

ABSTRACT
We analyzed the 16S rRNA amplicon composition in fecal samples of selected patients during their prolonged stay in an intensive care unit (ICU) and observed the emergence of ultra-low-diversity communities (1 to 4 bacterial taxa) in 30% of the patients. Bacteria associated with the genera Enterococcus and Staphylococcus and the family Enterobacteriaceae comprised the majority of these communities. The composition of cultured species from stool samples correlated to the 16S rRNA analysis and additionally revealed the emergence of Candida albicans and Candida glabrata in ~75% of cases. Four of 14 ICU patients harbored 2-member pathogen communities consisting of one Candida taxon and one bacterial taxon. Bacterial members displayed a high degree of resistance to multiple antibiotics. The virulence potential of the 2-member communities was examined in C. elegans during nutrient deprivation and exposure to opioids in order to mimic local conditions in the gut during critical illness. Under conditions of nutrient deprivation, the bacterial members attenuated the virulence of fungal members, leading to a “commensal lifestyle.” However, exposure to opioids led to a breakdown in this commensalism in 2 of the ultra-low-diversity communities. Application of a novel antivirulence agent (phosphate-polyethylene glycol [Pi-PEG]) that creates local phosphate abundance prevented opioid-induced virulence among these pathogen communities, thus rescuing the commensal lifestyle. To conclude, the gut microflora in critically ill patients can consist of ultra-low-diversity communities of multidrug-resistant pathogenic microbes. Local environmental conditions in gut may direct pathogen communities to adapt to either a commensal style or a pathogenic style.
IMPORTANCE
During critical illness, the normal gut microbiota becomes disrupted in response to host physiologic stress and antibiotic treatment. Here we demonstrate that the community structure of the gut microbiota during prolonged critical illness is dramatically changed such that in many cases only two-member pathogen communities remain. Most of these ultra-low-membership communities display low virulence when grouped together (i.e., a commensal lifestyle); individually, however, they can express highly harmful behaviors (i.e., a pathogenic lifestyle). The commensal lifestyle of the whole community can be shifted to a pathogenic one in response to host factors such as opioids that are released during physiologic stress and critical illness. This shift can be prevented by using compounds such as Pi-PEG15-20 that interrupt bacterial virulence expression. Taking the data together, this report characterizes the plasticity seen with respect to the choice between a commensal lifestyle and a pathogenic lifestyle among ultra-low-diversity pathogen communities that predominate in the gut during critical illness and offers novel strategies for prevention of sepsis.
INTRODUCTION
The gastrointestinal tract reservoir is the primary site of colonization of health care-associated pathogens and the site from which most pathogens disseminate to cause serious infections (1–5). In addition, the intestinal tract has been considered to be the key site for the emergence of antibiotic resistance and virulence expression among health care-associated pathogens that ultimately cause life-threatening sepsis (6–8). This situation is especially prevalent during prolonged critical illness, when the gut undergoes nearly complete ecological collapse owing to the selective pressures imposed by modern intensive care therapy, including multiple antibiotic exposure, provision of all nutrients exclusively through the intravenous route (total parenteral nutrition), and the use of vasoactive agents that alter the intestinal blood supply, acid-reducing agents, and opioids. As a result, a syndrome of gut-derived sepsis can be observed to occur late in the course of critical illness as microbes struggle to survive. Emergence of antibiotic resistance under such circumstances is proposed to occur in the gut due to these aggregate selective pressures. Thus, the term “late-onset sepsis” is being used to describe sepsis late in the course of critical illness involving multidrug-resistant (MDR) health care-acquired pathogens (9–14). A better understanding of the ecological perturbations that develop in the microbiota during critical illness, in terms of both composition and function, is crucial to prevent late-onset sepsis during the often-unanticipated prolonged course of recovery from extreme medical interventions such as organ transplant, cancer chemotherapy, and burn injury.
Here we characterized the composition and function of microbial communities from several sets of fecal samples from patients suffering from prolonged critical illness. Our results indicate that prolonged critical illness results in near-complete disruption of the normal microbiota, the members of which are replaced by ultra-low-diversity communities of highly resistant pathogens whose virulent or nonvirulent behavior is dependent on the interactions between its members and provocative host factors (i.e., opioids). A more complete understanding of the threat of highly virulent and resistant pathobiomes that emerge as members of the normal microbiota disappear over the course of prolonged critical illness is warranted to develop new strategies to prevent late-onset sepsis in hospitalized patients.
RESULTS
16S rRNA analysis of the bacterial composition in stool samples of ICU patients. (i) Phylum level.
Stool samples from healthy volunteers demonstrated dominance of Firmicutes and Bacteroidetes among the four stool samples (H1, H2, H4, and H5). A predominance of Firmicutes and very low levels of Bacteroidetes were found in one (H3) stool sample. Proteobacterial abundance remained below 1% in healthy volunteers (Fig. 1A). In 50% of intensive care unit (ICU) patients (ICU1, ICU4, ICU6, ICU9, ICU11, and ICU15), either Proteobacteria or Firmicutes organisms were totally dominant from at least one time point of stool collection (Fig. 1B and C). In patients ICU1, ICU11, and ICU15, during their course of hospitalization, we observed dramatic changes in microbial composition, with Firmicutes being completely replaced by Proteobacteria. Proteobacteria-dominated communities that persisted during the course of hospitalization were found in the stool samples of patients ICU6 (Fig. 1B) and ICU4 (Fig. 1C). Importantly, Proteobacteria and Firmicutes dominated in the stool of both deceased (Fig. 1B) and discharged (Fig. 1C) ICU patients.
FIG 1 .

The taxonomic composition of the gut microbiome at the phylum level determined by 16S rRNA analysis of stool samples collected from healthy volunteers (A), ICU patients dying with signs of severe sepsis (as indicated by black circles on the time line) (B), and ICU patients who had recovered (as indicated by green circles on the time line) (C). Dates of stool collection are displayed in numbered quadrants.
(ii) Genus level.
In the group of healthy volunteers, the microbial communities comprised ~40 bacterial genera (see Fig. S1 in the supplemental material), including Coprococcus (phylum Firmicutes [p_Firmicutes], family Lachnospiraceae [f_Lachnospiraceae]), Bacteroides (p_Bacteroidetes, f_Bacteroidaceae), Roseburia (p_Firmicutes, f_Lachnospiraceae), Prevotella (p_Bacteroidetes, f_Prevotellaceae), a nonidentified genus of f_Lachnospiraceae (p_Firmicutes), a nonidentified genus of f_Coprobacillaceae (p_Firmicutes), and Blautia (p_Firmicutes, f_Lachnospiraceae) as the seven most abundant. The microbial composition in critically ill patients was profoundly different from that in healthy volunteers. We observed decreased diversity in >80% ICU stool samples as seen by Chao1 index values below 50 and extremely low diversity in ~50% of ICU stool samples as seen by Chao1 diversity index values below 10 (Fig. 2). Unusually high abundances of Pseudomonadaceae were observed in the stool of ICU2 and ICU3 (10% and 30%, respectively) (see Fig. S2). Decreased diversity and domination by Escherichia and Enterococcus were found in seven patients (ICU5, ICU8, ICU10, and ICU12 to ICU15) (see Fig. S3).
FIG 2 .
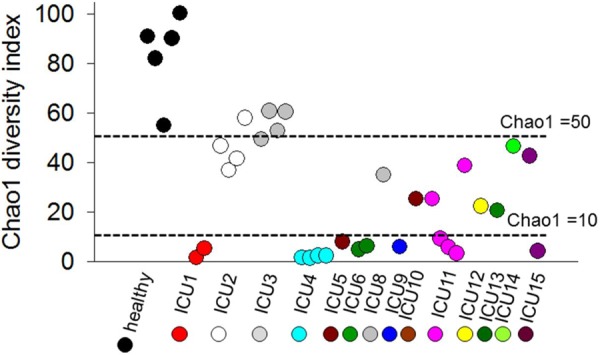
Chao1 diversity index of bacterial composition in fecal samples. Chao1 index values for stool samples of healthy volunteers (black circles) and ICU patients (colored circles) are ordered as follows (left to right): 1 to 5, healthy volunteers; 6 and 7, ICU1-1 and ICU1-2; 8 to 11, ICU2-1 to ICU2-4; 12 to 15, ICU3-1 to ICU3-4; 16 to 19, ICU4-1 to ICU4-4; 20, ICU5-1; 21 and 22, ICU6-1 and ICU6-2; 23, ICU8-1; 24, ICU9-1; 25, ICU10-1; 26 to 30, ICU11-1 to ICU11-5; 31, ICU12-1; 32, ICU13-1; 33, ICU14-1; and 34 and 35, ICU15-1 and ICU15-2. The horizontal dashed lines corresponding to Chao1 index values <50 and <10 represent arbitrary cutoffs. For the samples in this study, 14 had Chao1 index values of <10, 12 had Chao1 index values of 10 to 15, and 9 had Chao1 index values of >50. Mean Chao1 index value, 33; median, 25; range, 1.46 to 100.24.
The emergence of extremely low bacterial diversity (>90% abundance of one bacterial taxon) was determined in samples from five patients (ICU1, ICU4, ICU6, ICU9, and ICU11) (Fig. 3; see also Fig. S4 in the supplemental material). A predominance (>90% abundance) of Enterococcus (p_Firmicutes, f_Enterococcaceae), Staphylococcus (p_Firmicutes, f_Staphylococcaceae), and f_Enterobacteriaceae with an absence of p_Bacteroidetes was the most striking feature of this group. Another striking feature of this group was fast dynamic replacement of one dominated phylum by another (i.e., Firmicutes by Proteobacteria). The taxonomic composition in the microbiomes in the members of these groups was found to be similar to the composition of cultured bacteria (Fig. 3; see also Fig. S4), a unique situation that may develop only when the microbiome diversity is profoundly decreased. We also noticed the presence of the eukaryotic pathogen Candida in four of five patients. One of the five patients (ICU9, who was discharged after 10 days in the ICU) did not harbor Candida species. Reads corresponding to Enterococcus determined by 16S rRNA amplicon sequencing in the stool of ICU9 comprised 98.82% of all sequence reads and represented 2 cultivable Enterococcus species, MDR E. faecium and E. gallinarum (see Fig. S4). Blood cultures from this patient yielded Escherichia coli but were culture negative for Enterococcus. The other 4 patients (ICU1, ICU4, ICU6, and ICU11) were admitted to the ICU for longer than a month; for these patients, a temporal series of stool samples were acquired. Thus, we could correlate the 16S rRNA amplicon profiles with the clinical histories, antibiotic therapy, and microbiological culture data from nongastrointestinal sources (i.e., blood, sputum, and urine) and gastrointestinal sources (feces) for each of these patients (Fig. 3).
FIG 3 .




Taxonomic and culturing composition of 2-member pathogen communities in correlation to antibiotic regimen, antibiotic resistance of stool isolates, and microbial data of cultured species from nonintestinal samples. (A) Patient ICU1. (B) Patient ICU6. (C) Patient ICU11. (D) Patient ICU4. Cultured species from sources other than the gut refer to species cultured from extraintestinal sources such as blood, urine, and lung. The arrows indicate the time points along the course of antibiotic exposure at which these organisms were cultured. Vanc, vancomycin; cipro, ciprofloxacin; difluca, fluconazole (Diflucan); AmphoB, amphotericin B; flagyl, metronidazole (Flagyl); Pip, piperacillin; TMP/SUX, trimethoprim/sulfamethoxazole; Quinupristin/dalf, quinupristin/dalfopristin; Staph, Staphylococcus.
The 16S rRNA profiling is in agreement with culture analysis of stool samples in ICU patients harboring low-diversity pathogen communities. (i) Patient ICU1 (Fig. 3A).
Patient ICU1 was admitted for a liver transplant but died from severe sepsis before undergoing transplantation following 8 weeks of ICU confinement. In this patient’s stool sample collected at time point 1 (i.e., ICU1-1), Enterococcus reads comprised 99.9% of the 16S rRNA sequenced reads. Culture analysis of the first stool samples (ICU1-1) presented a 2-member community of MDR E. faecium and Candida albicans. A retrospective analysis of the clinical history of this patient demonstrated that the urine and paracentesis fluid of this patient were culture positive for C. albicans and E. faecium (Fig. 3A). In the second stool sample (ICU1-2) taken 23 days later, Enterobacteriaceae and Enterococcus reads comprised 96% and 3.3% of the 16S rRNA reads, respectively. Culture results from the feces demonstrated a 4-member community consisting of MDR Serratia marcescens, MDR Klebsiella oxytoca, tetracycline-resistant (Tetr) E. faecalis, and C. albicans. At that time, the urine and blood of the patient were culture positive for C. albicans and Enterococcus. The bacterial members of pathogen communities were found to be resistant to multiple antibiotics (Fig. 3A). A retrospective analysis of the clinical history demonstrated that antibiotics were aggressively administered to treat the presumed infection with combinations of up to six antibiotics administered daily. C. albicans was sensitive to the fungal antibiotics used, but the bacteria present were resistant to several of the antibiotics used, including ciprofloxacin, piperacillin-tazobactam (Zosyn), and linezolid. The observation that microbial pathogens persisted in the gut suggests that they might have been a continuing source of infection.
(ii) Patient ICU6 (Fig. 3B).
Patient ICU6 was transferred from an outside hospital, where he was treated for a presumed pneumonia without any improvement. He was suspected of having a postprocedure esophageal perforation. The patient was placed on broad-spectrum antibiotics during his hospital stay because of mediastinitis and bilateral empyema. Throughout his stay, methicillin-resistant Staphylococcus aureus (MRSA) was cultured from his sputum as well as from his chest tube sites bilaterally and from his pleural fluid, and he eventually died of severe sepsis. Two stool samples (ICU6-1 and ICU6-2) collected 25 days apart were analyzed using 16S rRNA amplicon sequencing. In sample ICU6-1, 86.83% of sequence reads showed the presence of Enterobacteriaceae, while 12.9% showed the presence of Staphylococcus; these were confirmed by culture to represent K. pneumoniae and methicillin-resistant S. aureus. In sample ICU6-2, 97.78% of the 16S rRNA reads indicated the presence of Enterobacteriaceae, which was in agreement with culture results indicating that the reads exclusively represented K. pneumoniae. The eukaryotic pathogen C. albicans was cultured in both stool samples (ICU6-1 and ICU6-2). Despite the sensitivity to fluconazole, C. albicans persisted in the gut of patient ICU6 throughout treatment with multiple fungal antibiotics, including fluconazole. K. pneumoniae was found to produce extended-spectrum beta-lactamase, defining it as strain ESBL, and also to be resistant to multiple antibiotics. It was sensitive, however, to tobramycin and imipenem, but despite the patient being treated with these antibiotics, K. pneumoniae ESBL persisted in his gut during his ICU stay. C. albicans, MRSA, and K. pneumoniae were cultured from the pleural fluid and tracheal aspirates over the course of hospitalization in patient ICU6.
(iii) Patient ICU11 (Fig. 3C).
Patient ICU11 suffered from intestinal bleeding, requiring several operations to control the bleeding. The patient had several underlying comorbidities, including a nonischemic cardiomyopathy. Following the last operation, the patient developed progressive decompensation and multiorgan system failure and eventually died from severe sepsis after being confined to the ICU for 2 months. Stool samples were collected at 5 time points (Fig. 3C). ICU11-1 and ICU11-2 16S rRNA reads indicated 89.7% and 99.4% levels of Staphylococcus, respectively, which was in agreement with culturing analyses yielding only coagulase-negative (CoA−) staphylococcus. Stool samples ICU11-3 and ICU11-4 demonstrated the presence of Enterobacteriaceae, corresponding to 99.26% and 94.98% of sequence reads, respectively, which was in agreement with culture identifying K. pneumoniae. Later in the course of the patient’s illness (ICU 11-5), coagulase-negative staphylococci emerged again. The eukaryotic pathogen Candida glabrata persisted at all points of stool collections from this patient. CoA− Staphylococcus, K. pneumoniae, and C. glabrata were cultured from blood and urine at various times during the course of ICU confinement. Similarly to the cases described above, the gut microbial pathogens persisted regardless of their sensitivity or resistance to antibiotics applied to the patient.
(iv) Patient ICU4 (Fig. 3D).
Patient ICU4 was the only patient of the four with ultra-low-diversity pathogen communities who survived and was discharged after 40 days in the ICU. This patient was admitted to the ICU with a history of anaplastic astrocytoma, including 2 weeks of decreased oral intake, altered mental status, and fevers. The analysis of four stool collections at various time points (Fig. 3D) demonstrated the persistence of the same composition microbial community dominated by Enterobacteriaceae, comprising 99.8% to 99.9% of the 16S rRNA sequences, which was in agreement with the culture results showing the presence of Enterobacter aerogenes resistant to multiple antibiotics. Additionally, the eukaryotic pathogen C. albicans was persistently cultured from feces of patient ICU4 during the ICU stay. C. albicans was found to be sensitive to fluconazole and E. aerogenes to be sensitive to piperacillin-tazobactam and imipenem—antibiotics with which the patient was treated. Despite the sensitivity to these antibiotics, the community comprising C. albicans plus E. aerogenes persisted during this course of treatment (Fig. 3D). E. aerogenes was recovered from the bronchoalveolar lavage samples and endotracheal aspirates of this patient.
To conclude, in all cases listed above (ICU1, ICU4, ICU6, and ICU11), there were similar striking features that included a strong agreement of 16S rRNA amplicon sequencing profiles with the bacterial composition shown by culture, an agreement of the results determined using microbes cultured from intestinal and nonintestinal sources, and a failure of aggressive antibiotic treatment to eliminate these pathogens regardless of their sensitivities to administered antibiotics. Antibiotic susceptibilities of all the cultured microbes from the patients in this study are displayed in Table S1 in the supplemental material. As might be predicted, a high frequency of antibiotic-resistant microorganisms colonizing the gut of critically ill patients was observed.
Ex vivo analysis of potential virulence of low-diversity intestinal communities.
Given the close correlation of 16S rRNA amplicon sequencing profiles to microbial composition revealed by culture, we decided to determine the virulence potential of these pathogen communities by reassembling them ex vivo and allowing C. elegans to feed on them. C. elegans normally feeds on bacteria as a food source, and therefore its response to pathogen communities can be viewed as a proxy for the virulence potential of the bacterium. Six communities were included in the analysis: ICU1-1 (C. albicans: Enterococcus faecium), ICU6-2 (C. albicans: Klebsiella pneumoniae), ICU4-4 (C. albicans: Enterobacter aerogenes), ICU11-2 (C. glabrata: CoA− Staphylococcus), ICU11-4 (C. glabrata: K. pneumoniae), and ICU9-1 (Enterococcus faecium: Enterococcus gallinarum). Based on results of C. elegans experiments, we made several important observations recorded below.
(i) Commensal lifestyle of 2-member pathogen communities.
In the first round of the experiments, to mimic the nutrient-limited environment typical of the gut of a critically ill host, we grew the individual members of the communities as well as assembled whole communities in nutrient-poor 0.1× TY liquid media (tryptone, 1 g/liter; yeast extract, 0.5 g/liter, potassium phosphate buffer, 0.1 mM [pH 6.0]) (referred to here as 0.1× TY). After 4 h of growth in 0.1× TY, prestarved and antibiotic-treated worms were transferred to the microbial cultures. Antibiotic treatment was used to eliminate all residual bacteria from the intestinal tubes of the worms. We found that all but one Candida species (C. albicans ICU4-4) caused high levels of mortality in C. elegans (Fig. 4). None of the individual bacteria demonstrated a high killing capacity, and they were able to attenuate the C. elegans mortality caused by the Candida species, resulting in a commensal assemblage.
FIG 4 .

The commensal behavior of the 2-member pathogen communities. Data represent Kaplan-Meier survival curves of C. elegans fed on 2-member pathogen communities and their individual members (n = 10/plate, 3 plates/group). Data represent the cumulative results of 2 experiments.
(ii) Replacement of a comember can switch commensal organisms to display pathogenic behavior.
As seen in Fig. 5, the killing ability of the ICU1-2 community significantly exceeded the killing ability of the ICU1-1 community. This correlated with the morphology of C. albicans inside the community, with the yeast form being prevalent in the ICU1-1 sample and the hyphal form being prevalent in the ICU1-2 sample. These data confirm our previous demonstration that the ICU1-2 community consisting of C. albicans plus S. marcescens plus K. oxytoca plus E. faecalis induces a high level of mortality in C. elegans and mouse models (15). Therefore, the sudden replacement of a comember may provoke pathogenic behavior in a newly created community.
FIG 5 .

Replacement of bacterial comember switches the commensal behavior to pathogenic. (A) Kaplan-Meier survival curves of C. elegans fed on ICU1-1 and ICU1-2 pathogen communities (n = 10/plate, 3 plates/group). Data represent the cumulative results of 2 experiments. (B and C) Scanning electron microscopy images of ICU1-1 (B) and ICU1-2 (C) grown for 20 h in 0.1× TY.
(iii) Critical care therapy can break commensalism.
As was previously demonstrated, critical care factors used for treatment of critically ill patients (i.e., steroids, hormones, and opioids) can induce bacterial virulence via interkingdom signaling through their interaction with quorum-sensing signaling systems (15–23). Opioids can also be released in the gut during physiological stress such as that seen following ischemia, which commonly occurs during critical illness (16). To determine if host-derived factors can affect the commensalism, we included a synthetic opioid (U-50,488) in the growth media. We have previously shown that this potent ligand of the kappa-opioid receptor acts similarly to the endogenous kappa-opioid dynorphin (16), which is released during intestinal ischemia. C. elegans was allowed to feed on bacteria grown under conditions of nutrient limitation (0.1× TY) and exposure to opioids (50 µM of U-50,488). In two communities, ICU6-2 and ICU11-2, the addition of 50 µM U-50,488 significantly increased the killing capacity of bacterial members K. pneumoniae ICU6-2 and CoA− Staphylococcus ICU11-2; accordingly, the virulence of each assemblage was shifted from a commensal behavior to a pathogenic one (Fig. 6). However, not all the pathogen communities responded with enhanced killing capacity during opioid exposure. There was no significant increase in the mortality of worms fed on ICU1-1, ICU11-4, ICU4-4, and ICU9-1 communities (Fig. 6; see also Fig. S5 in the supplemental material), a result similar to the baseline control of mortality seen with C. elegans fed on a nonpathogen food source such as E. coli OP 50 (see Fig. S6).
FIG 6 .

Opioids can break the commensalism of 2-member pathogen communities. Kaplan-Meier survival curves of C. elegans fed on ICU6-2 (A), ICU11-2 (B), ICU11-4 (C), and ICU4-4 (D) 2-member communities in the presence of an opioid (50 µM U-50,488) (n = 10/plate, 3 plates/group; P < 0.01). Data represent the cumulative results of 2 experiments. Exposure to opioids broke the commensalism in the ICU6-2 and ICU11-2 communities.
We have summarized the community compositions, their behavior in the C. elegans model, and the outcomes of patients in Table 1. Although this study was not powered to determine if C. elegans mortality readouts can predict patient outcome, there appears to be an association between whether a community displays commensal versus pathogenic behavior and an outcome. For example, community ICU1-1, which displayed commensal behavior, became replaced by community ICU1-2, which displayed a pathogenic behavior, and this patient died. The proteodominant ICU4-4 community displayed commensal behavior even under provocative conditions (i.e., opioid exposure), and the patient recovered and was discharged. In patient ICU11, pathogenic community ICU11-2 (CoA− Staphylococcus plus C. glabrata) was replaced by a nonpathogenic community, ICU11-4 (K. pneumoniae plus C. glabrata); however, reemergence of CoA− Staphylococcus was observed later, at time point 5 (ICU11-5), and this patient died. These data (although not powered) do suggest that the development of sepsis may not necessarily be a function of the specific taxa present but rather of whether the remaining species behave commensally or pathogenically as a community.
TABLE 1 .
Compositions of microbial communities, their behavior in the C. elegans model, and patient outcomes
| Patient | Time point of feces collection | Cultured community | Behavior in C. elegans model | Patient outcome |
|---|---|---|---|---|
| ICU1 | 1 | E. faecium ICU1-1, C. albicans ICU1-1 | Commensal | Deceased |
| ICU1 | 2 | E. faecalis ICU1-2, C. albicans ICU1-2, S. marcescens ICU1-2, K. oxytoca ICU1-2 | Pathogenic | |
| ICU4 | 1 | E. aerogenes ICU4-1, C. albicans ICU4-1 | NTb | Discharged |
| ICU4 | 2 | E. aerogenes ICU4-2, C. albicans ICU4-2 | NT | |
| ICU4 | 3 | E. aerogenes ICU4-3, C. albicans ICU4-3 | NT | |
| ICU4 | 4 | E. aerogenes ICU4-4, C. albicans ICU4-4 | Commensal | |
| ICU6 | 1 | S. aureus ICU6-1, K. pneumoniae ICU6-1, C. albicans ICU6-1 | NT | Deceased |
| ICU6 | 2 | K. pneumoniae ICU6-2, C. albicans ICU6-2 | Pathogenic | |
| ICU9 | 1 | E. faecium ICU9-1, E. gallinarum ICU9-1 | Commensal | Discharged |
| ICU11 | 1 | CoA− Staphylococcus ICU11-1, C. glabrata ICU11-1 | NT | Deceased |
| ICU11 | 2 | CoA− Staphylococcus ICU11-2, C. glabrata ICU11-2 | Pathogenic | |
| ICU11 | 3 | K. pneumoniae ICU11-3, C. glabrata ICU11-3 | NT | |
| ICU11 | 4 | K. pneumoniae ICU11-4, C. glabrata ICU11-4 | Commensal | |
| ICU11 | 5 | CoA− Staphylococcus ICU11-5,a K. pneumoniae ICU11-5, C. glabrata ICU11-5 | NT |
Note the reemergence of CoA− Staphylococcus at time point 5 of stool collection.
NT, not tested.
(iv) Phosphate abundance in the form of Pi-PEG can promote commensalism.
Recent work from our laboratory demonstrated that extracellular phosphate (Pi) regulates bacterial virulence across a wide variety of pathogens (24). When phosphate is abundant, pathogen virulence is suppressed even in the presence of opioids; conversely, when phosphate is depleted, virulence expression is enhanced (17, 25). In order to preserve phosphate abundance in the gut during physiologic stress in a durable and bioavailable form, we covalently bound phosphate onto a high-molecular-weight polyethylene glycol (PEG) carrier compound to create Pi-PEG (15). We recently reported the efficacy and mechanism by which Pi-PEG can attenuate the virulence and lethality of highly resistant and virulent intestinal pathogens recovered from critically ill patients (15). In the current study, synthesized Pi-PEG15-20 was observed to prevent C. elegans mortality when C. elegans was feeding on whole communities ICU6-2 and ICU11-2 during exposure to the opioid compound U-50,488 (Fig. 7).
FIG 7 .
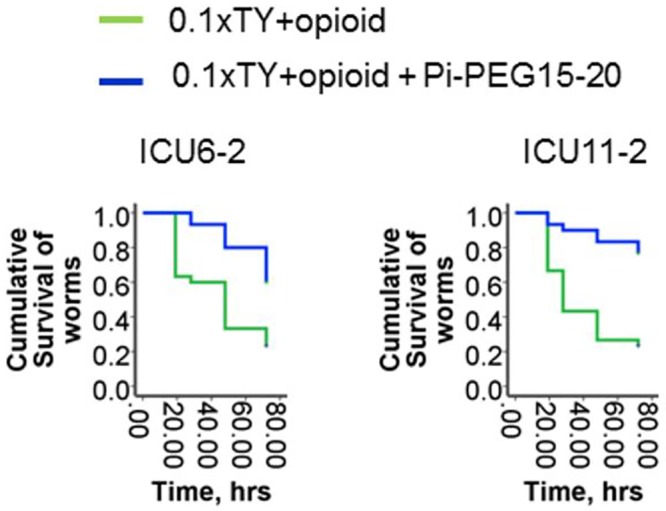
Pi-PEG15-20 stabilizes the commensal behavior of 2-member communities. Kaplan-Meier survival curves of C. elegans fed on ICU6-2 (left panel) and ICU11-2 (right panel) 2-member communities in the presence of an opioid (50 µM U-50,488) with or without 10% Pi-PEG15-20 (n = 10/plate, 3 plates/group; P < 0.01). Data represent cumulative results of 2 experiments.
DISCUSSION
The composition, community structure, and behavior of microbes that colonize the human gut during prolonged critical illness remain poorly characterized. Most patients who enter an intensive care unit remain there, on average, 2 to 3 days until they are stabilized, transferred to the recovery floor, and discharged to home. However, the members of the gut microbiota of a patient confined for a prolonged period in the ICU may be subjected to unprecedented environmental circumstances where they have to compete for limited resources and survive under harsh and hostile conditions. Microbial diversity can be lost and resistance genes and virulence traits can be selected for as the remaining pathogenic communities struggle to survive conditions of nutrient deprivation and exposure to drugs and host compensatory signals. There exists little information regarding which members of the pathobiota remain in the gut following prolonged critical illness and how they function in this hostile microenvironment. Advantages in next-generation sequencing provide exciting opportunities to determine the composition of the gut microbiome in individual patients as a “microbial fingerprint.” The challenge, however, will be to predict the behavior of a given pathogen community in the context of altering the clinical course of the patient.
Here we found that critically ill patients subjected to prolonged confinement in the ICU can harbor multidrug-resistant ultra-low-diversity microbial communities consisting of 2 to 4 pathogens whose individual virulence capacities are dependent on their composition and ability to respond to compensatory host factors released during stress. Candida albicans was the most prevalent and most stable member in 2-member pathogen communities despite patients being treated with a variety of antifungals to which it was susceptible. Infections due to C. albicans are known to originate from the gut during critical illness and are a major cause of late-onset sepsis and mortality in the ICU (26–28). The C. albicans virulence in the dual communities was found to be suppressed by the presence of bacterial comembers such as E. faecium, K. pneumoniae, and CoA− Staphylococcus. Such commensal-like behavior has been recently described by others for C. albicans and E. faecalis (29, 30), and yet we show that C. albicans-bacterium commensalism can be disrupted when the organisms are exposed to opioids, which raises the possibility that the degree of host stress may play a role in shifting communities toward a more pathogenic state. We show for the first time that 2-member communities of C. albicans plus bacteria can superdominate the normal gut microbiota during prolonged critical illness. There was strong agreement between 16S rRNA amplicon profiling and the community composition identified on the basis of culture, which provided confidence that the cultured communities could be used ex vivo to understand virulence potential by using C. elegans models. Previous work in our laboratory has demonstrated that opioids can enter the cytoplasm of bacteria such as P. aeruginosa and activate their quorum-sensing signaling system, leading to enhanced virulence (16, 18). Opioids were assessed in this study because they represent major stress-mediated signaling compounds in the gut (16, 31–35). The molecular details of the means by which opioids disrupt commensalism between C. albicans and other bacteria that persist in the gut during critical illness remain to be elucidated and are likely complex.
We have previously reported the efficacy of Pi-PEG in suppression of the virulence and lethality of the ICU1-2 community via mechanisms that involve phosphate-regulated pathways of virulence expression and maintenance of the normal indigenous microbiota (15). Studies were carried out both in C. elegans and in a mouse model of lethal gut-derived sepsis. In both C. elegans and mice, Pi-PEG was highly protective against mortality, and that protection was due in part to its capacity to create local phosphate abundance, which attenuates virulence. We had previously shown that phosphate abundance could prevent virulence of C. albicans and override the virulence-inducing effect of opioids in P. aeruginosa (17, 25). The present study confirmed that this effect of Pi-PEG is preserved among multidrug-resistant ultra-low-diversity pathogen communities.
A major challenge in treating critically ill patients is the overuse of antibiotics, a practice that is often unavoidable with patients exposed to multiple invasive procedures and extreme physiologic stress. A strategy to promote commensalism among the remaining pathogens by the use of agents such as Pi-PEG, which itself does not affect bacterial growth, may be useful. For example, data from the present study suggest that loss of the bacterial partner from a C. albicans community can derepress its virulence, a possible unanticipated consequence of aggressive antibiotic therapy applied to ICU patients. Our methods and results suggest that such a loss of these core commensal organisms can be deleterious, especially when critically ill patients harbor ultra-low-diversity pathogens. Use of stabilizing agents such as Pi-PEG may offer a mechanism to preserve commensalism in the gut when aggressive antibiotics are needed to treat extraintestinal infections.
It is curious that communities that were otherwise seemingly avirulent (i.e., C. albicans plus Enterobacter aerogenes in patient ICU4 and E. faecium plus E. gallinarum in patient ICU9) were present in the feces of patients who were ultimately discharged from the hospital. These communities could not be induced to express virulence against C. elegans even under provocative conditions. In fact, we have recently reported that C. albicans isolated from patient ICU4 did not respond to phosphate limitation with enhanced virulence compared to that seen with C. albicans isolated from patients ICU1 and ICU6 (25). The nonvirulent behavior of C. albicans isolated from patient ICU4 was again seen in the present study. A study in which dynamic and repeated fecal microbial analysis is coupled to physiologic variables in the host might more accurately inform outcome measure under the highly oscillatory conditions of critical illness.
In summary, here we demonstrate that the intestinal microbiome in critically ill patients can be considered a “damaged organ” given that its main cellular mass, the normal microbiota, is disrupted and dominated by pathobiota which may be an ever-threatening source for disseminating pathogens. The emergence of ultra-low-diversity pathogen communities and the activation of virulence in the response to host cues released during physiologic stress are characteristic features of this organ in some critically ill patients. Dynamic assessment of the composition and virulence potential of the intestinal microbiota or pathobiota over the course of a critical illness may allow a more calibrated use of antibiotics or, alternatively, the development of novel strategies to preserve the core intestinal microbiome when multiple antibiotics are needed to treat life-threatening extraintestinal infections.
MATERIALS AND METHODS
Stool sample collection.
Stool sampling was performed in accordance with the Institutional Review Board (IRB) protocol approved by University of Chicago. In order to establish baseline normal controls, samples were collected from five healthy volunteers who were not treated with any antibiotics for 1 year prior to sample collection. We next consecutively selected 14 patients admitted to the intensive care units (ICUs) at the University of Chicago hospitals from whom consent could be obtained for stool collection. All patients signed consent forms to participate in the study. The 14 patients represented a diverse group of ICU patients with various lengths of stay, including those who were resident for less than 10 days (3 of 14 patients), those who were resident for between 20 and 30 days (3 of 14), and those who were resident for >30 days (6 of 14). When possible, stool samples were collected at several time points during the ICU course.
Demographic and clinical history.
Demographic and clinical histories of ICU patients, including past medical history, admitting diagnosis, hospital course, therapy, especially antibiotic therapy, and discharge summary, were available in the patients’ charts in accordance with the IRB protocol. Among the 14 ICU patients, 10 were admitted to the ICU for a period of 20 days or longer. Of these 10 patients, 7 died despite continuous intensive care. The patients who died all died with signs of severe sepsis, although there was considerable variability in their comorbidities and underlying illnesses.
16S rRNA analysis of stool samples.
The microbial composition of stool samples was analyzed by 16S rRNA V4 iTAG amplicon sequencing of DNA recovered from stool samples. A BiOstic Bacteremia DNA isolation kit from MoBio Laboratories was used to extract DNA. DNA was analyzed using next-generation Illumina sequencing. Reads containing low-confidence base calls (phred value, <20) were dropped, and the remaining 150-bp-long sequences were clustered into 97%-similarity operational taxonomic units (OTUs) using QIIME 1.6.0’s pick_otus.py script with default parameters (36, 37). OTUs composed of fewer than 10 sequences were discarded, and the remaining OTUs were assigned taxonomic annotations by searching representative members of each cluster against the Greengenes 12_10 (38) database using the RDP classifier (39). Sequencing depth among samples was normalized by rarefaction to 1,200 sequences per sample prior to calculating taxonomic relative abundances. Microbial-community diversity was calculated for each sample using the Chao1 alpha diversity metric, as implemented by the QIIME alpha_rarefaction.py script.
The samples isolated from ICU patients (patients ICU1-1, ICU1-2, ICU2-1, ICU2-2, ICU3-1, ICU3-2, ICU4-1, ICU4-2, ICU4-3, and ICU4-4) were independently processed for 16S rRNA analysis at Michigan State University. DNA was extracted from the stool samples using a PowerSoil DNA isolation kit from MoBio Laboratories following the manufacturer’s instructions with the exception that Inhibitor Removal Solution from MoBio’s Ultra-Clean Fecal DNA isolation kit was added to the PowerBead Tubes along with solution C1. PCR amplification and purification were similar to those described by Rosenzweig et al. (40). The amplification and purification procedures differed in that samples were first amplified for 15 cycles using primers 577f and 927r without bar codes or adaptors, and then 2 µl of each reaction mixture was amplified for 15 more cycles with primers containing the bar codes and Roche adaptors. The thermocycler program for the first PCR was 95°C for 3 min and then 15 cycles of 95°C for 45 s, an annealing temperature of 44°C (determined from optimization experiments) for 45 s, and 72°C for 90 s, followed by elongation at 72°C for 7 min before the reaction mixture was held at 4°C. The thermocycler program for the second PCR was 95°C for 3 min and then 15 cycles of 95°C for 45 s, 56°C for 45 s, and 72°C for 90 s, followed by elongation at 72°C for 7 min before the reaction mixture was held at 4°C. Equal mass amounts of the amplicon libraries were mixed and submitted for sequencing to the Research Technology Support Facility at Michigan State University.
The sequence results were obtained and processed using the tools on the Ribosomal Database Project’s Pyrosequencing Pipeline at http://pyro.cme.msu.edu/. Initial processing parameters were a forward primer maximum edit distance of 2, a reverse primer maximum edit distance of 0, a maximum number of N’s of 0, a minimum read Q score of 20, and a minimum sequence length of 300. Trimmed sequences averaging 331 bp were aligned and clustered at a distance of 3%, and the cluster results were reformatted to be read into R (41) as an OTU table. Representative sequences for each OTU were submitted to the RDP Classifier for identification and to the Decipher Find Chimeras tool of the University of Wisconsin—Madison at http://decipher.cee.wisc.edu/ using the Short-Length Sequences option. OTUs identified as chimeric sequences were removed from the data. Further analysis was done using the BioDiversityR (42) package within the R program.
The 16S rRNA analysis of the aliquots of stool samples from the same patients was performed by the Argonne National Laboratories and Michigan State University with almost identical results, confirming extremely low diversity of the stool samples from patients ICU1 and ICU4.
Isolation, identification, and antibiotic resistance of cultured microorganisms.
Microbial species were isolated by plating bacterial communities on plates selective for Gram-negative (−) bacteria (MacConkey II), for Gram-positive (+) bacteria (Columbia CNA agar with 5% sheep’s blood [SB]) (Becton and Dickinson), for Pseudomonas aeruginosa (Pseudomonas isolation agar [PIA]), and for fungi (saturated dextrose agar with chloramphenicol and gentamicin) and on Trypticase soy agar with 5% sheep blood (TSA II 5% SB) for cultivating fastidious microorganisms. Gram (−) bacteria were identified by the use of a Vitek2 system (bioMérieux, Inc., Durham, NC). Gram (+) bacteria were identified by standard manual methods. Enterococcus species identification was performed by the use of a Vitek2 system. A positive germ tube test identified Candida albicans. Susceptibility testing was performed by testing on a Vitek2 system or by disk diffusion. Susceptibility testing of Gram-negative bacilli was performed by the use of a Vitek2 system or by disk diffusion. Susceptibility testing of MRSA and enterococci was performed by the use of a Vitek2 system. Susceptibility testing of the other Gram-positive cocci was performed using a combination of disk diffusion and E test strips. Susceptibility testing of Candida was performed using a Sensititre YeastOne MIC panel (TREK Diagnostic Systems Inc., Cleveland, OH). Susceptibility results were interpreted using Clinical and Laboratory Standards Institute (CLSI) guidelines.
Caenorhabditis elegans killing assay.
The 2-member whole communities were compared to their individual members for killing capacity by assessing mortality of C. elegans transferred into liquid microbial cultures as in our previously described experiments (15, 17). Nematodes of C. elegans N2 provided by the Caenorhabditis Genetic Center (CGC), University of Minnesota, were used in all experiments. Nematodes were prepared as follows: synchronized nematodes (larval stage 4 [L4] to young adults) were transferred from E. coli OP 50 stock plates onto plain agarized plates, followed by a second transfer onto new plain agarized plates (Falcon) (60-mm diameter). Next, 1 ml of 100 µg/ml kanamycin was poured on the agar surface; after 3 h, worms were retransferred into experimental microbial cultures.
Microbial cultures were prepared as follows: microbes from frozen individual stocks were plated on Trypticase soy broth (TSB) solid plates and incubated at 37°C overnight. Cells grown overnight were used to prepare stock microbial suspensions in 0.1× TY liquid media (tryptone, 1 g/liter; yeast extract, 0.5 g/liter, potassium phosphate buffer, 0.1 mM [pH 6.0]) (referred to here as 0.1× TY) to reach an absorbance of 1.0 at an optical density of 600 (OD600). Stock microbial suspensions were diluted in fresh 0.1× TY to an OD600 of 0.1 to prepare individual microbial cultures. To prepare community cultures, the respective stocks were mixed to reach an OD600 of 0.1 for each individual microbe in the mixture. In the experiments performed with Pi-PEG15-20, the stock microbial suspensions and final microbial cultures were prepared in 0.1× TY supplemented with 10% Pi-PEG15-20, and the pH was adjusted with KOH to that of 0.1× TY (pH 5.8). Microbial cultures were grown individually and as a mixture for 5 h at 37°C with shaking at 200 rpm. Then, 2 ml of the microbial culture was poured into a 35-mm-diameter empty sterile plate and adjusted to room temperature, and 5 to 10 worms were transferred into each plate. For each variant, 3 to 5 plates were prepared. C. elegans were fed on microbial cultures for up to 72 h under static conditions at room temperature (~23°C). The mortality of worms was followed dynamically.
SUPPLEMENTAL MATERIAL
The taxonomic composition of the gut microbiome at the genus level determined by 16S rRNA analysis of stool samples collected from healthy volunteers. The top 7 genera, consisting of Coprococcus (no. 36, p_Firmicutes, f_Lachnospiraceae), Bacteroides (no. 6, p_Bacteroidetes, f_Bacteroidaceae), Roseburia (no. 40, p_Firmicutes, f_Lachnospiraceae), Prevotella (no. 8, p_Bacteroidetes, f_Prevotellaceae), a nonidentified genus of Lachnospiraceae (no. 32, p_Firmicutes, f_Lachnospiraceae), a nonidentified genus of Coprobacillaceae (no. 62, p_Firmicutes, f_Coprobacillaceae), and Blautia (no. 35, p_Firmicutes, f_Lachnospiraceae), are shown. Download
The taxonomic composition of the gut microbiome in stool samples collected from patients ICU2 and ICU3. Pseudomonadaceae (no. 77) reached levels of 10% (ICU2-4) and 30% (ICU3-4). Download
The taxonomic composition of gut microbiome in stool samples with decreased bacterial diversity. Escherichia (no. 74) and Enterococcus (no. 20) were major pathogens in this group. Download
Taxonomic and culturing composition of the ICU4 communities in correlation to antibiotic regimen, antibiotic resistance of stool isolates, and microbial data of cultured species from nonintestinal samples. Download
Kaplan-Meier survival curves of C. elegans fed on 2-member communities whose response to an opioid (50 µM U-50,488) was negligible (n = 10/plate, 3 plates/group; P < 0.01). Data represent cumulative results of 2 experiments. Download
Kaplan-Meier survival curves of C. elegans fed on E. coli OP 50 with and without exposure to an opioid (50 µM U-50,488) (n = 10/plate, 3 plates/group). Download
Antibiotic susceptibility of microbes isolated from ICU patients and healthy individuals.
ACKNOWLEDGMENTS
This study was funded by NIH grant RO1 2R01GM062344-13A1 (J.C.A.). This work was supported in part by the U.S. Dept. of Energy under contract DE-AC02-06CH11357 (J.G.) and by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences, and an Engineering Division (M.K. and M.T.).
Footnotes
Citation Zaborin A, Smith D, Garfield K, Quensen J, Shakhsheer B, Kade M, Tirrell M, Tiedje J, Gilbert JA, Zaborina O, Alverdy JC. 2014. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. mBio 5(5):e01361-14. doi:10.1128/mBio.01361-14.
REFERENCES
- 1. Gosain A, Gamelli RL. 2005. Role of the gastrointestinal tract in burn sepsis. J. Burn Care Rehabil. 26:85–91. 10.1097/01.BCR.0000150212.21651.79 [DOI] [PubMed] [Google Scholar]
- 2. Rowlands BJ, Soong CV, Gardiner KR. 1999. The gastrointestinal tract as a barrier in sepsis. Br. Med. Bull. 55:196–211. 10.1258/0007142991902213 [DOI] [PubMed] [Google Scholar]
- 3. Soeorg H, Huik K, Parm U, Ilmoja ML, Metelskaja N, Metsvaht T, Lutsar I. 2013. Genetic relatedness of coagulase-negative staphylococci from gastrointestinal tract and blood of preterm neonates with late-onset sepsis. Pediatr. Infect. Dis. J. 32:389–393. 10.1097/INF.0b013e3182791abd [DOI] [PubMed] [Google Scholar]
- 4. Alverdy JC, Chang EB. 2008. The re-emerging role of the intestinal microflora in critical illness and inflammation: why the gut hypothesis of sepsis syndrome will not go away. J. Leukoc. Biol. 83:461–466. 10.1189/jlb.0607372 [DOI] [PubMed] [Google Scholar]
- 5. Alverdy J, Holbrook C, Rocha F, Seiden L, Wu RL, Musch M, Chang E, Ohman D, Suh S. 2000. Gut-derived sepsis occurs when the right pathogen with the right virulence genes meets the right host: evidence for in vivo virulence expression in Pseudomonas aeruginosa. Ann. Surg. 232:480–489. 10.1097/00000658-200010000-00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salyers AA, Gupta A, Wang Y. 2004. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol. 12:412–416. 10.1016/j.tim.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 7. Shimizu K, Ogura H, Hamasaki T, Goto M, Tasaki O, Asahara T, Nomoto K, Morotomi M, Matsushima A, Kuwagata Y, Sugimoto H. 2011. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig. Dis. Sci. 56:1171–1177. 10.1007/s10620-010-1418-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deitch EA. 2012. Gut-origin sepsis: evolution of a concept. Surgeon 10:350–356. 10.1016/j.surge.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, Burnham CA, Weinstock ES, Weinstock GM, Wylie TN, Mitreva M, Abubucker S, Zhou Y, Stevens HJ, Hall-Moore C, Julian S, Shaikh N, Warner BB, Tarr PI. 2014. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin. Infect. Dis. 58:1211–1218. 10.1093/cid/ciu084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Samuelsson A, Isaksson B, Hanberger H, Olhager E. 2014. Late-onset neonatal sepsis, risk factors and interventions: an analysis of recurrent outbreaks of Serratia marcescens, 2006-2011. J. Hosp. Infect. 86:57–63. 10.1016/j.jhin.2013.09.017 [DOI] [PubMed] [Google Scholar]
- 11. Blackburn RM, Verlander NQ, Heath PT, Muller-Pebody B. 2014. The changing antibiotic susceptibility of bloodstream infections in the first month of life: informing antibiotic policies for early- and late-onset neonatal sepsis. Epidemiol. Infect. 142:803–811. 10.1017/S0950268813001520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saleem AF, Qamar FN, Shahzad H, Qadir M, Zaidi AK. 2013. Trends in antibiotic susceptibility and incidence of late-onset Klebsiella pneumoniae neonatal sepsis over a six-year period in a neonatal intensive care unit in Karachi, Pakistan. Int. J. Infect. Dis. 17:e961–e965. 10.1016/j.ijid.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 13. Gray J, Arvelo W, McCracken J, Lopez B, Lessa FC, Kitchel B, Wong B, Reyes L, Lindblade K. 2012. An outbreak of Klebsiella pneumoniae late-onset sepsis in a neonatal intensive care unit in Guatemala. Am. J. Infect. Control 40:516–520. 10.1016/j.ajic.2012.02.031 [DOI] [PubMed] [Google Scholar]
- 14. Rasigade JP, Raulin O, Picaud JC, Tellini C, Bes M, Grando J, Ben Saïd M, Claris O, Etienne J, Tigaud S, Laurent F. 2012. Methicillin-resistant Staphylococcus capitis with reduced vancomycin susceptibility causes late-onset sepsis in intensive care neonates. PLoS One 7:e31548. 10.1371/journal.pone.0031548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaborin A, Defazio JR, Kade M, Kaiser BL, Belogortseva N, Camp DG, II, Smith RD, Adkins JN, Kim SM, Alverdy A, Goldfeld D, Firestone MA, Collier JH, Jabri B, Tirrell M, Zaborina O, Alverdy JC. 2014. Phosphate-containing polyethylene glycol polymers prevent lethal sepsis by multidrug-resistant pathogens. Antimicrob. Agents Chemother. 58:966–977. 10.1128/AAC.02183-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zaborina O, Lepine F, Xiao G, Valuckaite V, Chen Y, Li T, Ciancio M, Zaborin A, Petrof EO, Turner JR, Rahme LG, Chang E, Alverdy JC. 2007. Dynorphin activates quorum sensing quinolone signaling in Pseudomonas aeruginosa. PLoS Pathog. 3:e35. 10.1371/journal.ppat.0030035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaborin A, Gerdes S, Holbrook C, Liu DC, Zaborina OY, Alverdy JC. 2012. Pseudomonas aeruginosa overrides the virulence inducing effect of opioids when it senses an abundance of phosphate. PLoS One 7:e34883. 10.1371/journal.pone.0034883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Babrowski T, Holbrook C, Moss J, Gottlieb L, Valuckaite V, Zaborin A, Poroyko V, Liu DC, Zaborina O, Alverdy JC. 2012. Pseudomonas aeruginosa virulence expression is directly activated by morphine and is capable of causing lethal gut-derived sepsis in mice during chronic morphine administration. Ann. Surg. 255:386–393. 10.1097/SLA.0b013e3182331870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. 2003. Bacteria-host communication: the language of hormones. Proc. Natl. Acad. Sci. U. S. A. 100:8951–8956. 10.1073/pnas.1537100100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmer AG, Blackwell HE. 2008. Deciphering a protolanguage for bacteria-host communication. Nat. Chem. Biol. 4:452–454. 10.1038/nchembio0808-452 [DOI] [PubMed] [Google Scholar]
- 21. Spencer H, Karavolos MH, Bulmer DM, Aldridge P, Chhabra SR, Winzer K, Williams P, Khan CM. 2010. Genome-wide transposon mutagenesis identifies a role for host neuroendocrine stress hormones in regulating the expression of virulence genes in Salmonella. J. Bacteriol. 192:714–724. 10.1128/JB.01329-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lyte M. 2013. Microbial endocrinology in the microbiome-gut-brain axis: how bacterial production and utilization of neurochemicals influence behavior. PLoS Pathog. 9:e1003726. 10.1371/journal.ppat.1003726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freestone PP, Lyte M. 2008. Microbial endocrinology: experimental design issues in the study of interkingdom signalling in infectious disease. Adv. Appl. Microbiol. 64:75–105. 10.1016/S0065-2164(08)00402-4 [DOI] [PubMed] [Google Scholar]
- 24. Zaborin A, Romanowski K, Gerdes S, Holbrook C, Lepine F, Long J, Poroyko V, Diggle SP, Wilke A, Righetti K, Morozova I, Babrowski T, Liu DC, Zaborina O, Alverdy JC. 2009. Red death in Caenorhabditis elegans caused by Pseudomonas aeruginosa PAO1. Proc. Natl. Acad. Sci. U. S. A. 106:6327–6332. 10.1073/pnas.0813199106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Romanowski K, Zaborin A, Valuckaite V, Rolfes RJ, Babrowski T, Bethel C, Olivas A, Zaborina O, Alverdy JC. 2012. Candida albicans isolates from the gut of critically ill patients respond to phosphate limitation by expressing filaments and a lethal phenotype. PLoS One 7:e30119. 10.1371/journal.pone.0030119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eloy O, Marque S, Botterel F, Stephan F, Costa JM, Lasserre V, Bretagne S. 2006. Uniform distribution of three Candida albicans microsatellite markers in two French ICU populations supports a lack of nosocomial cross-contamination. BMC Infect. Dis. 6:162. 10.1186/1471-2334-6-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmid J, Tortorano AM, Jones G, Lazzarini C, Zhang N, Bendall MJ, Cogliati M, Wattimena S, Klingspor L, ECMM Survey. French, Mycoses Study Group 2011. Increased mortality in young candidemia patients associated with presence of a Candida albicans general-purpose genotype. J. Clin. Microbiol. 49:3250–3256. 10.1128/JCM.00941-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schelenz S. 2008. Management of candidiasis in the intensive care unit. J. Antimicrob. Chemother. 61(Suppl 1):i31–i34. 10.1093/jac/dkm430 [DOI] [PubMed] [Google Scholar]
- 29. Garsin DA, Lorenz MC. 2013. Candida albicans and Enterococcus faecalis in the gut: synergy in commensalism? Gut Microbes 4:409–415. 10.4161/gmic.26040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cruz MR, Graham CE, Gagliano BC, Lorenz MC, Garsin DA. 2013. Enterococcus faecalis inhibits hyphal morphogenesis and virulence of Candida albicans. Infect. Immun. 81:189–200. 10.1128/IAI.00914-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stefano GB, Zhu W, Cadet P, Bilfinger TV, Mantione K. 2004. Morphine enhances nitric oxide release in the mammalian gastrointestinal tract via the micro(3) opiate receptor subtype: a hormonal role for endogenous morphine. J. Physiol. Pharmacol. 55(Pt 2):279–288 [PubMed] [Google Scholar]
- 32. Manara L, Bianchetti A. 1985. The central and peripheral influences of opioids on gastrointestinal propulsion. Annu. Rev. Pharmacol. Toxicol. 25:249–273. 10.1146/annurev.pa.25.040185.001341 [DOI] [PubMed] [Google Scholar]
- 33. Holzer P. 2009. Opioid receptors in the gastrointestinal tract. Regul. Pept. 155:11–17. 10.1016/j.regpep.2009.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sobczak M, Sałaga M, Storr MA, Fichna J. 2014. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: current concepts and future perspectives. J. Gastroenterol. 49:24–45. 10.1007/s00535-013-0753-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spampinato S, Ferri GL, Candeletti S, Romualdi P, Cavicchini E, Soimero L, Labò G, Ferri S. 1988. Regional distribution of immunoreactive dynorphin A in the human gastrointestinal tract. Neuropeptides 11:101–105. 10.1016/0143-4179(88)90077-7 [DOI] [PubMed] [Google Scholar]
- 36. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 38. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosenzweig N, Tiedje JM, Quensen JF, Meng QX, Hao JJ. 2012. Microbial communities associated with potato common scab-suppressive soil determined by pyrosequencing analyses. Plant Dis. 96:718–725. 10.1094/PDIS-07-11-0571 [DOI] [PubMed] [Google Scholar]
- 41. Core R. Team; 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 42. Kindt R, Coe R. 2005. Tree diversity analysis. A manual and software for common statistical methods for ecological and biodiversity studies. World Agroforestry Centre (ICRAF), Nairobi, Nairobi, Kenya [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The taxonomic composition of the gut microbiome at the genus level determined by 16S rRNA analysis of stool samples collected from healthy volunteers. The top 7 genera, consisting of Coprococcus (no. 36, p_Firmicutes, f_Lachnospiraceae), Bacteroides (no. 6, p_Bacteroidetes, f_Bacteroidaceae), Roseburia (no. 40, p_Firmicutes, f_Lachnospiraceae), Prevotella (no. 8, p_Bacteroidetes, f_Prevotellaceae), a nonidentified genus of Lachnospiraceae (no. 32, p_Firmicutes, f_Lachnospiraceae), a nonidentified genus of Coprobacillaceae (no. 62, p_Firmicutes, f_Coprobacillaceae), and Blautia (no. 35, p_Firmicutes, f_Lachnospiraceae), are shown. Download
The taxonomic composition of the gut microbiome in stool samples collected from patients ICU2 and ICU3. Pseudomonadaceae (no. 77) reached levels of 10% (ICU2-4) and 30% (ICU3-4). Download
The taxonomic composition of gut microbiome in stool samples with decreased bacterial diversity. Escherichia (no. 74) and Enterococcus (no. 20) were major pathogens in this group. Download
Taxonomic and culturing composition of the ICU4 communities in correlation to antibiotic regimen, antibiotic resistance of stool isolates, and microbial data of cultured species from nonintestinal samples. Download
Kaplan-Meier survival curves of C. elegans fed on 2-member communities whose response to an opioid (50 µM U-50,488) was negligible (n = 10/plate, 3 plates/group; P < 0.01). Data represent cumulative results of 2 experiments. Download
Kaplan-Meier survival curves of C. elegans fed on E. coli OP 50 with and without exposure to an opioid (50 µM U-50,488) (n = 10/plate, 3 plates/group). Download
Antibiotic susceptibility of microbes isolated from ICU patients and healthy individuals.