

ABSTRACT
Kaposi’s sarcoma (KS), caused by KS-associated herpesvirus (KSHV), is the most common cancer among HIV-infected patients in Malawi and in the United States today. In Malawi, KSHV is endemic. We conducted a cross-sectional study of patients with HIV infection and KS with no history of chemo- or antiretroviral therapy (ART). Seventy patients were enrolled. Eighty-one percent had T1 (advanced) KS. Median CD4 and HIV RNA levels were 181 cells/mm3 and 138,641 copies/ml, respectively. We had complete information and suitable plasma and biopsy samples for 66 patients. For 59/66 (89%) patients, a detectable KSHV load was found in plasma (median, 2,291 copies/ml; interquartile range [IQR], 741 to 5,623). We utilized a novel KSHV real-time quantitative PCR (qPCR) array with multiple primers per open reading frame to examine KSHV transcription. Seventeen samples exhibited only minimal levels of KSHV mRNAs, presumably due to the limited number of infected cells. For all other biopsy samples, the viral latency locus (LANA, vCyc, vFLIP, kaposin, and microRNAs [miRNAs]) was transcribed abundantly, as was K15 mRNA. We could identify two subtypes of treatment-naive KS: lesions that transcribed viral RNAs across the length of the viral genome and lesions that displayed only limited transcription restricted to the latency locus. This finding demonstrates for the first time the existence of multiple subtypes of KS lesions in HIV- and KS-treatment naive patients.
IMPORTANCE
KS is the leading cancer in people infected with HIV worldwide and is causally linked to KSHV infection. Using viral transcription profiling, we have demonstrated the existence of multiple subtypes of KS lesions for the first time in HIV- and KS-treatment-naive patients. A substantial number of lesions transcribe mRNAs which encode the viral kinases and hence could be targeted by the antiviral drugs ganciclovir or AZT in addition to chemotherapy.
INTRODUCTION
Kaposi’s sarcoma (KS) is the most common malignancy associated with HIV infection (1). KS is a leading cancer among both men and women in countries where KS-associated herpesvirus (KSHV) is endemic and HIV has become epidemic (1, 2). In the United States and Europe, KS remains the most common cancer in HIV patients, even after active antiretroviral therapy (ART) has become widely available (3–6). The decline in KS, which followed the initial introduction of ART for HIV in the United States and Europe, has plateaued, and it is anticipated that KS will remain the leading cancer for persons living with or at high risk for acquiring HIV infection.
The clinical course of KS can range from an indolent state to a severe, progressive disease leading to significant morbidity and mortality. Advances have been made in the treatment of KS (reviewed in references 7 and 8). However, optimal therapy and long-term management, particularly in resource-limited settings, are not well defined. Standard cytotoxic chemotherapy (liposomal polyethylene glycol (PEG)-doxorubicin, or “Doxil”) is curative in only a subset of KS patients and is limited by the cumulative lifetime dose of doxorubicin and its derivatives. Access to liposomal PEG-doxorubicin and the supportive care needed for chemotherapy remain problematic in resource-limited settings, which includes all countries where KS and KSHV are endemic. ART is essential for HIV-infected KS patients, but despite ART, up to a third of KS patients have disease recurrence or do not respond to ART alone (9, 10). Ten to twenty percent of KS patients initiating ART may develop worsening KS disease, a condition called KS inflammation reconstitution syndrome (KS-IRIS) (11–14). KS also develops in HIV patients despite ART, i.e., in patients with no detectable HIV load and near-normal CD4 levels.
KS is a tumor of endothelial cell lineage, which is characterized histologically by slit-like vascular spaces, extravasated red blood cells, elongated “spindle”-like endothelial cells, and an infiltrate of lymphocytes or other inflammatory cells. The discovery of KSHV was a major advance in our understanding of this disease (15), since essentially all cases of KS carry KSHV and the continued presence of KSHV is required for KS tumorigenesis. KSHV is a human gammaherpesvirus that encodes more than 84 proteins that mediate viral replication and virus-host interactions (reviewed in reference 16). Theoretically, all KSHV proteins can be considered potential therapeutic targets. The viral proteins are expressed only in the tumor cells and probably also in preneoplastic cells, which form the latent reservoir that can progress to tumor cells. Two viral proteins that exhibit kinase activity are the viral thymidine kinase Orf21 and the viral protein kinase Orf36. These proteins have been shown to convert antiherpesvirus drugs, specifically ganciclovir, into their functional, toxic forms (17). Ganciclovir inhibits KSHV viral replication (18–21) and at high concentrations can inhibit tumor growth (17, 22–24).
The clinical experience of antiherpesvirus drugs in KS has yielded mixed results. We believe that this was in part because patients were enrolled without knowing if their lesions expressed the viral kinases (25–28). Knowing whether and which lesions express the viral kinases represents a gap in our current understanding. Increased understanding of KSHV gene expression in different types of KS may lead to patient intervention and stratification as a form of “personalized” KS therapy.
We found that treatment-naive AIDS-KS lesions differed significantly in their degree of KSHV transcription, allowing for a stratification of AIDS-KS cases according to the degree of so-called “lytic” gene expression. Such molecular biomarker-based division may be useful in optimizing treatment for KS patients. Previous studies evaluating this principle were small. Our group has prior experience in KSHV profiling of primary KS tissue. Those prior studies were based on archival samples from HIV-infected AIDS patients in the United States, which were heavily pretreated with chemotherapy, ART, or both (29, 30). To expand our understanding of KSHV gene expression, we conducted a cross-sectional study of 70 ART- and chemotherapy treatment-naive KS patients attending the Lighthouse (LH) clinic in Lilongwe, Malawi. We describe the KSHV viral load and KSHV mRNA transcription pattern in KS lesions from these patients so as to develop novel stratification methods that may provide prognostic and/or predictive information.
RESULTS
Study population and clinical characterization.
From November 2008 through August 2010, we screened 217 HIV-infected KS patients. Seventy (32%) of the screened patients were eligible and enrolled in the profiling project. Fifty percent of patients were enrolled in the first 100 days, attesting to the high prevalence of KS in Malawi (see Fig. S1 in the supplemental material). Primary reasons for exclusion included previous use of ART (26%), chemotherapy (9.5%), or both (48%) and disease restricted to the oral cavity (5.5%). In total, 105 patients were excluded. Among enrolled clients, 71% were male. The median (interquartile range [IQR]) age, body mass index (BMI), CD4 level, and HIV RNA level were 32 years (29 to 38), 22.4 kg/m2 (20.5 to 24.3), 181 cells/mm3 (85 to 261), and 138,641 copies/ml (54,900 to 285,000), respectively. Eighty-one percent presented with T1 (poor-risk) KS. Fifteen (22%) patients had 1 to 10 KS lesions, 42 (60%) patients had 10 to 50 lesions, and 12 (17%) patients had more than 50 lesions. Thirty-two (46%) patients had edema, 6 (9%) had concurrent oral KS, and 4 (6%) showed evidence of visceral KS (because of the absence of computed tomography scans, we cannot exclude that more of the patients had visceral KS) (see Table S1).
KSHV load did not correlate with CD4 count or HIV load.
We observed no significant correlation between the log HIV load and CD4 counts per microliter (Fig. 1A). We observed no significant correlation between log KSHV load and disease stage (limited KS [T0] versus T1) or any other clinical parameters by multivariate linear regression. We chose to measure cell-free KSHV load rather than KSHV DNA in peripheral blood mononuclear cells (PBMC), since this is directly related to viral replication. The median plasma KSHV DNA load was 3.36 log copies (IQR, 2.87 to 3.75). In 7/66 (11%) samples, the KSHV load was below our limit of detection (200 copies/ml) even though we could detect KSHV mRNA in biopsy specimens from 6 of these 7 patients. Since we were able to amplify the internal control in these serum samples, they represent cases of clinically apparent KS in the absence of detectable systemic KSHV viral load.
FIG 1 .
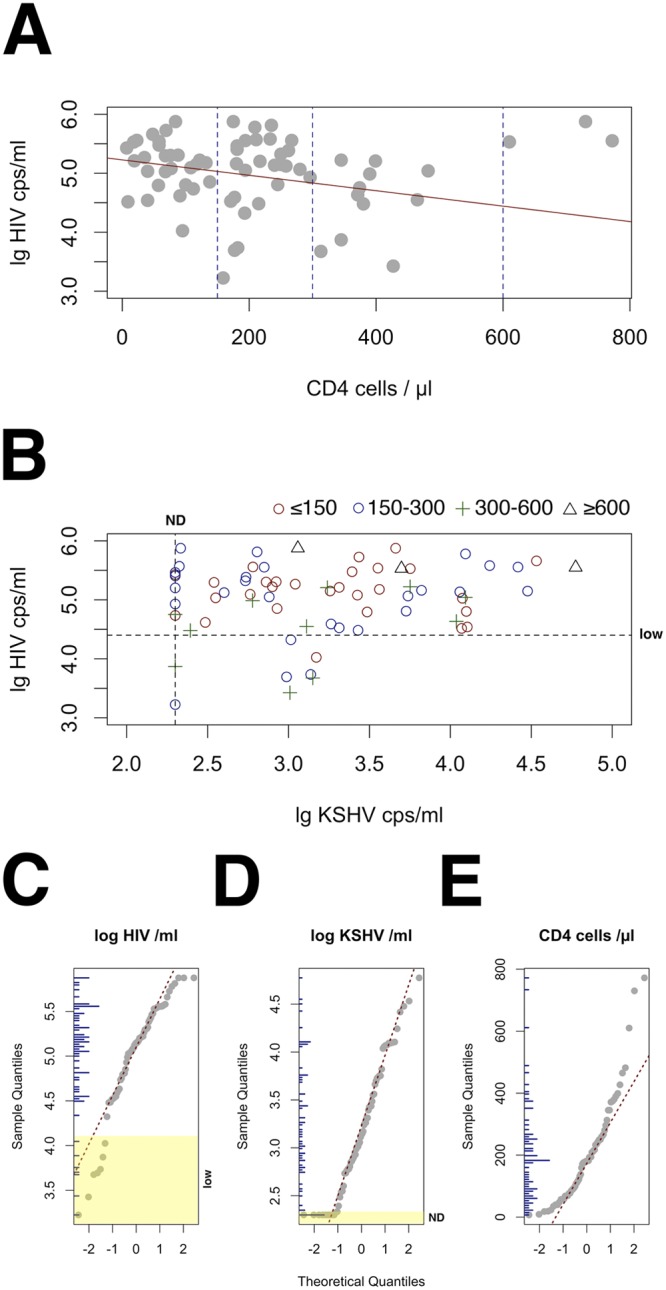
Lack of correlation between KSHV load, CD4 count, and HIV load in ART-naive patients presenting with HIV-associated KS in an area of KSHV endemicity. (A) Scatter plot of log10 HIV load (in copies per ml) on the vertical axis versus CD4 cell count per microliter on the horizontal axis. The solid red line indicates a linear regression taking into account only data points with a CD4 level of <550. The blue striped lines indicate the cutoff levels of CD4 counts used in panel B. (B) Scatter plot of log10 KSHV load (in copies per ml) on the vertical axis versus log10 HIV load (in copies per ml) on the horizontal axis. The symbols are coded by CD4 group as indicated above the panel. The black striped lines indicate the limit of detection for KSHV (200 copies /ml) and a cutoff for low HIV load (4.4 log10 copies/ml). The HIV load assay had a limit of detection of 50 copies/ml). (C) QQplot of distribution of log HIV copies/ml. Theoretical quantiles are shown on the horizontal axis and sample quantiles on the vertical axis. Vertical blue bars on the inner vertical axis represent the distribution histogram, and yellow highlights the region designated low HIV load (low). (D) QQplot of distribution of log KSHV copies/ml. Yellow highlights the region designated nondetectable KSHV in plasma (ND). (E) QQplot of CD4 count/µl distribution. Note the curvature of the points, which denotes systematic difference from a normal distribution (P ≤ 0.05 by Shapiro-Wilk test).
After excluding the 7 samples with a KSHV load below the limit of detection, the log10 KSHV load and log10 HIV load were normally distributed (Fig. 1C and D). Since the CD4 count was not normally distributed, (Fig. 1E), we used square-root-transformed CD4 counts for all subsequent analyses. We observed no correlation between the log10 KSHV load and log10 HIV load or between the log10 KSHV load and square-root-transformed CD4 count. Three patients (triangles in Fig. 1B) had a CD4 count of >600 cells per microliter, high HIV load, and substantial KSHV loads in plasma at the same time. Overall, our data demonstrate that the KSHV load, HIV load, and CD4 counts are independent of each other in therapy-naive, HIV+ KS patients in regions to which KSHV is endemic.
Two subtypes of KS.
One aim of this research was to determine whether molecular profiling of KSHV transcription in a suspected lesion was feasible and could provide an alternative to traditional pathology in resource-limited settings. We found RNAlater to be a suitable fixation and transport medium at tropical room temperature. Figure 2A describes the data analysis plan. First, we excluded any cases with incomplete clinical data. Five out of 66 (8%) biopsy specimens had “housekeeping” mRNAs with abnormally low levels (threshold cycle CT > 32), suggestive of RNA degradation. These samples were excluded from further analysis. We could not detect the mRNA for the KSHV latent genes in 10 biopsy specimens for which we could detect “housekeeping” mRNAs. This suggests that very few cells in the lesions were infected or that the level of latent gene expression per cell was minimal. Nine of these ten patients had a detectable KSHV load in plasma and multiple KS lesions. We cannot exclude that the wrong lesions or a highly fibrous or necrotic lesion was biopsied. These 10 samples were therefore excluded from further analysis. To arrive at the final high quality data set, we removed additional samples, which had KSHV latent mRNA levels that were detectable but much lower than the mean of the set. This provided us with 48 high-quality samples, which were split into a discovery set and a validation set. These samples were run on different days and with different lots of reagents, i.e., as true experimental replicates. We used a new, second-generation real-time quantitative PCR (qPCR) array consisting of 188 individual qPCR primer pairs (see Fig. S2 in the supplemental material).
FIG 2 .

(A) The data and sample flow into two cohorts: “discovery” and “validation.” (B) The distribution of the raw data (CT). Real-time qPCR outputs CT values, which represent the number of cycles needed to yield a positive signal. The cycle number CT is shown on the horizontal axis for each of 12 different samples from the “discovery” cohort. Shown in red is the distribution of the result of qPCR with KSHV mRNA-specific primers, and in blue the distribution of the result of qPCR with human/housekeeping mRNA-specific primers is shown. The yellow background indicates samples with very limited KSHV transcription; the gray background highlights samples with significant KSHV transcription.
Figure 2B shows the density curves for 12 samples from the discovery set, i.e., the fraction of primers with indicated cycle number CT. A higher CT corresponds to lower mRNA abundance. The housekeeping mRNAs (blue) were more abundant and similar across all samples than the viral mRNAs (red). Two “bulges” were observed for the “housekeeping” mRNAs, representing highly abundant (CT < 30; actin, gapdh, and β2m) and less abundant (CT > 30; hprt) “housekeeping” mRNAs. In contrast, many viral mRNAs were undetectable, leading to the sharp peak at a CT of 45. A peak at CT 45 indicates the end of the PCR. Such a mRNA was not detectable, and no PCR product was formed. We could distinguish two subtypes of KS: samples with a significant peak at a CT = 45, i.e., very limited viral transcription (yellow background), and samples with a lower peak at a CT of 45 and a significant, second “bulge” at a CT of <40, i.e., many viral mRNAs were detectable (gray background).
Figure 3 shows a heat map of viral transcription in the n = 35 sample validation set (a heat map of the discovery set is shown in Fig. S3 in the supplemental material; also provided, as Fig. S5, is a high-resolution image of Fig. 3). This unsupervised approach uncovered two subtypes: a and b. Subtype a contained biopsy specimens with a highly restricted, latent pattern of KSHV transcription; subtype b contained biopsy specimens with a more extended pattern of KSHV transcription. Note that mRNAs with marginal mean expression or those which did not change across samples were trimmed prior to clustering. The KSHV primer pairs (and underlying mRNA levels) fell into 3 different clusters. Note that both the overall levels and the pattern of transcription influence cluster membership. The ordering within any cluster was not significant. Whereas the samples could be divided into two subtypes, a and b, the various KSHV mRNAs, as measured by real-time qPCR primer pairs, could be divided into multiple classes or clusters, labeled i, ii, and iii. Members of cluster i were highly expressed in many samples. Members of cluster ii were expressed at high levels in only a few samples. Members of cluster iii were expressed at low levels in only a few samples. KSHV mRNA classes are further analyzed using more accurate methods detailed below. In sum, on the basis of unsupervised hierarchical clustering, KS lesions could be divided into two subtypes: those with limited viral gene transcription and those with evidence for widespread KSHV gene transcription.
FIG 3 .
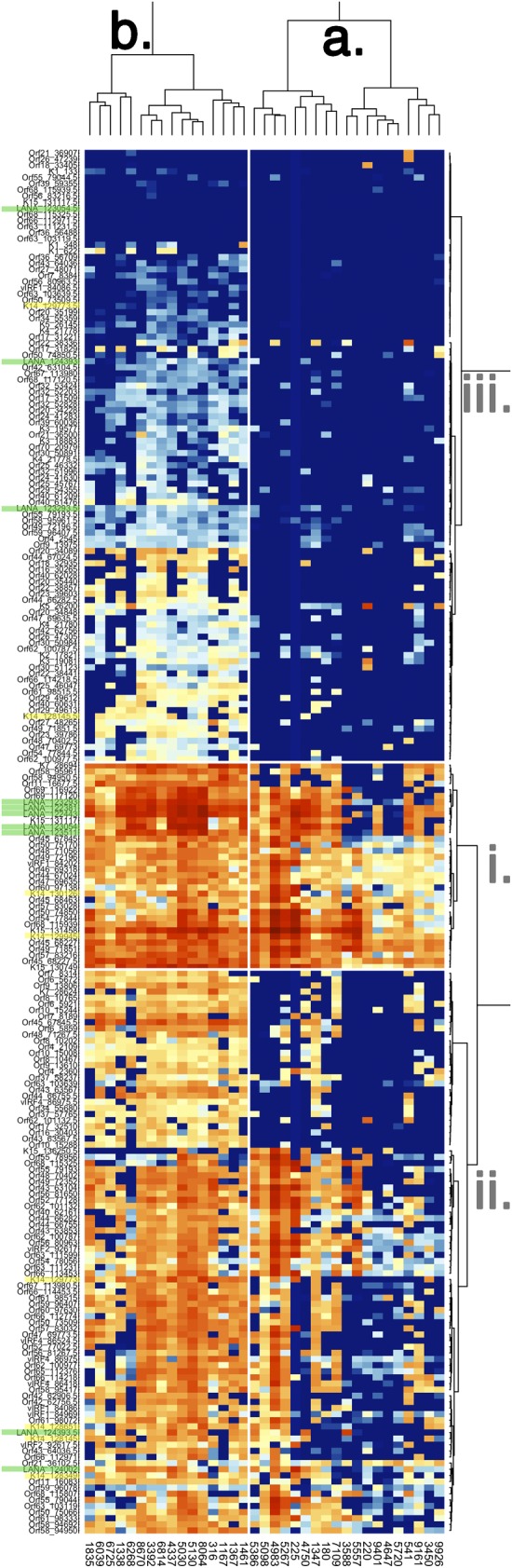
Heat map representation of two-way unsupervised clustering of the “validation” set of KS biopsy specimens. Before clustering, those mRNAs which did not change or which were not detectable in any of the samples were removed. Red indicates the highest, yellow indicates intermediate, and blue indicates no or low signal for a given primer pair on the vertical axis. Each PCR primer pair is named on the left and coded by the Orf name and forward primer position. The green overlay indicates mRNAs that originate in the KSHV latency region, and the yellow overlay highlights primer pairs that detect expression of the KSHV K14/vGPCR transcript. The dendrogram on top showing clustering of KS biopsy specimens indicates two clusters of samples, a and b. Sample, i.e., biopsy specimen identities are listed on the bottom. The larger number of samples allowed for more detailed clustering of KSHV transcription. Three clusters of KSHV transcripts could be identified, and those are labeled on the right as i, ii, and iii. A high-resolution figure with exact primer locations can be found in the supplemental material.
KSHV mRNA patterns in KS.
We are confident about the existence of different subtypes of KS, because the number of variables, i.e., primer pairs (n = 188), exceeded the number of samples (n = 35). Independent corroboration using principal-component analysis (PCA) and analysis of normalization methods is presented in Fig. S4 in the supplemental material. Identifying clusters of differentially regulated viral mRNAs was much more challenging, because here the design was underpowered. Ideally, one would like to have as many samples as genes. This would require 188 biopsy specimens. We attempted to identify mRNAs that differ among the KS subtypes and thus may be developed into prognostic and/or predictive biomarker signatures by making the assumption that latency locus mRNAs do not change in levels and could therefore be used to normalize all data (ddCT) (30). This also normalizes for tumor cell content per sample, since all KS tumor cells and only the KS tumor cells transcribe viral latent mRNAs, as previously shown by in situ methods (31, 32). Figure 4A shows the relative expression (ddCT) at each position on the genome. Red indicates members of cluster b., i.e., biopsy specimens with evidence of extended viral transcription, and blue indicates members of cluster a, i.e., biopsy specimens with limited transcription.
FIG 4 .

Analysis of individual KSHV transcripts across two KS subtypes in the validation set (n = 35). (A) Dot plot of the ddCT values on the vertical axis versus KSHV genome position on the horizontal axis. The ddCT values were obtained by first normalizing to the geometric mean of a set of “housekeeping mRNAs” and then the level of vFLIP mRNA (indicated at ddCT = 0). Red indicates samples from the “expanded” subtype, and blue indicates samples from the “restricted” KS subtype. All samples with a ddCT value of <−20 were set to ddCT = −20 and considered background. (B) Box plot of the ddCT values on the vertical axis versus KSHV Orf on the horizontal axis. All primers within the same Orf were averaged to give the “white” median value. The extent of the box indicates the 25th to 75th percentiles of the data. Outliers are not shown. Red indicates samples from the “expanded” subtype, and blue indicates samples from the “restricted” KS subtype. The vFLIP measurements used for normalization are shown on the far right. (C) P value distribution of Wilcoxon nonparametric comparison of relative mRNA levels by Orf between “red” and “blue” KS subtypes. (D) q value distribution, which represents adjustment for multiple comparison. A significance level of log(q) < 1.5, i.e., q < 0.03, is shown. (E) Distribution of the expected number of significant tests at a given q value cutoff. (F) Distribution of the expected number of false-positive comparisons based on the number of significant tests. Table S2 lists these mRNAs.
Due to the fact that poly(A) mRNA was isolated from the tumor samples in order to increase sensitivity and to decrease background due to residual DNA, we noticed a poly(A) site position effect: the signal for the vFLIP primers, which are located proximal to the poly(A) site for the tricistronic LANA/vCYC/vFLIP latency mRNA (as well as the bicistronic vCYC/ vFLIP mRNA), yielded a higher signal than the more distal vCYC and LANA primers. Likewise, the most poly(A)-site-proximal vGPCR primers yielded a higher signal than the more distally located K14 primers, even though both measure the same mRNA. This did not affect the comparison between samples, however, since those comparisons were done on a primer-by-primer basis.
Next, we averaged all primer pairs located within the same Orf together (Fig. 4B). Again, red indicates biopsy specimens with evidence for extended KSHV transcription, and blue indicates biopsy specimens with limited KSHV transcription. For some genes, e.g., K3 and K4, no mRNA was detectable in the latent KS subtypes. For most KSHV genes, e.g., Orf10 and Orf6, mRNA levels were significantly higher in the “extended” transcription cluster (red) than in the tightly latent cluster (blue). Orf58 represents an example of an mRNA with consistently high levels in the “extended” transcription subtype, as indicated by the short red bar, and extremely variable mRNA levels within the “latent” subtype, as indicated by the very long blue bar. K15 represents an example of an mRNA with consistently variable expression in both KS subtypes. In the case of K1 and K15, some of the variability may be due to strain differences, since these two genes are highly polymorphic.
The large number of samples and primers allowed us to test the hypothesis that transcription differed for specific orfs among the subtypes (Fig. 4C). To adjust for multiple comparisons, we controlled the false discovery rate to a significance level of 0.05. Figure 4E shows that approximately 30 KSHV mRNAs differed significantly between the two KS subtypes (also see Table S2). Figure 4F shows that among the top 30 differently regulated KSHV mRNAs, we would expect no more than 2 false-positive results. Table S2 lists these mRNAs.
In sum, using multiple analytical approaches, more than 180 individual markers, and two independent biological data sets, we established the existence of at least two KS subtypes in chemotherapy- and ART treatment-naive KS lesions. Potentially, up to 30 KSHV genes could be developed into either individual biomarkers or a composite biomarker signature for the purpose of prognosis and treatment stratification of KS prior to therapy.
DISCUSSION
Advances in molecular profiling and in histopathology have taught us the value of tumor subtyping and stratification in understanding pathology and designing better treatment regimens, as has been seen, e.g., in breast cancer. The same rationale applies to KS. Clinicians have long distinguished plaque, patch, and nodular KS lesions. Pathologists have long distinguished plaque, patch, and nodular histological stages (33, 34), though the histology does not always correspond to the clinical notation.
For HIV+ KS patients, ART represents first-line therapy since ART alone can lead to the resolution of KS in some patients. It is unclear, however, which KS patients respond to ART alone, which KS patients benefit from immediate, ART-concurrent chemotherapy, and which KS patients benefit from ART-concurrent, anti-KSHV therapy (ganciclovir, valganciclovir, and cidofovir) (10, 35–37). Some patients, particularly in regions to which KS is endemic, respond to ART initiation with exaggerated disease, termed KS immune reconstitution syndrome (KS-IRIS) (11–14).
The current KS staging is based on immune status (CD4 count), KS disease stage, and systemic involvement (T0, T1) (38). In the post-ART era, CD4 count may no longer provide prognostic information (39). It is unclear whether the same KS staging criteria that were developed in the United States and Europe during the initial phase of the AIDS epidemic, where KSHV was acquired late in life and predominantly by sexual contact (40, 41), also apply to KS that develops in HIV+ patients in areas of KSHV endemicity, where KSHV is often acquired before the onset of sexual maturity.
We observed only marginal correlations between extents of disease, systemic KSHV load, HIV load, and CD4 counts. In the post-ART era, approximately 30% of KS in the United States manifested itself in long-term ART-controlled individuals (9, 42, 43) with no detectable HIV load and >250 CD4 cells/µl. An implication of this study is that in areas to which KS is endemic, KS disease is observed in the setting of low as well as moderate CD4 counts (>250 CD4 cells/µl). Cases of extensive KS, despite high CD4 counts, were also seen in an ART-naive South African cohort (44) and in Uganda (38). This may be a unique feature of AIDS-KS in regions of KSHV endemicity.
The diagnosis of KS is a challenge in areas where pathology is limited. A molecular assay would greatly improve diagnosis. We observed 7 cases of KS with no detectable systemic KSHV load. For 6 of these patients, the KS lesions nevertheless tested positive for KSHV mRNA. Others also failed to detect KSHV in blood in as many as 25% of patients (45, 46). Thus, detecting KSHV mRNA in lesions may improve diagnosis. An advantage of molecular assays, particularly if they can be developed into robust point-of-care assays, is that they obviate the need for evaluation by pathologists, who are not affordable or not present in many low- and middle-income countries.
We previously demonstrated that lesions from advanced AIDS-KS cases from the pre-ART era and KS that developed in HIV-suppressed individuals on successful ART separated into two groups based on their KSHV transcription profiles (29, 30). Evidence of “extended” KS kinase transcription was virtually absent in classic KS, where ganciclovir monotherapy did not induce KS regression (25). These earlier studies were limited in size and comprised of groups of heavily pretreated patients. Those limitations were overcome in the current study, which involved only treatment-naive patients. Here, we identified multiple subtypes of KS transcriptional signatures. Half the samples exhibited a “latent” transcription pattern, which was limited to transcripts across the KSHV latency locus (LANA/vCyc/vFLIP/miRNAs). This kind of transcription predominates in primary effusion lymphoma (PEL) grown in culture (47), as well as most latently KSHV-infected endothelial cells (48). The other half of samples exhibited extended but incomplete viral transcription. The mRNAs of known signaling/pathogenesis molecules, such as K1, K15, and some of the viral interferon regulatory factors (vIRFs), were detectable, as were mRNAs encoding Orf21/thymidine kinase and Orf36/protein kinase. Similar “extended” transcription has been observed in PEL xenografts (49), in some PEL with substantial rates of spontaneous lytic reactivation (50), and other KSHV-infected endothelial cells (51–53). Other studies now also find extended viral transcripts under conditions of incomplete/abortive gammaherpesvirus replication (50, 54, 55).
Only one of the samples exhibited a complete (within the sensitivity of detection) lytic transcription profile, as would be seen in virus-producing cell lines (47, 56, 57). For this cross-sectional study, we do not have follow-up data on the patients and thus cannot directly assess the prognostic or predictive value of transcriptional profiling. Two larger clinical trials, A5263/AMC066 for advanced (T1) KS and A5264/AMC067 for limited (T0) KS, are currently enrolling patients across sub-Saharan Africa. These trials will also profile KS transcription and will allow us to correlate the molecular profile to disease progression and treatment responses. Even this limited cohort suggests that ART-naive KS patients may benefit from antiherpesvirus therapy in addition to ART and/or anticancer therapy and that these benefits would be most pronounced if patients were stratified based on transcriptional profiling of KS biopsy specimens.
MATERIALS AND METHODS
Study setting.
The Lighthouse (LH) ART clinic at Kamuzu Central Hospital serves as the Center of Excellence for the Central Region of Malawi. In addition to providing primary care and general ART services for 20,000 ART clients according to Malawi ART guidelines, the LH provides specialized services for referred cases, including combination chemotherapy for KS, evaluation of severe toxicities and ART treatment failure, and modification of ART for second line and nonstandard regimens. All services at LH are provided for free.
Design and participants.
A cross-sectional study was designed. All adult (age ≥ 18 years) KS patients presenting to the LH clinic between November 2008 and August 2010 were evaluated for potential enrollment. Patients with prior chemotherapy or antiretroviral therapy use were excluded, as were individuals with disease limited to the oral cavity (due to an inability to safely biopsy such lesions in the clinical setting).
Clinical procedures.
KS clinical staging was performed according to AIDS Clinical Trials Group (ACTG) criteria (T1, advanced stage, versus T0, limited disease); blood was obtained for CD4 cell count and KSHV quantification, and a punch biopsy was performed for evaluation of viral transcription. The 2- by 2-mm punch biopsy samples were immersed in RNAlater (Ambion Inc.) due to its suitability for collection at the bedside, local storage, intercontinental transport (samples could be shipped at room temperature), and automated RNA isolation.
Clinical laboratory procedures.
CD4 counts and HIV RNA determination were performed in real time at the UNC Project laboratory using a FACSCount system and Roche Amplicor assay 1.5. KSHV DNA was isolated using the MagnaPure system (Roche Inc.). The KSHV load was determined in plasma DNA by real-time qPCR as described, using the LANA78 primers (5′-GGAAGAGCCCATAATCTTGC and 5′-GCCTCATACGAACTCCAGGT) and SYBR green (Roche Inc.) as the method of detection.
Real-time qPCR profiling.
KSHV mRNA levels were determined in batch at the UNC—Chapel Hill Vironomics Core as per our published procedures (30, 58). These are available at http://www.med.unc.edu/vironomics/protocols.
Statistical analysis.
We performed basic descriptive statistics with bivariate comparisons according to ACTG tumor status using χ2 methodology or Student’s t test as appropriate. Further statistical analysis was performed using the software environment R, version 2.15.1, as indicated in Text S1 (Methods) in the supplemental material.
Ethics.
The study was IRB approved in Malawi and at the University of North Carolina (UNC) (IRB no. 08-0567) as “AMC-S001: CID 0802—a pilot study of Kaposi sarcoma-associated herpesvirus (KSHV) gene expression in patients with newly diagnosed Kaposi sarcoma (KS) in Malawi.” All subjects provided written informed consent.
SUPPLEMENTAL MATERIAL
Cumulative enrollment into AMC-S001/“A pilot study of Kaposi sarcoma-associated herpesvirus (KSHV) gene expression in patients with newly diagnosed Kaposi sarcoma (KS) in Malawi,” IRB no. 08-0567, over time since opening. Shown on the vertical axis is the number of subjects and on the horizontal axis the number of days since trial opening. Download
Design and validation of a 188-primer real-time quantitative PCR array to measure KSHV transcription genome-wide. (A) Positions of all primer pairs across the viral genome. (B) Heat map of the array validation data: red indicated the highest, yellow intermediate, and blue no signal for a given primer pair on the vertical axis. The samples (horizontal axis) are BC-1 cell DNA at indicated dilutions, cDNA made from KSHV-negative Namalwa cells with (RT+) or without (RT−) the inclusion of reverse transcriptase, and two water, i.e., nontemplate, control reactions. Download
Heat map representation of two-way unsupervised clustering of 15 samples of the “discovery” set of KS biopsy specimens. The red color indicates abundantly present mRNAs, while the blue color represents almost absent mRNAs. Orange indicates mRNAs at intermediate levels. To arrive at this figure, the CT′ data were normalized to a set of host mRNAs to yield dCT′. Any qPCR target mRNA, which did not significantly change across the different samples (standard deviation, <1 CT unit) or which was expressed at very low levels (mean CT′ > 45) was excluded from further analysis. The remaining, significantly expressed and variable mRNAs were clustered using Ward’s algorithm and the “Manhattan/city-block” distance metric. This unsupervised approach uncovered two general clusters, a and b. Download
For each set separately, the discovery and validation data sets. Panel A shows the correlation among the three housekeeping mRNAs (beta-actin, beta-2-microglobulin, and actin with a different primer set) used in the discovery set; panel C shows the correlation among the six host mRNAs that were used as the basis for normalization of the validation set in Fig. 4. (B and D) Principal-component analysis (PCA) of mRNA expression values. We displayed the first two principal components and then projected the highs for each heat map dendrogram onto the first two principal components. For the discovery set (B), the very lytic sample no. 8239 was clearly evident as an outlier and differed from the two main clusters. The first two dimensions of the PCA explained 27.96% + 11.95% (~40%) of the variation. For panel D, the first two dimensions of the PCA explained 43.46% + 20.56% (~65%) of the variation. Download
High-resolution, scalable image of Fig. 4. Download
Cohort characteristics.
Differentially regulated genes from Fig. 4 with exact p and adjusted P values; significant genes are shown in red.
Copy of the data collection tool. Download
Detailed description of methods. Download
ACKNOWLEDGMENTS
This study was supported by the AIDS Malignancy Clinical Trials Consortium (CA121947), NCI supplement to the UNC Lineberger Comprehensive Cancer Center (CA016086), the UNC Center for AIDS Research (AI050410), and NIH grants CA109232 and CA019014 to D.D. and CA096500, AI107810, and DE023946 to B.D. K.T. was supported by T32 GM07092-34 and by a grant to the University of North Carolina at Chapel Hill from Howard Hughes Medical Institute (HHMI) through the Med into Grad Initiative.
Footnotes
Citation Hosseinipour MC, Sweet KM, Xiong J, Namarika D, Mwafongo A, Nyirenda M, Chiwoko L, Kamwendo D, Hoffman I, Lee J, Phiri S, Vahrson W, Damania B, Dittmer DP. 2014. Viral profiling identifies multiple subtypes of Kaposi’s sarcoma. mBio 5(5):e01633-14. doi:10.1128/mBio.01633-14.
REFERENCES
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. 2011. Global cancer statistics. CA Cancer J. Clin. 61:69–90. 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 2. Semeere AS, Busakhala N, Martin JN. 2012. Impact of antiretroviral therapy on the incidence of Kaposi’s sarcoma in resource-rich and resource-limited settings. Curr. Opin. Oncol. 24:522–530. 10.1097/CCO.0b013e328355e14b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gopal S, Achenbach CJ, Yanik EL, Dittmer DP, Eron JJ, Engels EA. 2014. Moving forward in HIV-associated cancer. J. Clin. Oncol. 32:876–880. 10.1200/JCO.2013.53.1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yanik EL, Napravnik S, Cole SR, Achenbach CJ, Gopal S, Olshan A, Dittmer DP, Kitahata MM, Mugavero MJ, Saag M, Moore RD, Mayer K, Mathews WC, Hunt PW, Rodriguez B, Eron JJ. 2013. Incidence and timing of cancer in HIV-infected individuals following initiation of combination antiretroviral therapy. Clin. Infect. Dis. 57:756–764. 10.1093/cid/cit369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yanik EL, Tamburro K, Eron JJ, Damania B, Napravnik S, Dittmer DP. 2013. Recent cancer incidence trends in an observational clinical cohort of HIV-infected patients in the US, 2000-2011. Infect. Agents Cancer 8:18. 10.1186/1750-9378-8-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shiels MS, Pfeiffer RM, Hall HI, Li J, Goedert JJ, Morton LM, Hartge P, Engels EA. 2011. Proportions of Kaposi sarcoma, selected non-Hodgkin lymphomas, and cervical cancer in the United States occurring in persons with AIDS, 1980-2007. JAMA 305:1450–1459. 10.1001/jama.2011.396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dittmer DP, Richards KL, Damania B. 2012. Treatment of Kaposi sarcoma-associated herpesvirus-associated cancers. Front. Microbiol. 3:141. 10.3389/fmicb.2012.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krown SE. 2011. Treatment strategies for Kaposi sarcoma in sub-Saharan Africa: challenges and opportunities. Curr. Opin. Oncol. 23:463–468. 10.1097/CCO.0b013e328349428d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krown SE, Lee JY, Dittmer DP, AIDS Malignancy Consortium 2008. More on HIV-associated Kaposi’s sarcoma. N. Engl. J. Med. 358:535–536; author reply, 536. 10.1056/NEJMc072994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen HQ, Magaret AS, Kitahata MM, Van Rompaey SE, Wald A, Casper C. 2008. Persistent Kaposi sarcoma in the era of highly active antiretroviral therapy: characterizing the predictors of clinical response. AIDS 22:937–945. 10.1097/QAD.0b013e3282ff6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cox CM, El-Mallawany NK, Kabue M, Kovarik C, Schutze GE, Kazembe PN, Mehta PS. 2013. Clinical characteristics and outcomes of HIV-infected children diagnosed with Kaposi sarcoma in Malawi and Botswana. Pediatr. Blood Cancer 60:1274–1280. 10.1002/pbc.24516 [DOI] [PubMed] [Google Scholar]
- 12. Letang E, Lewis JJ, Bower M, Mosam A, Borok M, Campbell TB, Naniche D, Newsom-Davis T, Shaik F, Fiorillo S, Miro JM, Schellenberg D, Easterbrook PJ. 2013. Immune reconstitution inflammatory syndrome associated with Kaposi sarcoma: higher incidence and mortality in Africa than in the UK. AIDS 27:1603–1613. 10.1097/QAD.0b013e328360a5a1 [DOI] [PubMed] [Google Scholar]
- 13. Achenbach CJ, Harrington RD, Dhanireddy S, Crane HM, Casper C, Kitahata MM. 2012. Paradoxical immune reconstitution inflammatory syndrome in HIV-infected patients treated with combination antiretroviral therapy after AIDS-defining opportunistic infection. Clin. Infect. Dis. 54:424–433. 10.1093/cid/cir802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bower M, Nelson M, Young AM, Thirlwell C, Newsom-Davis T, Mandalia S, Dhillon T, Holmes P, Gazzard BG, Stebbing J. 2005. Immune reconstitution inflammatory syndrome associated with Kaposi’s sarcoma. J. Clin. Oncol. 23:5224–5228. 10.1200/JCO.2005.14.597 [DOI] [PubMed] [Google Scholar]
- 15. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 16. Dittmer DP, Damania B. 2013. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr. Opin. Virol. 3:238–244. 10.1016/j.coviro.2013.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davis DA, Singer KE, Reynolds IP, Haque M, Yarchoan R. 2007. Hypoxia enhances the phosphorylation and cytotoxicity of ganciclovir and zidovudine in Kaposi’s sarcoma-associated herpesvirus infected cells. Cancer Res. 67:7003–7010. 10.1158/0008-5472.CAN-07-0939 [DOI] [PubMed] [Google Scholar]
- 18. Kedes DH, Ganem D. 1997. Sensitivity of Kaposi’s sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J. Clin. Invest. 99:2082–2086. 10.1172/JCI119380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neyts J, De Clercq E. 1997. Antiviral drug susceptibility of human herpesvirus 8. Antimicrob. Agents Chemother. 41:2754–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dittmer D, Stoddart C, Renne R, Linquist-Stepps V, Moreno ME, Bare C, McCune JM, Ganem D. 1999. Experimental transmission of Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 190:1857–1868. 10.1084/jem.190.12.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cannon JS, Hamzeh F, Moore S, Nicholas J, Ambinder RF. 1999. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J. Virol. 73:4786–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fu DX, Tanhehco Y, Chen J, Foss CA, Fox JJ, Chong JM, Hobbs RF, Fukayama M, Sgouros G, Kowalski J, Pomper MG, Ambinder RF. 2008. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nat. Med. 14:1118–1122. 10.1038/nm.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kurokawa M, Ghosh SK, Ramos JC, Mian AM, Toomey NL, Cabral L, Whitby D, Barber GN, Dittmer DP, Harrington WJ., Jr. 2005. Azidothymidine inhibits NF-kappaB and induces Epstein-Barr virus gene expression in Burkitt lymphoma. Blood 106:235–240. 10.1182/blood-2004-09-3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. 2002. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. 62:1920–1926 [PubMed] [Google Scholar]
- 25. Krown SE, Dittmer DP, Cesarman E. 2011. Pilot study of oral valganciclovir therapy in patients with classic Kaposi sarcoma. J. Infect. Dis. 203:1082–1086. 10.1093/infdis/jiq177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Little RF, Merced-Galindez F, Staskus K, Whitby D, Aoki Y, Humphrey R, Pluda JM, Marshall V, Walters M, Welles L, Rodriguez-Chavez IR, Pittaluga S, Tosato G, Yarchoan R. 2003. A pilot study of cidofovir in patients with Kaposi sarcoma. J. Infect. Dis. 187:149–153. 10.1086/346159 [DOI] [PubMed] [Google Scholar]
- 27. Uldrick TS, Polizzotto MN, Aleman K, O’Mahony D, Wyvill KM, Wang V, Marshall V, Pittaluga S, Steinberg SM, Tosato G, Whitby D, Little RF, Yarchoan R. 2011. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood 117:6977–6986. 10.1182/blood-2010-11-317610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pantanowitz L, Früh K, Marconi S, Moses AV, Dezube BJ. 2008. Pathology of rituximab-induced Kaposi sarcoma flare. BMC Clin. Pathol. 8:7. 10.1186/1472-6890-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dittmer DP. 2003. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 [PubMed] [Google Scholar]
- 30. Dittmer DP. 2011. Restricted Kaposi’s sarcoma (KS) herpesvirus transcription in KS lesions from patients on successful antiretroviral therapy. mBio 2(6):e00138-11. 10.1128/mBio.00138-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551. 10.1073/pnas.96.8.4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 72:8309–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pantanowitz L, Otis CN, Dezube BJ. 2010. Immunohistochemistry in Kaposi’s sarcoma. Clin. Exp. Dermatol. 35:68–72. 10.1111/j.1365-2230.2009.03707.x [DOI] [PubMed] [Google Scholar]
- 34. Stürzl M, Brandstetter H, Roth WK. 1992. Kaposi’s sarcoma: a review of gene expression and ultrastructure of KS spindle cells in vivo. AIDS Res. Hum. Retroviruses 8:1753–1763. 10.1089/aid.1992.8.1753 [DOI] [PubMed] [Google Scholar]
- 35. Borok M, Fiorillo S, Gudza I, Putnam B, Ndemera B, White IE, Gwanzura L, Schooley RT, Campbell TB. 2010. Evaluation of plasma human herpesvirus 8 DNA as a marker of clinical outcomes during antiretroviral therapy for AIDS-related Kaposi sarcoma in Zimbabwe. Clin. Infect. Dis. 51:342–349. 10.1086/654800 [DOI] [PubMed] [Google Scholar]
- 36. Mosam A, Shaik F, Uldrick TS, Esterhuizen T, Friedland GH, Scadden DT, Aboobaker J, Coovadia HM. 2012. A randomized controlled trial of highly active antiretroviral therapy versus highly active antiretroviral therapy and chemotherapy in therapy-naive patients with HIV-associated Kaposi sarcoma in South Africa. J. Acquir. Immune Defic. Syndr. 60:150–157. 10.1097/QAI.0b013e318251aedd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bihl F, Mosam A, Henry LN, Chisholm JV, III, Dollard S, Gumbi P, Cassol E, Page T, Mueller N, Kiepiela P, Martin JN, Coovadia HM, Scadden DT, Brander C. 2007. Kaposi’s sarcoma-associated herpesvirus-specific immune reconstitution and antiviral effect of combined HAART/chemotherapy in HIV clade C-infected individuals with Kaposi’s sarcoma. AIDS 21:1245–1252. 10.1097/QAD.0b013e328182df03 [DOI] [PubMed] [Google Scholar]
- 38. Johnston C, Orem J, Okuku F, Kalinaki M, Saracino M, Katongole-Mbidde E, Sande M, Ronald A, McAdam K, Huang ML, Drolette L, Selke S, Wald A, Corey L, Casper C. 2009. Impact of HIV infection and Kaposi sarcoma on human herpesvirus-8 mucosal replication and dissemination in Uganda. PLoS One 4:e4222. 10.1371/journal.pone.0004222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nasti G, Martellotta F, Berretta M, Mena M, Fasan M, Di Perri G, Talamini R, Pagano G, Montroni M, Cinelli R, Vaccher E, D’Arminio Monforte A, Tirelli U, GICAT ICONA. 2003. Impact of highly active antiretroviral therapy on the presenting features and outcome of patients with acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer 98:2440–2446. 10.1002/cncr.11816 [DOI] [PubMed] [Google Scholar]
- 40. Martin JN, Ganem DE, Osmond DH, Page-Shafer KA, Macrae D, Kedes DH. 1998. Sexual transmission and the natural history of human herpesvirus 8 infection. N. Engl. J. Med. 338:948–954. 10.1056/NEJM199804023381403 [DOI] [PubMed] [Google Scholar]
- 41. Butler LM, Dorsey G, Hladik W, Rosenthal PJ, Brander C, Neilands TB, Mbisa G, Whitby D, Kiepiela P, Mosam A, Mzolo S, Dollard SC, Martin JN. 2009. Kaposi sarcoma-associated herpesvirus (KSHV) seroprevalence in population-based samples of African children: evidence for at least 2 patterns of KSHV transmission. J. Infect. Dis. 200:430–438. 10.1086/600103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maurer T, Ponte M, Leslie K. 2007. HIV-associated Kaposi’s sarcoma with a high CD4 count and a low viral load. N. Engl. J. Med. 357:1352–1353. 10.1056/NEJMc070508 [DOI] [PubMed] [Google Scholar]
- 43. Cianfrocca M, Lee S, Von Roenn J, Tulpule A, Dezube BJ, Aboulafia DM, Ambinder RF, Lee JY, Krown SE, Sparano JA. 2010. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 116:3969–3977. 10.1002/cncr.25362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cassol E, Page T, Mosam A, Friedland G, Jack C, Lalloo U, Kopetka J, Patterson B, Esterhuizen T, Coovadia HM. 2005. Therapeutic response of HIV-1 subtype C in African patients coinfected with either Mycobacterium tuberculosis or human herpesvirus-8. J. Infect. Dis. 191:324–332. 10.1086/427337 [DOI] [PubMed] [Google Scholar]
- 45. Martín-Carbonero L, Palacios R, Valencia E, Saballs P, Sirera G, Santos I, Baldobí F, Alegre M, Goyenechea A, Pedreira J, González del Castillo J, Martínez-Lacasa J, Ocampo A, Alsina M, Santos J, Podzamczer D, González-Lahoz J, Caelyx, Kaposi's Sarcoma Spanish Group 2008. Long-term prognosis of HIV-infected patients with Kaposi sarcoma treated with pegylated liposomal doxorubicin. Clin. Infect. Dis. 47:410–417. 10.1086/589865 [DOI] [PubMed] [Google Scholar]
- 46. Bower M, Dalla Pria A, Coyle C, Andrews E, Tittle V, Dhoot S, Nelson M. 2014. Prospective stage-stratified approach to AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 32:409–414. 10.1200/JCO.2013.51.6757 [DOI] [PubMed] [Google Scholar]
- 47. Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213–6223. 10.1128/JVI.76.12.6213-6223.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, Fakhari FD, Dittmer DP, Renne R. 2006. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J. Virol. 80:4833–4846. 10.1128/JVI.80.10.4833-4846.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Staudt MR, Kanan Y, Jeong JH, Papin JF, Hines-Boykin R, Dittmer DP. 2004. The tumor microenvironment controls primary effusion lymphoma growth in vivo. Cancer Res. 64:4790–4799. 10.1158/0008-5472.CAN-03-3835 [DOI] [PubMed] [Google Scholar]
- 50. Chandriani S, Ganem D. 2010. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 84:5565–5573. 10.1128/JVI.02723-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chang HH, Ganem D. 2013. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 13:429–440. 10.1016/j.chom.2013.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. 2006. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 66:3658–3666. 10.1158/0008-5472.CAN-05-3680 [DOI] [PubMed] [Google Scholar]
- 53. Mutlu AD, Cavallin LE, Vincent L, Chiozzini C, Eroles P, Duran EM, Asgari Z, Hooper AT, La Perle KM, Hilsher C, Gao SJ, Dittmer DP, Rafii S, Mesri EA. 2007. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 11:245–258. 10.1016/j.ccr.2007.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D. 2014. KSHV 2.0: a comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 10:e1003847. 10.1371/journal.ppat.1003847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Canny SP, Reese TA, Johnson LS, Zhang X, Kambal A, Duan E, Liu CY, Virgin HW. 2014. Pervasive transcription of a herpesvirus genome generates functionally important RNAs. mBio 5(2):e01033-13. 10.1128/mBio.01033-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jenner RG, Albà MM, Boshoff C, Kellam P. 2001. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891–902. 10.1128/JVI.75.2.891-902.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Paulose-Murphy M, Ha NK, Xiang C, Chen Y, Gillim L, Yarchoan R, Meltzer P, Bittner M, Trent J, Zeichner S. 2001. Transcription program of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus). J. Virol. 75:4843–4853. 10.1128/JVI.75.10.4843-4853.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Papin J, Vahrson W, Hines-Boykin R, Dittmer DP. 2005. Real-time quantitative PCR analysis of viral transcription. Methods Mol. Biol. 292:449–480 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cumulative enrollment into AMC-S001/“A pilot study of Kaposi sarcoma-associated herpesvirus (KSHV) gene expression in patients with newly diagnosed Kaposi sarcoma (KS) in Malawi,” IRB no. 08-0567, over time since opening. Shown on the vertical axis is the number of subjects and on the horizontal axis the number of days since trial opening. Download
Design and validation of a 188-primer real-time quantitative PCR array to measure KSHV transcription genome-wide. (A) Positions of all primer pairs across the viral genome. (B) Heat map of the array validation data: red indicated the highest, yellow intermediate, and blue no signal for a given primer pair on the vertical axis. The samples (horizontal axis) are BC-1 cell DNA at indicated dilutions, cDNA made from KSHV-negative Namalwa cells with (RT+) or without (RT−) the inclusion of reverse transcriptase, and two water, i.e., nontemplate, control reactions. Download
Heat map representation of two-way unsupervised clustering of 15 samples of the “discovery” set of KS biopsy specimens. The red color indicates abundantly present mRNAs, while the blue color represents almost absent mRNAs. Orange indicates mRNAs at intermediate levels. To arrive at this figure, the CT′ data were normalized to a set of host mRNAs to yield dCT′. Any qPCR target mRNA, which did not significantly change across the different samples (standard deviation, <1 CT unit) or which was expressed at very low levels (mean CT′ > 45) was excluded from further analysis. The remaining, significantly expressed and variable mRNAs were clustered using Ward’s algorithm and the “Manhattan/city-block” distance metric. This unsupervised approach uncovered two general clusters, a and b. Download
For each set separately, the discovery and validation data sets. Panel A shows the correlation among the three housekeeping mRNAs (beta-actin, beta-2-microglobulin, and actin with a different primer set) used in the discovery set; panel C shows the correlation among the six host mRNAs that were used as the basis for normalization of the validation set in Fig. 4. (B and D) Principal-component analysis (PCA) of mRNA expression values. We displayed the first two principal components and then projected the highs for each heat map dendrogram onto the first two principal components. For the discovery set (B), the very lytic sample no. 8239 was clearly evident as an outlier and differed from the two main clusters. The first two dimensions of the PCA explained 27.96% + 11.95% (~40%) of the variation. For panel D, the first two dimensions of the PCA explained 43.46% + 20.56% (~65%) of the variation. Download
High-resolution, scalable image of Fig. 4. Download
Cohort characteristics.
Differentially regulated genes from Fig. 4 with exact p and adjusted P values; significant genes are shown in red.
Copy of the data collection tool. Download
Detailed description of methods. Download