreased to reach baseline levels by day 7 after AP challenge (day 21 after the initial AP sensitization step) (Fig. 4B). Importantly, hASC and hASC-sST2 treatments fully reduced the early neutrophilia from the BALFs of the asthmatic mice to baseline levels. Conversely, we observed in the BALFs from both hASC-treated and hASC-sST2-treated mice an increase in the percentage of lymphocytes late after AP challenge (day 21) (Fig. 4C), concomitantly with a complementary decrease in the percentage of alveolar macrophages, the major cell type present in the BALFs of OA mice (Fig. 4D). As previously reported, no eosinophils were recruited into the AP-treated lungs [16].
FIG. 4.

Inflammatory cell dynamics in the bronchoalveolar lavage fluid (BALF) from the OA mouse model. Total leukocyte counts were determined using a hemocytometer (A). Relevant BALF cell populations were further evaluated using Diff-Quick-stained cytospin preparations, and displayed as percentage of neutrophils (B), lymphocytes (C), and macrophages (D). Results are shown at 16, 18, and 21 days after the initial AP sensitization step. Experimental groups are DMSO/SAL–PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2. Both DMSO/SAL–hASC* and DMSO/SAL–hASC-sST2* groups were analyzed only at day 21. Values are means±SDs (n=6–10 mice per group). In B, *P<0.05 comparing AP/AP–PBS respect to all other groups; in C, ***P<0.001 comparing AP/AP–hASC, DMSO/SAL–hASC* and DMSO/SAL–hASC-sST2* respect to AP/AP–PBS, and #P<0.01 comparing AP/AP–hASC-sST2 respect to AP/AP–PBS; in (D), **P<0.01 comparing AP/AP–hASC, DMSO/SAL–hASC* and DMSO/SAL–hASC-sST2* respect to AP/AP–PBS. Color images available online at www.liebertpub.com/scd
The IL-13 (Th2-type) and the IFN-γ (Th1-type) cytokine levels did not vary between groups, while IL-6 (Th17-inducing) was reproducibly increased in the BALFs of AP-sensitized and -challenged (AP/AP–PBS) asthmatic mice at day 16 after the initial AP sensitization step, although it did not reach statistical significance (Supplementary Fig. S4).
Conversely, we confirmed a significant early increase of serum total IgE levels in the AP-sensitized and -challenged animals (AP/AP–PBS group), which, however, could be fully reversed by hASC treatment (AP/AP–hASC and AP/AP–hASC-sST2 groups) (Fig. 5).
FIG. 5.

Total serum IgE levels in the OA mouse model. Total IgE levels were quantified by ELISA in both treated and nontreated mice sera (1:5 dilution). Total serum IgE at day 16 after the initial AP sensitization step. Experimental groups are DMSO/SAL–PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2. Values are means±SDs (n=5–7 mice per group). *P<0.05 compared with AP/AP–hASC and AP/AP–hASC-sST2 groups.
hASCs resolve AP-induced airway hyperreactivity in the OA mouse model
AP-sensitized and -challenged mice (AP/AP–PBS) had a significant decline in pulmonary function [assessed by measuring total respiratory system elastance (E) and resistance (R)] compared with control nonsensitized, unchallenged or with nonsensitized, AP-challenged mice (DMSO/SAL–PBS and DMSO/AP–PBS) both at day 16 and 21 after the initial AP sensitization step (Fig. 6). Moreover, hASC infusion (both AP/AP–hASC and AP/AP–hASC-sST2 groups) was able to fully resolve persistent AHR. Consequently, at both day 16 and 21 after the initial AP sensitization step, the mean AUC of R and E from the untreated asthmatic group (AP/AP–PBS) was significantly higher than the mean AUC of R and E from both hASC-treated groups (AP/AP–hASC and AP/AP–hASC-sST2), which matched those from the control nonasthmatic groups (DMSO/SAL–PBS and DMSO/AP–PBS). We also discarded a potential immune stimulatory hASC-based contribution on lung function because the mean AUC of R and E from the control hASC-instilled groups (DMSO/SAL–hASC and DMSO/SAL–hASC-sST2) at day 21 was identical to that from the control untreated groups (DMSO/SAL–PBS and DMSO/AP–PBS).
FIG. 6.

Airway hyperreactivity (AHR) to methacholine in the OA mouse model. AHR expressed as resistance (R) and elastance (E) was measured at day 16 (A) and day 21 (B) after the initial AP sensitization step by the forced oscillation technique. Left panels show the mean R values±SEM resulting from increased concentrations of methacholine, and the individual values of the area under the curve (AUC) of R. Right panels show the mean E values±SEM resulting from increased concentrations of methacholine, and the individual values of the AUC of E. Experimental groups are at days 16 and 21: DMSO/SAL–PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2, plus at day 21 only: DMSO/SAL–hASC and DMSO/SAL–hASC-sST2 (n=4–7 mice per group). *P<0.05, **P<0.01, and ***P<0.001 compared with AP/AP–hASC and AP/AP–hASC-sST2 groups; #P<0.001 compared with the AP/AP–hASC-sST2 group.
Airway histopathological alterations induced by AP in the OA mice are ameliorated by hASC treatment
A blinded histopathological examination of lung tissue sections from the asthmatic AP-sensitized and -challenged mice (AP/AP–PBS) revealed marked hyperemia, moderate leukostasis and inflammatory cell infiltration, and peribronchiolar smooth muscle hyperplasia/hypertrophy (Fig. 7; left panels). Conversely, lungs from both hASC-treated mice (AP/AP–hASC group and AP/AP–hASC-sST2 group) (Fig. 7; middle panels) showed preserved alveolar architecture with only slight hyperemia, few lymphoplasmacytic infiltrates, and nearly negligible smooth muscle hyperplasia/hypertrophy in the peribronchiolar areas, approaching the tissue appearance from control, nonsensitized untreated groups DMSO/SAL–PBS (Fig. 7; right panels) and DMSO/AP–PBS (not shown). Thus, hASC treatment attenuated perivascular and peribronchial inflammation and remodeling in the AP-based OA mouse model (Table 1).
FIG. 7.

Lung histopathology in the mouse OA model. Histological evaluation of the therapeutic potential of hASC and hASC-sST2 in murine OA at 21 days after the initial AP sensitization step. Representative images of hematoxylin-and-eosin-stained, 4-μm lung sections are shown at low (upper panels) and high (lower panels) magnification. AP/AP, AP-sensitized and -challenged, untreated mice; AP/AP–hASC, AP-sensitized and -challenged, hASC-treated mice; AP/AP–hASC-sST2, AP-sensitized and -challenged, hASC-sST2-treated mice; DMSO/SAL, control noninduced, untreated mice (n=4–7 mice per group). In the left panels and denoted by arrows, AP/AP mice show a marked hyperemia, moderate leukostasis in the middle-size vessels, and hyperplasia/hypertrophy of the peribronchial musculature, when compared with the mild changes present in the AP/AP–hASC and AP/AP–hASC-sST2 mice. Scale bars=20 μm. Color images available online at www.liebertpub.com/scd
Table 1.
Histopathological Scoring of Lung Injury in the Ammonium-Persulfate-Induced Occupational Asthma Mouse Model
| Mouse groupsa | Vascular hyperemia | Pulmonary leukostasis | Smooth muscle hyperplasia/hypertrophy |
|---|---|---|---|
| DMSO/SAL–PBS | +/− | − | − |
| DMSO/AP–PBS | +/− | − | − |
| AP/AP–PBS | +++ | +++ | +++ |
| AP/AP–hASC | + | + | + |
| AP/AP–hASC-sST2 | + | +/− | + |
Experimental groups are DMSO/SAL- PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2.
DMSO, dimethylsulfoxide; SAL, saline; PBS, phosphate-buffered saline; AP, ammonium persulfate; hASC, hASCs overexpressing EGFP; hASC-sST2, hASCs overexpressing EGFP and sST2. Histopathological scoring relied on the examination of 10 different lung tissue sections/mouse from four animals per group by a blinded expert pathologist.
−, negative; +, mild; ++, moderate; +++, severe.
Immune reactivity of xenotransplanted hASCs in the AP-induced OA mouse model
We reported earlier a significant increase in the percentage of lymphocytes from the BALFs of both hASC-treated groups (AP/AP–hASC and AP/AP–hASC-sST2) late (7 days) at 21 days after the initial AP sensitization step (Fig. 4C). Accordingly, at 18 days after the initial AP sensitization step, we noticed a three- to fourfold increase of the IL-2 levels in the BALFs of the hASC-treated groups (AP/AP–hASC and AP/AP–hASC-sST2) compared with the nontreated groups (DMSO/SAL–PBS, DMSO/AP–PBS, and AP/AP–PBS) (Fig. 8A). A more systematic assessment of the increased presence of lymphocytes in the BALFs of hASC- and hASC-sST2-treated mice suggested the induction of an immune response against xenogeneic hASCs regardless of AP sensitization and/or challenge (Fig. 8B). An independent study confirmed that the significant lymphocyte boost occurring 1 week after hASC treatment was indeed induced by the infused hASC xenocells, because a parallel infusion of syngeneic murine ASCs was unable to raise the percentage of lymphocytes in the recipient BALFs (Fig. 8C). Moreover, a significant reduction in the percentage of BALF lymphocytes in the AP/AP–hASC and AP/AP–hASC-sST2 groups compared with the DMSO/SAL–hASC and DMSO/SAL–hASC-sST2 groups, respectively, highlighted the superior immune evasiveness of activated hASCs within the proinflammatory AP-induced milieu of the lungs, respect to hASCs reaching the control (DMSO/SAL), noninflamed lungs (Fig. 8B).
FIG. 8.

Immunogenicity of xenogeneic hASCs in the mouse background. (A) IL-2 levels in the BALFs measured by multiplex ELISA at day 18 after the initial AP sensitization step (n=3–5 mice per group). Experimental groups are DMSO/SAL–PBS, DMSO/AP–PBS, AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2. *P<0.05 compared with the DMSO/SAL–PBS and DMSO/AP–PBS groups. (B) Percentage of lymphocytes in the BALFs evaluated at day 21 after the initial AP sensitization step using Diff-Quick-stained cytospin preparations. Experimental groups are DMSO/SAL–PBS, DMSO/SAL–hASC, and DMSO/SAL–hASC-sST2; DMSO/AP–PBS, DMSO/AP–hASC, and DMSO/AP–hASC-sST2; AP/AP–PBS, AP/AP–hASC, and AP/AP–hASC-sST2. Values are means±SDs (n=5–7 mice per group). **P<0.01 and ***P<0.001 compared with the DMSO/SAL–PBS, DMSO/AP–PBS, and AP/AP–PBS groups. #P<0.05 compared with the DMSO/SAL–hASC and DMSO/SAL–hASC-sST2 groups. (C) Percentage of lymphocytes in the BALFs at 7 days after intravenous infusion of nonmanipulated syngeneic mouse (mASC) or xenogeneic human adipose-tissue-derived mesenchymal stem cells, either nonmanipulated (hASC), or genetically engineered to overexpress EGFP (hASC-EGFP). Control: PBS-injected mice. **P<0.01 and ***P<0.001 compared with the control PBS group. (D) Dynamics of the percentage of lymphocytes in the BALFs of hASC-treated mice. (E) Weight evolution of mice treated with the above ASC types.
Specific immunostaining of the major lymphocyte subpopulations present in the BALFs uncovered fourfold increase in the CD3+CD8+ T cell subpopulation from the hASC-transplanted mice (DMSO/SAL–hASC and AP/AP–hASC) compared with nontransplanted mice (DMSO/SAL–PBS and AP/AP–PBS) (Table 2). Nonetheless, a follow-up of the lymphocyte content in the BALFs of hASC recipient mice showed that the increased presence of lymphocytes by day 7 returned toward baseline levels by day 10, indicating that the immune response elicited by the xenotransplanted hASCs was transient (Fig. 8D). Further, the weight of the treated animals, indicative of their overall health, was normalized by day 10 after cell infusion (Fig. 8E).
Table 2.
Relative Number of BALF Lymphocyte Subpopulations in the OA Mouse Model After Xenogeneic hASC Infusion
| Experimental groupsa | % viable CD3+ | % CD3+CD4+ | % CD3+CD8+ | CD3+CD4+/CD3+CD8+ |
|---|---|---|---|---|
| DMSO/SAL–PBS | 0.2 | 36 | 11.8 | 3.1 |
| DMSO/SAL–hASC | 5.4 | 28.6 | 50.5 | 0.6 |
| AP/AP–PBS | 0.5 | 44.9 | 21.4 | 2.1 |
| AP/AP–hASC | 8.8 | 29.9 | 48.5 | 0.6 |
The percentage of the different lymphocyte subpopulations in the pooled BALFs from 10 mice per group receiving an intravenous injection of 1×106 hASCs or PBS was assessed 6 days later by flow cytometry with CD-specific antibodies.
BALF, bronchoalveolar lavage fluid.
Discussion
This study demonstrates that the intravenous administration of hASCs has a therapeutic action in established OA pathology, restoring the basal levels of both neutrophils in BALF and total IgE in serum, resolving the AHR, and attenuating early lung remodeling.
Recent reports have demonstrated the anti-inflammatory potential of MSCs in several murine asthma models induced by high-molecular-weight allergens (ovalbumin, ragweed, and aspergillus hyphal extract) [11,13,22–28] and in a low-molecular-weight, toluene diisocyanate (TDI)–induced asthma model [14]. However, in most of these models, the MSCs were administered preventively, that is, during the immunization phase or concomitantly with the induction of asthma. We transferred the hASCs to the mice 24 h after AP challenge, when the main pathological features of OA had already developed [16], which increases the therapeutic window for hASC intervention and, therefore, their clinical potential.
The immediate consequence of systemic hASC administration at the molecular level was the activation of an immune modulatory profile in these cells upon reaching the proinflammatory context of the AP-induced asthmatic lung. This was characterized by early upregulation of ICAM-1, a critical adhesion molecule involved in cell–cell contact–mediated immune regulation [20], and further overexpression of immunosuppressive molecules such as COX-2, responsible for the paracrine release of prostaglandin E2 [21]. The therapeutic potential of hASCs is related to their immediate anti-inflammatory and immunomodulatory actions [17,29], which indirectly influence epithelial repair and regeneration of the injured lungs, instead to their intrinsic regenerative potential, because of their limited engraftment and transient presence in the hASC-infused mouse lungs. A very recent study in a mouse model of OVA-induced allergic asthma has shown that hASCs activate the predominant effectors in the pulmonary system, alveolar macrophages, providing a compelling mechanism for their durable effects despite limited engraftment [27].
Unlike most Th2-driven and eosinophil-mediated classical asthma models, we assessed the anti-inflammatory effect of hASCs in an atypical AP-induced and neutrophil-mediated asthma context, demonstrating baseline neutrophil levels in the BALFs from hASC-treated mice early after AP challenge. Accordingly, a recent report also highlights the anti-inflammatory effects of bone-marrow-derived MSCs decreasing neutrophils and, to a lesser extension, eosinophils, in an experimental TDI-induced OA model [14]. These results underlay the plasticity of MSCs depending on the inflammatory context in which they are introduced.
Moreover, we have confirmed the outcome of another atypical trait from the AP-based OA murine model: the preservation of BALF's total immune cell counts despite AP sensitization and challenge [16]. It has been shown that dermal exposure leads to respiratory tract sensitization, and several groups have reported in mice that the magnitude of respiratory responses to inhaled isocyanates (an analogous low-molecular-weight chemical OA inducer) depends on prior frequency and concentration of dermal sensitization [30–32]. Relatedly, we found no statistical differences in IL-13 and IFN-γ cytokine levels measured in the BALFs regardless of AP-mediated asthma induction. In fact, in a preceding report establishing the AP-mediated OA model, significant differences in cytokine levels among the AP-sensitized and -challenged group and the control nonsensitized and/or unchallenged groups were found only in the undiluted supernatants of in-vitro-cultured auricular lymphocytes [16]. To note, the slightly increased, although nonsignificant, level of IL-6 found early after AP challenge only in the BALFs of the asthmatic untreated mice might suggest the involvement of the IL-17 axis in the AP-induced OA phenotype. IL-6 has recently emerged as main regulator of the differentiation and function of Th17 cells [33]. Thus, further research is needed to confirm the occurrence of Th17-driven airway inflammation in our AP-induced neutrophilic asthma model and its therapeutic reduction by hASCs, analogously to other recently reported Th17-driven neutrophilic asthma models [28].
On the other hand, the involvement of IgE to the pathogenesis of OA induced by high-molecular-weight agents has been well defined, although its contribution to persulfate-induced OA is still unclear. It has been recently shown that in vivo intranasal challenge with AP in sensitized mice leads to an immediate increase in total serum IgE [16]. Here we revealed that hASC treatment at 24 h after AP inhalation, when there is already a significant increase of IgE levels [16], was able to revert IgE production to basal levels. The mechanisms by which IgE normalization is achieved are not well understood, although the immune regulatory actions of hASCs inhibiting B cell proliferation, differentiation to antibody secreting cells, and chemotaxis may play a pivotal role [34].
Further, AP-induced asthmatic mice exhibited a marked hyperemia with polymorphonuclear cell infiltrates and thickening of the peribronchial musculature. In contrast, pulmonary histology of hASC-treated mice revealed a significant reduction or even absence of asthmatic pathology, likely due to both paracrine anti-inflammatory and indirect “regenerative” actions of hASCs, administered after the entire inflammatory response was triggered and, hence, once the remodeling of the airways was initiated.
One of the most relevant symptoms of asthma is the existence of AHR to different stimuli such as methacholine. There are several studies that have analyzed the effect of MSCs on the AHR in mouse asthma models [see, for example, 14,21,35]. Nearly all of them performed the analysis 2 days after asthma induction. In our study, hASC-treated animals developed neither airway resistance nor airway elastance in response to methacholine compared with untreated animals at both 2 and 7 days after AP-mediated asthma induction. This observation is clinically relevant because a single hASC administration enabled the maintenance of lung unresponsiveness for at least 1 week, which demonstrates the therapeutic utility of hASCs abolishing inflammation, preventing early lung remodeling, and, consequently, resolving AHR [36]. Nevertheless, a more comprehensive airway function study will be needed to assess additional parameters, such as upper airway resistance (Rn), tissue damping (G), and tissue elastance (H), allowing to distinguish central and peripheral respiratory mechanics and to provide information about the heterogeneity of the respiratory response in the AP-induced OA model.
To assess whether inhibiting IL-33 could further contribute to the therapeutic potential of the hASCs in our AP-induced OA model, we engineered these cells to overexpress sST2, the soluble decoy for IL-33 (hASC-sST2). Although hASC-sST2-based therapy was able to prevent IL-33 induction, the fact that unmodified hASCs already resolved the principal features of the asthmatic pathology in our model would explain the minor enhancement of the beneficial effect observed after hASC-sST2 administration. Thus, additional blockade of the IL-33-T1/ST2 pathway by sustained overexpression of sST2 from the engineered hASCs might be able to abrogate the persistence of AHR, as suggested recently using either this decoy receptor, a monoclonal antibody against IL-33 [37], or a monoclonal antibody against T1/ST2 [38]. It remains to be seen whether hASC-sST2 would be able to further extend the therapeutic benefit provided by unmodified hASCs at later time points after AP challenge.
Finally, a significant rise in CD3+ T cell numbers was observed uniquely in the BALF of the hASC- and hASC-sST2-treated animals up to day 7 after treatment regardless of asthma induction, with an increased presence of CD3+CD8+ T cells, considered to have a major role in graft rejection [39]. In fact, a recent study has shown that allogeneic ASCs are susceptible for lysis by both cytotoxic CD8+ T cells and NK cells [40]. This suggests the occurrence of a host immune response against xenogeneic hASCs. However, this response was transient and did appear to compromise neither the health of the treated mice nor the therapeutic potential of hASCs. We and others have demonstrated the effectiveness of human ASC/MSC infusion within a xenogeneic environment in murine models of lung injury [13,17,24,27,30,41], although the present study highlights for the first time the development of a short host immune response against the infused xenogeneic cells. Nevertheless, from a clinical standpoint, the immune-mediated clearance of allogeneic hASCs, particularly genetically engineered hASCs, in the treated lungs should reduce the safety concerns regarding the persistence of these implanted cells longer than required. Therefore, in several pathological settings, hASCs may exert therapeutic function through a brief “hit-and-run” mechanism, although in chronic conditions protecting these cells from immune detection and prolonging their persistence in vivo may improve clinical outcomes and prevent patient sensitization toward donor antigens.
Supplementary Material
Acknowledgments
This work was supported by grants from the “Federación Española de Fibrosis Quística,” and from 2009SGR1490 (Generalitat de Catalunya) to J.M.A., by fellowships from “Associació Catalana de Fibrosi Quística” to I.M., and from FIS PI10/00782, SEPAR, and FUCAP. J.M.A. is sponsored by the “Researchers Stabilization Program” from the SNS-Dpt. Salut Generalitat de Catalunya (Exp. CES06/012). The authors wish to thank Dr. Xavier Tintoré (Capio Hospital General de Catalunya) for supplying the lipoaspirates, and Esther Castaño (CCiT-UB) for her aid in the flow cytometry assays.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Szram J. and Cullinan P. (2012). Occupational asthma. Semin Respir Crit Care Med 33:653–665 [DOI] [PubMed] [Google Scholar]
- 2.Moscato G. and Galdi E. (2006). Asthma and hairdressers. Curr Opin Allergy Clin Immunol 6:91–95 [DOI] [PubMed] [Google Scholar]
- 3.Orriols R, Isidro I, Abu-Shams K, Costa R, Boldu J, Rego G, Zock JP; other Members of the Enfermedades Respiratorias Ocupacionales y Medioambientales (EROM) Group. (2010). Reported occupational respiratory diseases in three Spanish regions. Am J Ind Med 53:922–930 [DOI] [PubMed] [Google Scholar]
- 4.Muñoz X, Cruz MJ, Orriols R, Bravo C, Espuga M. and Morell F. (2003) Occupational asthma due to persulfate salts: diagnosis and follow-up. Chest 123:2124–2129 [DOI] [PubMed] [Google Scholar]
- 5.Vandenplas O, Dressel H, Wilken D, Jamart J, Heederik D, Maestrelli P, Sigsgaard T, Henneberger P. and Baur X. (2011). Management of occupational asthma: cessation or reduction of exposure?. A systematic review of available evidence. Eur Respir J 38:804–811 [DOI] [PubMed] [Google Scholar]
- 6.Rachiotis G, Savani R, Brant A, MacNeill SJ, Newman Taylor A. and Cullinan P. (2007). Outcome of occupational asthma after cessation of exposure: a systematic review. Thorax 62:147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muñoz X, Gómez-Ollés S, Cruz MJ, Untoria MD, Orriols R. and Morell F. (2008). Course of bronchial hyperresponsiveness in patients with occupational asthma caused by exposure to persulfate salts. Arch Bronconeumol 44:140–145 [DOI] [PubMed] [Google Scholar]
- 8.Prockop DJ. and Oh JY. (2012). Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther 20:14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knight DA, Rossi FM. and Hackett TL. (2010). Mesenchymal stem cells for repair of the airway epithelium in asthma. Expert Rev Respir Med 4:747–758 [DOI] [PubMed] [Google Scholar]
- 10.Strioga M, Viswanathan S, Darinskas A, Slaby O. and Michalek J. (2012). Same or not the same?. Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev 21:2724–2752 [DOI] [PubMed] [Google Scholar]
- 11.Németh K, Keane-Myers A, Brown JM, Metcalfe DD, Gorham JD, Bundoc VG, Hodges MG, Jelinek I, Madala S, Karpati S. and Mezey E. (2010). Bone marrow stromal cells use TGF-beta to suppress allergic responses in a mouse model of ragweed-induced asthma. Proc Natl Acad Sci U S A 107:5652–5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanz L, Compt M, Guijarro-Muñoz I. and Álvarez-Vallina L. (2012). Nonhematopoietic stem cells as factories for in vivo therapeutic protein production. Gene Ther 19:1–7 [DOI] [PubMed] [Google Scholar]
- 13.Bonfield TL, Koloze M, Lennon DP, Zuchowski B, Yang SE. and Caplan AI. (2010). Human mesenchymal stem cells suppress chronic airway inflammation in the murine ovalbumin asthma model. Am J Physiol Lung Cell Mol Physiol 299:L760–L770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SH, Jang AS, Kwon JH, Park SK, Won JH. and Park CS. (2011). Mesenchymal stem cell transfer suppresses airway remodeling in a toluene diisocyanate-induced murine asthma model. Allergy Asthma Immunol Res 3:205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC. and Stewart DJ. (2007). Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med 4:e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Vooght V, Cruz MJ, Haenen S, Wijnhoven K, Muñoz X, Hoet PH, Morell F, Nemery B. and Vanoirbeek JA. (2010). Ammonium persulfate can initiate an asthmatic response in mice. Thorax 65:252–257 [DOI] [PubMed] [Google Scholar]
- 17.Martínez-González I, Roca O, Masclans JR, Moreno R, Salcedo MT, Baekelandt V, Cruz MJ, Rello J. and Aran JM. (2013). Human mesenchymal stem cells overexpressing the IL-33 antagonist soluble IL-1 receptor-like-1 attenuate endotoxin-induced acute lung injury. Am J Respir Cell Mol Biol 49:552–562 [DOI] [PubMed] [Google Scholar]
- 18.Lloyd CM. (2010). IL-33 family members and asthma—bridging innate and adaptive immune responses. Curr Opin Immunol 22:800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno R, Martínez I, Petriz J, Nadal M, Tintoré X, Gonzalez JR, Gratacós E. and Aran JM. (2011). The β-interferon scaffold attachment region confers high-level transgene expression and avoids extinction by epigenetic modifications of integrated provirus in adipose tissue-derived human mesenchymal stem cells. Tissue Eng Part C Methods 17:275–287 [DOI] [PubMed] [Google Scholar]
- 20.Ren G, Zhao X, Zhang L, Zhang J, L'Huillier A, Ling W, Roberts AI, Le AD, Shi S, Shao C. and Shi Y. (2010). Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol 184:2321–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, et al. (2009). Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med 15:42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kavanagh H. and Mahon BP. (2010). Allogeneic mesenchymal stem cells prevent allergic airway inflammation by inducing murine regulatory T cells. Allergy 66:523–531 [DOI] [PubMed] [Google Scholar]
- 23.Goodwin M, Sueblinvong V, Eisenhauer P, Ziats NP, LeClair L, Poynter ME, Steele C, Rincon M. and Weiss DJ. (2011). Bone marrow-derived mesenchymal stromal cells inhibit Th2-mediated allergic airways inflammation in mice. Stem Cells 29:1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun YQ, Deng MX, He J, Zeng QX, Wen W, Wong DS, Tse HF, Xu G, Lian Q, Shi J. and Fu QL. (2012). Human pluripotent stem cell-derived mesenchymal stem cells prevent allergic airway inflammation in mice. Stem Cells 30:2692–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abreu SC, Antunes MA, de Castro JC, de Oliveira MV, Bandeira E, Ornellas DS, Diaz BL, Morales MM, Xisto DG. and Rocco PRM. (2013). Bone marrow-derived mononuclear cells vs. mesenchymal stromal cells in experimental allergic asthma. Respir Physiol Neurobiol 187:190–198 [DOI] [PubMed] [Google Scholar]
- 26.Ge X, Bai C, Yang J, Lou G, Li Q. and Chen R. (2013). Effect of mesenchymal stem cells on inhibiting airway remodeling and airway inflammation in chronic asthma. J Cell Biochem 114:1595–1605 [DOI] [PubMed] [Google Scholar]
- 27.Mathias LJ, Khong SML, Spyroglou L, Payne NL, Siatskas C, Thorburn AN, Boyd RL. and Heng TSP. (2013). Alveolar macrophages are critical for the inhibition of allergic asthma by mesenchymal stromal cells. J Immunol 191:5914–5924 [DOI] [PubMed] [Google Scholar]
- 28.Lathrop MJ, Brooks EM, Bonenfant NR, Sokocevic D, Borg ZD, Goodwin M, Loi R, Cruz F, Dunaway CW, Steele C. and Weiss DJ. (2014). Mesenchymal stromal cells mediate Aspergillus hyphal extract-induced allergic airway inflammation by inhibition of the Th17 signaling pathway. Stem Cells Transl Med 3:194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caplan AI. and Correa D. (2011). The MSC: an injury drugstore. Cell Stem Cell 9:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarkowski M, Vanoirbeek JA, Vanhooren HM, De Vooght V, Mercier CM, Ceuppens J, Nemery B. and Hoet PH. (2007). Immunological determinants of ventilatory changes induced in mice by dermal sensitization and respiratory challenge with toluene diisocyanate. Am J Physiol Lung Cell Mol Physiol 292:L207–L214 [DOI] [PubMed] [Google Scholar]
- 31.Vanoirbeek JA, De Vooght V, Vanhooren HM, Nawrot TS, Nemery B. and Hoet PH. (2008). How long do the systemic and ventilatory responses to toluene diisocyanate persist in dermally sensitized mice?. J Allergy Clin Immunol 121:456–463 [DOI] [PubMed] [Google Scholar]
- 32.Pauluhn J. (2008). Brown Norway rat asthma model of diphenylmethane-4,4′-diisocyanate (MDI): analysis of the elicitation dose-response relationship. Toxicol Sci 104:320–331 [DOI] [PubMed] [Google Scholar]
- 33.Camporeale A. and Poli V. (2012). IL-6, IL-17 and STAT3: a holy trinity in auto-immunity?. Front Biosci 17:2306–2326 [DOI] [PubMed] [Google Scholar]
- 34.Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V. and Uccelli A. (2006). Human mesenchymal stem cells modulate B-cell functions. Blood 107:367–372 [DOI] [PubMed] [Google Scholar]
- 35.Park HK, Cho KS, Park HY, Shin DH, Kim YK, Jung JS, Park SK. and Roh HJ. (2010). Adipose-derived stromal cells inhibit allergic airway inflammation in mice. Stem Cells Dev 19:1811–1818 [DOI] [PubMed] [Google Scholar]
- 36.Busse WW. (2010). The relationship of airway hyperresponsiveness and airway inflammation: airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest 138:4S–10S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, Kim YK, Kwon SS. and Lee SY. (2014). Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res 40:66–76 [DOI] [PubMed] [Google Scholar]
- 38.Kearley J, Buckland KF, Mathie SA. and Lloyd CM. (2009). Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am J Respir Crit Care Med 179:772–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rocha PN, Plumb TJ, Crowley SD. and Coffman TM. (2003). Effector mechanisms in transplant rejection. Immunol Rev 196:51–64 [DOI] [PubMed] [Google Scholar]
- 40.Crop MJ, Korevaar SS, de Kuiper R, IJzermans JN, van Besouw NM, Baan CC, Weimar W. and Hoogduijn MJ. (2011). Human mesenchymal stem cells are susceptible to lysis by CD8(+) T cells and NK cells. Cell Transplant 20:1547–1559 [DOI] [PubMed] [Google Scholar]
- 41.Meyerrose TE, Roberts M, Ohlemiller KK, Vogler CA, Wirthlin L, Nolta JA. and Sands MS. (2008). Lentiviral-transduced human mesenchymal stem cells persistently express therapeutic levels of enzyme in a xenotransplantation model of human disease. Stem Cells 26:1713–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.