

ABSTRACT
Kaposi’s sarcoma (KS) is an unusual neoplasia wherein the tumor consists primarily of endothelial cells infected with human herpesvirus 8 (HHV-8; Kaposi’s sarcoma-associated herpesvirus) that are not fully transformed but are instead driven to excess proliferation by inflammatory and angiogenic factors. This oncogenic process has been postulated but unproven to depend on a paracrine effect of an abnormal excess of host cytokines and chemokines produced by HHV-8-infected B lymphocytes. Using newly developed measures for intracellular detection of lytic cycle proteins and expression of cytokines and chemokines, we show that HHV-8 targets a range of naive B cell, IgM memory B cell, and plasma cell-like populations for infection and induction of interleukin-6, tumor necrosis factor alpha, macrophage inhibitory protein 1α, macrophage inhibitory protein 1β, and interleukin-8 in vitro and in the blood of HHV-8/HIV-1-coinfected subjects with KS. These B cell lineage subsets that support HHV-8 infection are highly polyfunctional, producing combinations of 2 to 5 of these cytokines and chemokines, with greater numbers in the blood of subjects with KS than in those without KS. Our study provides a new paradigm of B cell polyfunctionality and supports a key role for B cell-derived cytokines and chemokines produced during HHV-8 infection in the development of KS.
IMPORTANCE
Kaposi’s sarcoma (KS) is the most common cancer in HIV-1-infected persons and is caused by one of only 7 human cancer viruses, i.e., human herpesvirus 8 (HHV-8). It is unclear how this virus causes neoplastic transformation. Development and outgrowth of endothelial cell lesions characteristic of KS are hypothesized to be dependent on virus replication and multiple immune mediators produced by the KS cells and inflammatory cells, yet the roles of these viral and cell factors have not been defined. The present study advances our understanding of KS in that it supports a central role for HHV-8 infection of B cells inducing multiple cytokines and chemokines that can drive development of the cancer. Notably, HIV-1-infected individuals who developed KS had greater numbers of such HHV-8-infected, polyfunctional B cells across a range of B cell phenotypic lineages than did HHV-8-infected persons without KS. This intriguing production of polyfunctional immune mediators by B cells serves as a new paradigm for B cell function and classification.
INTRODUCTION
Human herpesvirus 8 (HHV-8, also termed Kaposi’s sarcoma-associated herpesvirus) is the etiologic agent of Kaposi’s sarcoma (KS) (1). How this herpesvirus causes KS is not clear. KS tumor cells are primarily of endothelial cell origin. Although HHV-8 infection of endothelial cells is necessary for development of KS, it is insufficient to drive the formation of KS lesions, and these cells are not fully transformed (2). Extensive studies suggest that this oncogenic process involves HHV-8 latency oncoproteins and microRNAs that cause cell proliferation and prevent apoptosis (3). Accumulating evidence, however, has incriminated lytic HHV-8 infection in driving HHV-8-associated cancers (4), with persistent latent HHV-8 infection being associated with ongoing lytic virus replication (5–7). Several HHV-8 lytic proteins with homology to human proteins are thought to contribute to endothelial cell survival and proliferation by mimicking host proteins that regulate the cell cycle as well as having immunomodulatory effects that favor virus replication. An unsolved enigma of KS is that HHV-8 latency and lytic cycle encoded factors, while unique among human oncogenic viruses, are insufficient to cause the cancer.
An emerging hypothesis is that KS is a paracrine neoplasia in which HHV-8-infected endothelial cells depend on an abnormal excess of host cytokines and chemokines for their outgrowth (2). We propose that B lymphocytes contribute to this process. Early studies found HHV-8 DNA associated with circulating B cells in patients with KS (2, 8). HHV-8-infected B cells are present in a large percentage of KS lesions (9). HHV-8 replicates in B cells in vitro, requiring preactivation of the cells with CD40 ligand (CD40L) and interleukin 4 (IL-4) (10), which are surrogates for CD4+ T helper cells (11). There is no information, however, on HHV-8 infection in relation to the cytokine and chemokine response of B cells in development of KS.
We assessed HHV-8 infection and production of cytokines and chemokines of B lymphocytes. Our results show that naive and IgM memory B cells, and plasma cell-like populations, support infection with HHV-8 both in vitro and in the blood of subjects with KS. Importantly, virus-infected B cells are highly polyfunctional, producing multiple cytokines and chemokines that have been postulated to enhance endothelial cell outgrowth (2). Our data support that HHV-8 driven, B cell cytokines and chemokines are central to the development of KS.
RESULTS
HHV-8 lytic infection induces a cytokine-chemokine response in B cells.
We previously showed that HHV-8 replication in B cells in vitro requires preactivation of the cells with CD40L and IL-4, which maintains B cell viability and increases expression of the HHV-8 receptor dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) (10). To extend this model, we developed new quantitative assays for measuring HHV-8 lytic proteins in purified B cells by flow cytometry, viral DNA by quantitative real-time PCR, and infectious virus based on the 50% tissue culture infectious dose (TCID50) (12). We found that HHV-8 productively replicated in a mean of 8.5% of CD40L- and IL-4-activated, HHV-8-naive CD19+ B cells infected in vitro by 48 h, as shown by staining with a monoclonal antibody (MAb) specific for the HHV-8 lytic protein ORF59, processivity factor 8 (PF-8) (Fig. 1A, left panel), which is necessary for processing of HHV-8 DNA polymerase and viral DNA replication (13). Similar results were found when a MAb specific for lytic cycle glycoprotein K8.1A/B was used (data not shown); this protein is part of the virion envelope (14), binds to heparan sulfate on cell surfaces (15), and regulates vascular endothelial growth factor (VEGF) production (16). This evidence of HHV-8 replication was supported by an approximate 0.7-log10 increase in HHV-8 DNA copies/500,000 cells (Fig. 1A, middle panel) and a >3-log10 increase in the TCID50 (Fig. 1A, right panel) by 48 h. Cell viability at 48 h was similar in HHV-8-exposed and -unexposed cultures, i.e., with average percentages of viable B cells of 80% and 83%, respectively (P value not significant), as determined by trypan blue dye exclusion (data not shown).
FIG 1 .

HHV-8 lytic infection in vitro and induction of a cytokine-chemokine response in B cells. (A, left panel) CD40L- and IL-4 activated, HHV-8-naive CD19+ B cells were infected in vitro for 48 h and assessed for intracellular expression of HHV-8 ORF59 PF-8. (Middle panel) HHV-8 DNA copies were determined in cell pellets over 48 h. (Right panel) The TCID50 in HHV-8-exposed B cell culture supernatants was determined (values are means ± SE; n = 4). (B) Supernatants from B cells either unexposed or exposed to HHV-8 were collected at 3, 24, and 48 h and used in a CBA to determine cytokine-chemokine production (values are means ± SE; n = 18). *, P < 0.05, Student’s t test. (C) RNA was extracted from B cells left unexposed or exposed to HHV-8 and hybridized to Illumina HT-12 v4 microchips (values are means ± SE of duplicate slides). Bars represent the fold increases in RNA extracted at the indicated times postinfection.
We next determined levels of 16 cytokines, chemokines, and growth factors that have been related to KS, in B cell supernatants by cytokine bead array (CBA), i.e., gamma interferon (IFN-γ), IL-1β, -2, -4, -6, -7, -8, -10, and -12, tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein 1 (MCP-1), MIP-1α (or CCL3), MIP-1β (or CCL4), the regulated on activation, normal T cell expressed and secreted protein (RANTES, or CCL5), IFN-inducible protein 10, and VEGF (data not shown). As expected, B cell activation with CD40L and IL-4 and without exposure to HHV-8 in vitro resulted in production of these 16 cytokines, chemokines, and growth factors (data not shown). In multiple experiments, however, we found significant levels of IL-6, TNF-α, MIP-1α, MIP-1β, and IL-8 during 48 h of virus exposure above those in the non-HHV-8-exposed, activated B cell cultures (Fig. 1B). This was confirmed in a transcriptome microarray in which ≥2-fold increases in mRNA were detected for IL-6, TNF-α, MIP-1α, CCL3-like (CCL3L), and CCL4L in virus-exposed compared to unexposed B cells at 3, 4, 6, 9, 15, and 27 h post-HHV-8 exposure (Fig. 1C).
Collectively, these results extend our previous finding (10) that a subset of approximately 8.5% of activated B cells from healthy, HHV-8-seronegative adults replicates HHV-8 in vitro and demonstrate a selective cytokine and chemokine response (IL-6, TNF-α, MIP-1α, MIP-1β, and IL-8) that is associated with lytic cycle viral replication.
HHV-8-infected B cells are polyfunctional.
We defined the intracellular pattern of cytokines and chemokines produced in B cells also expressing viral ORF59 PF-8 and compared the pattern to that of B cells not supporting HHV-8 infection in the same cultures by using multiparameter flow cytometry (see Fig. S1 in the supplemental material). We first determined that the proportion of cytokines and chemokines produced on a single-cell basis was not significantly different between the bulk cultures of unexposed and virus-exposed B cells (Fig. 2A, upper row of pie charts). We therefore examined cytokines and chemokines produced within the 8.5% of HHV-8-exposed B cells that supported HHV-8 infection, i.e., expressed virus lytic protein ORF59 PF-8 at 48 h, compared to B cells in the same cultures that were not replicating virus, i.e., negative for these viral proteins (Fig. 2A, middle row, black and gray pie sections, respectively). The results indicated that, although not significantly different by SPICE analysis, HHV-8-infected B cells were more polyfunctional than virus-exposed B cells that were not infected with HHV-8, i.e., more of the ORF59 PF-8-positive B cells produced 3, 4, and 5 cytokines and chemokines than HHV-8-exposed, ORF59 PF-8-negative B cells (Fig. 2A, middle row; note the larger orange, yellow, and red sections in the pie charts for ORF59-positive than ORF59-negative B cells). For example, approximately 40% of virus-infected, ORF59 PF-8-positive B cells produced 4 to 5 mediators, compared to 4% of uninfected B cells. Conversely, <1% of uninfected B cells in the virus-exposed cultures produced all 5 cytokines and chemokines, with a large portion being monofunctional (28%) or not producing any cytokines and chemokines (20%). These intracellular data for the virus-exposed, total CD19+ B cell population support that B cells expressing HHV-8 lytic proteins in vitro are polyfunctional, whereas uninfected, virus-exposed B cells have less polyfunctionality.
FIG 2 .

HHV-8 targets naive and IgM memory B cells and plasma cell-like subsets for lytic infection and induction of polyfunctional responses in vitro. (A, top row) Cytokine and chemokine responses were assessed at 48 h in unexposed and HHV-8-exposed B cells that were derived from HHV-8-naive, CD19+ B cells activated with CD40L and IL-4 by intracellular staining of ORF59 PF-8 and 5 cytokines and chemokines by polychromatic flow cytometry. The colored pie charts show the relative production of 0 to 5 cytokines and chemokines in HHV-8-unexposed (left) and -exposed (right) B cell cultures. (Middle row) Further gating was done on the virus-exposed B cell cultures to distinguish infected from uninfected cells. The black and gray pie chart indicates the proportion of infected ORF59 PF-8pos (black) and uninfected ORF59 PF-8neg (gray) B cells. The colored pie charts display the proportion of uninfected (left) and infected (right) B cells within the virus-exposed cultures that expressed 0 to 5 cytokines and chemokines. (Bottom row) Production of cytokines and chemokines in B cell lineage phenotypes: naive (CD19+ CD20+ IgM+ IgD+ CD27¯ CD138¯), IgM memory (CD19+ CD20+ IgM+ IgD± CD27+ CD138¯), and plasma cell-like (CD138+ CD20± CD38±) subsets within the ORF59 PF-8neg (left charts) and ORF59 PF-8pos (right charts). (B) HHV-8-exposed B cells were stained for phenotypic markers, ORF59 PF-8, and intracellular cytokines at 48 h. Cytokine and chemokine production was determined for naive, IgM memory, and plasma cell-like populations among ORF59 PF-8neg and ORF59 PF-8pos cells. Values are means ± SE; n = 4. SPICE was used to derive P values. Fig. 2B: *, P < 0.05 comparing HHV-8 exposed ORF59 negative to HHV-8 exposed ORF59 positive.
HHV-8 targets naive B cells, IgM memory B cells, and plasma cell-like subsets for infection and induction of polyfunctional responses in vitro.
We hypothesized that a more distinct differential in cytokine and chemokine production induced by HHV-8 could be revealed at the level of B cell lineage subsets than was evident in the total CD19+ B cell population. For this, initial flow cytometry panels included IgM and IgD heavy chains to represent non-isotype-switched cells, as well as the IgG heavy chain to represent class-switched isotypes. Since we did not observe significant differences in the number of CD19+ B cells expressing either ORF59 PF-8 or K8.1A/B (data not shown), we only used MAb specific for ORF59 PF-8 in these and all further flow cytometry experiments. As ORF59 PF-8 expression was not detected in class-switched B cells (data not shown), further assessment of cellular IgG was not included in this study. B cell subsets were classified as naive (CD19+ CD20+ IgM+ IgD+ CD27¯ CD138¯), IgM memory (CD19+ CD20+ IgM+ IgD± CD27+ CD138¯) and plasma cell-like (CD138+ CD20± CD38±) (17, 18) (see Fig. S2 in the supplemental material). We found that at 24 h, HHV-8-infected, ORF59 PF-8-expressing B cells consisted of 74% naive, 14% IgM memory, <5% plasma cell-like subsets, and 7% other phenotypes (data not shown). By 72 h, these infected subsets were 54%, 21%, 7%, and 18%, respectively. There was, however, a similar shift in B cell subsets in the ORF59 PF-8-negative cells in these virus-exposed cultures. Furthermore, a higher proportion of naive and plasma-like cells within the ORF59+ population expressed DC-SIGN than did ORF59¯ cells (data not shown).
This analysis revealed that HHV-8 infection of all 3 lineage subsets, i.e., naive B cells, IgM memory B cells, and plasma cell-like cells, induced significantly more polyfunctionality than that in virus-exposed but uninfected B cells in the same cultures. This is visually apparent based on the increase in the size of the red (3 functions), yellow (4 functions), and orange (5 functions) sections of the pie charts and decreases in the green (2 functions), blue (1 function), and white (0 functions) sections in Fig. 2A (HHV-8-exposed cultures, bottom row, ORF59 positive compared to ORF59 negative). Individual patterns of cytokine and chemokine polyfunctional data are shown in Fig. 2B. An increase in a shift to the left of predominant black bars is notable for naive and IgM memory B cell and plasma cell-like subsets. Of the HHV-8-infected cells, 35 to 41% produced at least 3 cytokines and chemokines, compared to 1.0 to 9.1% of uninfected cells (P < 0.01).
Thus, analysis of B cell lineage phenotypes revealed that HHV-8 targets naive and IgM memory B cells and plasma cell-like subsets for infection in vitro. Each of these B cell subsets expressed a variety of combinations of cytokines and chemokines that were greater than those produced by HHV-8-exposed, uninfected B cell subsets.
Circulating serum and B cell-associated cytokines and chemokines are enhanced in subjects with KS.
We next investigated the role of virus replication and production of cytokines and chemokines in KS based on our in vitro B cell model. We examined archived serum or plasma and peripheral blood mononuclear cells (PBMC) from participants in the Multicenter AIDS Cohort Study (MACS) who were coinfected with HIV-1 and HHV-8 for similar time periods prior to the advent of effective combination antiretroviral therapy and who did (KS positives) or did not (KS negatives) develop KS (Table 1). Asymptomatic, HHV-8 antibody-positive, HIV-1-negative MACS subjects served as healthy controls. As expected (19, 20), KS positives had lower CD3+ and CD4+ T cell counts and higher plasma HIV-1 and HHV-8 viral loads than KS negatives and healthy controls. HHV-8 DNA was detected in plasma of 61% of KS positives, 23% of KS negatives, and 0% of HHV-8-seropositive, HIV-1-negative healthy controls.
TABLE 1 .
T cell counts and viral loads in KS-positive and KS-negative study subjectsa
| Study group | Study visit age (yrs) | Median (range) time of visit in relation to KS diagnosis | T cell counts |
Plasma viral load |
|||
|---|---|---|---|---|---|---|---|
| CD3 | CD4 | CD8 | HHV-8 | HIV-1 | |||
| HHV-8+ HIV-1− healthy controls (n = 7) | 54 ± 6 | NAb | 1,348 ± 198 | 797 ± 113 | 552 ± 102 | 0 | 0 |
| HHV-8+ HIV-1+ KS negatives (n = 13) | 39 ± 2 | NA | 1,529 ± 79 | 543 ± 39 | 957 ± 62 | 1,050 ± 803 (3/13) | 61,846 ± 35,577 |
| HHV-8+ HIV-1+ KS positives (n = 13) | 40 ± 2 | 4.5 mos prior (18 mos prior to 3 mos after) | 1,111 ± 180 | 180 ± 38 | 881 ± 134 | 2,333 ± 1,938 (8/13) | 187,442 ± 32,142 |
| P value | NSc | NA | 0.03 | 3.9 × 10−7 | NS | NS | 0.007 |
Each visit corresponds to a MACS clinic visit when blood was collected and used for this study. Although some samples included time points after KS diagnosis, these subjects were not receiving treatment for KS. P values were determined by a paired t test between KS positives and KS negatives. For HHV-8 plasma viral loads, the numbers of positive donors per donors tested are given in parentheses.
NA, not applicable.
NS, not significant.
Analysis of serum collected within 1 year prior to KS diagnosis showed that levels of TNF-α, MIP-1α, and IL-8 were nearly 2-fold higher in KS positives than KS negatives (P = 0.01, 0.03, and 0.02, respectively) (Fig. 3). There was a trend for increased MIP-1β and decreased IL-6 levels in KS positives compared to KS negatives (P = not significant). Collectively, these data showed that systemic levels of IL-8, TNF-α, MIP-1α, and MIP-1β, but not IL-6, were increased within a year prior to KS diagnosis.
FIG 3 .
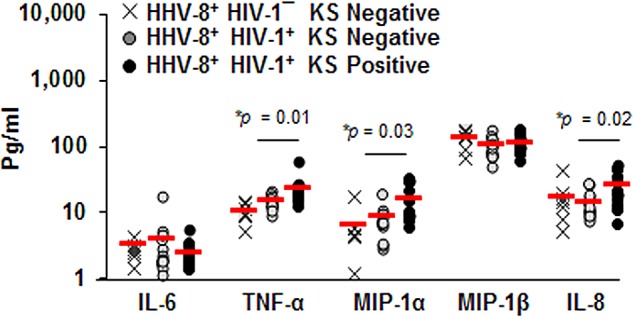
Levels of serum cytokines and chemokines are enhanced in KS. Concentrations of IL-6, TNF-α, MIP-1α, MIP-1β, and IL-8 were determined in serum from 7 HIV-1¯/HHV-8+ healthy controls, 13 HIV-1+/HHV-8+ KS negatives, and 13 HIV-1+/HHV-8+ KS positives. Each symbol represents an individual; red horizontal lines are the means.
To examine these factors at the cellular level in blood, HHV-8 infection was directly assessed within circulating, purified CD19+ B cells in KS positives and KS negatives (Table 2). Overall, KS positives displayed 32-fold more HHV-8 DNA in their B cells than KS negatives, although HHV-8 DNA was not detected in B cells from every KS-positive patient studied. By the same token, one donor in the KS-negative group had detectable HHV-8 DNA in his B cells. Furthermore, a mean of 2.3% of B cells expressed ORF59 PF-8 among KS positives, compared to 0.64% in KS negatives, confirming that KS positives had the greatest percentage of infected, circulating B cells.
TABLE 2 .
Viral loads and protein expression in KS-negative and KS-positive study subjectsa
| Study group | Subject no. | Plasma HHV-8 viral load (copies/ml) | B cell HHV-8 viral load (copies/500,000 cells) | % ORF59 PF-8+ B cellsa | Plasma HIV-1 viral load (copies/ml) |
|---|---|---|---|---|---|
| HHV-8+ HIV+ KS negatives (KS controls) | 1 | 0 | 0 | 0.37 | 300 |
| 2 | 0 | NDb | 0.11 | 22,810 | |
| 3 | 10,154 | 0 | 0.94 | 24,409 | |
| 4 | 0 | 6,835 | 1.00 | 891,846 | |
| 5 | 0 | ND | 0.90 | 593 | |
| 6 | 0 | 0 | 0.60 | 34,227 | |
| 7 | 0 | 0 | 0.64 | 19,552 | |
| Mean ± SE | 2,038 ± 2,038 | 1,367 ± 1,367 | 0.65 ± 0.12 | 141,962 ± 125,819 | |
| HHV-8+ HIV+ KS positives | 8 | 25,447 | 183,307 | 3.60 | 243,333 |
| 9 | 4,779 | ND | 2.10 | 144,161 | |
| 10 | 0 | 0 | 1.75 | 87,037 | |
| 11 | 0 | 20,884 | 2.10 | 69,444 | |
| 12 | 1,009 | ND | 2.85 | 68,585 | |
| 13 | 0 | 0 | 1.28 | 89,909 | |
| 14 | 0 | 14,914 | 2.44 | 12,090 | |
| Mean | 4,462 ± 3,567 | 43,821 ± 35,208 | 2.3 ± 0.28 | 102,080 ± 27,934 |
The flow cytometry percentages were normalized to the isotype controls of each experiment.
ND, not done.
To determine whether B cells infected with HHV-8 in vivo were a source of cytokines and chemokines, we examined purified CD19+ B cells directly without in vitro culture for cytokine and chemokine mRNA by real-time reverse transcription-PCR (RT-PCR). These B cells were obtained from HIV-1/HHV-8-coinfected patients that either did or did not develop KS and were not superinfected with HHV-8 in vitro. We found a consistent gradation in the levels of the 5 cytokine and chemokine mRNAs from the lowest amounts expressed in B cells from HHV8+ HIV-1¯ controls, to higher levels in HHV8+ HIV-1+ KS negatives, to the highest levels in the HHV8+ HIV-1+ KS positives (Fig. 4), although these differences were not statistically significant by a one-way analysis of variance (ANOVA). The latter group also had the greatest increases, of 4.1-, 3.6-, and 3.1-fold, in MIP-1α, MIP-1β, and IL-8, respectively, compared to KS negatives.
FIG 4 .
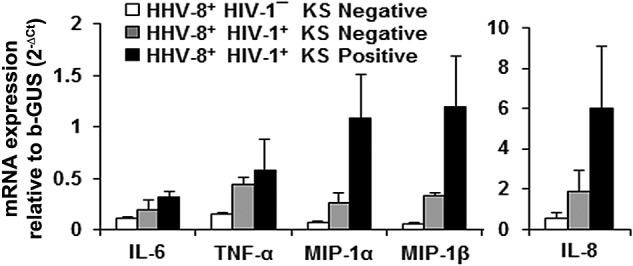
Levels of B cell cytokine and chemokine mRNA. RNA was extracted from purified CD19+ B cells of 2 healthy controls, 3 KS negatives, and 4 KS positives and used in a real-time RT-PCR assay to measure the expression levels of the indicated cytokine mRNAs relative to an endogenous control gene, the gene for β-glucuronidase. Data shown in the graphs were calculated as means ± SE. One-way ANOVA was used for statistical analysis, and no significant differences were observed among groups.
Direct analysis of purified B cells from the blood of HHV-8-infected subjects for 31 combinations of intracellular cytokine and chemokines by flow cytometry showed that more ORF59 PF-8-positive B cells from both the KS negatives and KS positives expressed 2 to 5 cytokines and chemokines than ORF59 PF-8-negative B cells, i.e., mean percentages (± standard errors [SE]) for KS negatives of 15.4% ± 4.5% compared to 1.3% ± 0.9% (P = 0.02), and in KS positives 17.2% ± 4.5% compared to 7.2% ± 5.8% (P = 0.01) (Fig. 5A, red, yellow and orange pieces, respectively, in ORF59 PF-8-positive compared to ORF59 PF-8-negative B cells). Lower numbers of ORF59 PF-8-negative B cells expressing each combination of 2 to 5 cytokines and chemokines for both the KS-negative and KS-positive groups are evident in Fig. 5B, compared to results for ORF59 PF-8-positive B cells in Fig. 5C.
FIG 5 .

HHV-8-infected, polyfunctional B cells detected in individuals with and without KS. (A) CD19+ B cells were stained for ORF59 PF-8 and IL-6, TNF-α, MIP-1α, MIP-1β, and IL-8. Percentages of B cells expressing 0 to 5 cytokines and chemokines and that were ORF59 PF-8¯ or ORF59 PF-8+ in 7 HHV-8+ HIV-1+ KS negatives (colored pies, top left) and 7 HHV-8+ HIV-1+ KS positives (colored pies, top right) are shown. (B) Percentages of B cells expressing different combinations of cytokines and chemokines within the ORF59 PF-8neg population of the study subjects (values are means ± SE; n = 7 HHV-8+ HIV-1+ KS negatives and 7 HHV-8+ HIV-1+ KS positives). (C) Percentages of B cells expressing different combinations of cytokines and chemokines within the ORF59 PF-8pos population of the 2 groups of subjects. Values are means ± SE. SPICE was used to derive P values. Fig. 5B: *, P < 0.05.
These data support that HHV-8-infected, CD19+ B cells from both HHV-8+ HIV-1+ individuals who did or did not develop KS had greater polyfunctionality than uninfected B cells.
HHV-8 infection is detected in highly polyfunctional naive and memory B cells and plasma cell-like subsets in subjects with KS.
Based on our in vitro findings, we postulated that HHV-8 induced greater production of cytokines and chemokines in lineage subsets of B cells in the blood of KS positives than KS negatives. Initial analysis of intracellular cytokine and chemokine production showed that HHV-8-infected cells among the IgM memory and plasma cell-like B cell lineage subsets were significantly more polyfunctional than HHV-8-uninfected cells in both KS-negative and KS-positive subjects (Fig. 6A, P values above pie charts). This was visually evident from the greater size of the orange (5 functions), yellow (4 functions), and red (3 functions) sections in the pie charts for ORF59 PF-8-positive versus ORF59 PF-8-negative subjects in Fig. 6A. Importantly, the 3 HHV-8-infected B cell subsets in the blood of the KS positives were more polyfunctional than those of the KS negatives (Fig. 6A, P values below pie charts).
FIG 6 .

HHV-8 targets naive and IgM memory B cells and plasma cell-like subsets for infection and induction of polyfunctional responses in KS positives and KS negatives. (A) HHV-8+ HIV-1+ KS negatives (top left) and HHV-8+ HIV-1+ KS positives (top right) were analyzed for ORF59 PF-8neg and ORF59 PF-8pos cells. Gates for B cells were used to derive naive and IgM memory B cell and plasma cell-like populations based on phenotypic markers and ORF59 PF-8 expression, as defined for Fig. 2. (B) HHV-8-infected B cells were stained for phenotypic markers, ORF59 PF-8, and intracellular cytokines. Cytokine production was determined for naive (top panel), IgM memory (middle panel), and plasma cell-like (bottom panel) subsets among ORF59 PF-8neg and ORF59 PF-8pos cells of the HHV-8+ HIV-1+ KS negatives and HHV-8+ HIV-1+ KS positives. Values are means ± SE; n = 7 in each group. SPICE was used to derive P values. Fig 6B: *, P < 0.05, where the color of the asterisk corresponds to the color of the KS negative/positive and ORF59 negative/positive group that is significantly different from the other 3 groups for that combination of cytokines and chemokines.
Notably, HHV-8-infected (ORF59 PF-8-positive) IgM memory B cells and plasma cell-like subsets had the greatest polyfunctionality, with 3, 4, and 5 combinations in both the KS positives and KS negatives (Fig. 6B). In sum, 61% to 88% of infected B cell subsets among KS positives were polyfunctional, compared to 46% to 75% of KS negatives (P < 0.01). In contrast, uninfected cells were more monofunctional in both KS positives and KS negatives. Furthermore, a higher percentage of naive, IgM memory, and plasma-like cells derived from KS-positive subjects expressed DC-SIGN than did cells obtained from KS-negative subjects (data not shown).
These data provide evidence that HHV-8 infection drives production of multiple cytokines and chemokines as revealed by analysis of B cell lineage subsets circulating in HHV-8+ HIV-1+ subjects with and without KS. Greater polyfunctional responses were found among the more-differentiated, IgM memory B cells and plasma cell-like subsets in the blood of subjects who developed KS.
DISCUSSION
B lymphocytes infected with HHV-8 are a potential source of infectious virus as well as inflammatory cytokines and chemokines that drive the oncogenic process of KS, yet little direct evidence exists to support this hypothesis. Here we have extended our previous results (10) to show that approximately 8.5% of CD19+ B cells derived from the blood of healthy HHV-8-seronegative adults support lytic replication of HHV-8 by demonstrating consistent increases in HHV-8 DNA, ORF59 PF-8 expression, and infectious virion production after in vitro infection. Virus replication requires preactivation of the B cells with CD40L and IL-4, surrogates of CD4+ T helper cells, and subsequent expression of the HHV-8 receptor DC-SIGN (10). Importantly, our new in vitro findings implicate less-differentiated naive and IgM memory B cells as predominant targets during 72 h of HHV-8 infection. This fits with classic, HIV-1-negative KS, where the preimmune/natural effector B cell compartment, including marginal zone-like memory and naive B cells, is expanded compared to healthy controls (21). Expansion of such B cell populations would provide targets for initial HHV-8 infection and lytic cycle replication. We also noted infection of plasma cell-like subsets during in vitro HHV-8 infection. It is not yet clear if there is a transition in HHV-8 infection from less-differentiated naive to memory B cells and finally to plasma cell-like subsets.
A similar yet distinct pattern of HHV-8 infection of B cells was evident in vivo in relation to KS. Using new flow cytometry methods to delineate infected B cells based on expression of ORF59 PF-8 directly in archived MACS samples, we found that B cells of KS positives had a higher percentage of viral lytic protein expression than KS negatives, i.e., approximately 2.3% and 0.64%, respectively. Expression of ORF59 PF-8 is a reliable marker of HHV-8 infection, having previously been observed in lymph node B cells of multicentric Castleman’s disease patients and KS tissue biopsy specimens (6). Interestingly, HHV-8 DNA was detected in B cells from the 3 of 5 KS positives and 1 of 5 KS negatives, supporting a greater presence of circulating, B cell-associated virus in KS positives. Two of the 5 KS positives displayed minimal expression of ORF59 PF-8 by flow cytometry, with undetectable levels of plasma or B cell-associated HHV-8 DNA. These data are consistent with those of earlier studies in which there was a range of HHV-8 DNA in the blood prior to development of KS (22) and detection in PBMC of only 50% of individuals with KS, compared 0 to 10% without KS (23). As detection of HHV-8 DNA is dependent on the number of PBMC used for DNA extraction (24), infected B cells with lower HHV-8 copy numbers could generate a negative PCR signal, while transcription/translation of the genome results in sufficient copies of ORF59 PF-8 for detection by flow cytometry.
Analysis of HHV-8 infection in subjects with KS provided evidence that virus infection was present in a more differentiated B cell, as 60% of the infected cells expressed the CD138+ plasma cell marker among KS positives. This is consistent with plasmablast gene expression profiles in HHV-8-associated, primary effusion lymphoma (25, 26). This supports that the virus drives plasma cell differentiation, as terminal B cell differentiation is associated with viral reactivation (27). Furthermore, our data suggest that HHV-8 coinfection could be a contributing factor in HIV-1-infected individuals who exhibit abnormal proportions of transitional immature, activated mature B cells and plasmablasts (28). Our combined in vitro and in vivo results expand on in vitro evidence that latent HHV-8 infection is detected in plasmablasts expressing the IgM heavy and lambda light chains (29). Collectively, these results support the hypothesis that B lymphocytes serve as a reservoir for HHV-8 lytic replication and/or latent reactivation. Although it has been demonstrated that plasma viral load, and not PBMC viral load, correlates with KS progression (30), B cell viral load and lytic protein expression together could serve as more sensitive prognostic factors for KS. We also need to determine if different subsets of B cells are targeted by HHV-8 in lymphoid compartments than in blood, similar to Epstein-Barr virus (31, 32), and also their relationships to KS.
Examination of the cytokines and chemokines produced by B cells infected with HHV-8 revealed multiple characteristics that could be important in HHV-8 pathogenesis and oncogenesis. When we analyzed this initially in our in vitro model, we found that among 16 cytokines and chemokines examined, HHV-8 infection of B cells induced the greatest amounts of mRNA and protein for 2 cytokines (IL-6 and TNF-α) and 3 chemokines (MIP-1α, MIP-1β, and IL-8). Within HHV-8-exposed B cell cultures, B cells supporting HHV-8 lytic infection exhibited the most significant polyfunctionality, i.e., concurrent production of 2 to 5 cytokines and chemokines, compared to both virus-negative B cells in HHV-8-exposed cultures and CD40L/IL-4-activated, uninfected B cell control cultures. In sum, these in vitro results indicate that actively replicating HHV-8 induces the highest levels and polyfunctionality of cytokines and chemokines in B cells over a background of that induced by the CD4+ T cell surrogates CD40L and IL-4.
Using this comprehensive approach that we developed in vitro, we were able for the first time to directly assess clinical correlates of HHV-8 infection and production of cytokines and chemokines in B cells of subjects who developed KS. We found enhanced mRNA expression for all of the cytokines and chemokines in circulating B cells of KS-positive compared to KS-negative subjects. Moreover, the proportion of infected cells among the KS positives was 2-fold higher than in KS negatives and was linked to a higher quantity of polyfunctional cytokine- and chemokine-producing B cells. Notably, a small portion of HHV-8-uninfected B cells among HIV-1-coinfected subjects was polyfunctional, an effect possibly due to activation by HIV-1 (28). Since the underlying HIV-1 effect on B cell activation is present in both KS-negative and KS-positive donors, we can postulate that the differences in B cell polyfunctionality are due to an HHV-8 effect. In addition to this overabundance of B cell activation, HHV-8 interactions with other professional antigen-presenting cells, including myeloid (33) and plasmacytoid (34) dendritic cells and monocytes/macrophages (35), as well as HHV-8 homologues of cellular cytokines, chemokines, and growth factors (2), could contribute to a systemic as well as localized cytokine and chemokine milieu that drives endothelial cell outgrowth in KS.
We present here a new paradigm of B cell biology wherein B cell subsets produce multiple cytokines and chemokines that mediate a variety of functions. Memory B cells have been shown to produce more IL-6, TNF-α, MIP-1α, and MIP-1β than naive B cells, which produce greater amounts of IL-10 (36, 37). However, little is known about the functional role of cytokines and chemokines produced by B cells. We propose that the presence of a small but highly active population of polyfunctional B cells in HHV-8 infection has detrimental rather than beneficial outcomes. HHV-8-infected B cells produced elevated levels of MIP-1α and -β, which are chemokines involved in B cell recruitment, activation, and immunoglobulin production (38, 39). MIP-1α and -β could increase the activated B cell population most capable of replicating HHV-8. Enhanced production of the B cell proliferation factor, IL-6, could also increase targets for HHV-8 replication (40). IL-6 is a proinflammatory cytokine that enhances TNF-α, and these cytokines together can create a rich inflammatory microenvironment that promotes KS tumor growth and vascularization (41). Our results indicating that IL-6 serum levels were lower prior to KS are similar to other reports (42) and imply that B cell-associated IL-6 production is more closely linked to development of KS than circulating IL-6 levels. Finally, IL-8 can serve as a ligand for the HHV-8-encoded viral G protein-coupled receptor (vGPCR), which after binding results in production of angiogenic factors VEGF, IL-6, and the chemokine growth-regulated oncogene α, as well as more IL-8 production (43, 44). As vGPCR expression upregulates the promoter for the lytic switch protein, leading to expression of ORF50 replication transcriptional activator (45), IL-8 could also act as an autocrine or paracrine factor to enhance HHV-8 replication via vGPCR-signaled enhancement of ORF50.
In conclusion, our study indicates that naive and IgM memory B cells, and a plasma cell-like population, serve as major targets for HHV-8 infection. HHV-8 infection of B cells is associated with production of a cytokine and chemokine milieu that is conducive to KS oncogenic cell proliferation. We propose that these virus-infected, polyfunctional B cells play a significant role in HHV-8 replication and dissemination, and also proliferation of the target cell populations of KS.
MATERIALS AND METHODS
Study participants and samples.
This study was approved by the University of Pittsburgh Institutional Review Board, with written informed consent by participants. In vitro studies used adult blood donors who were HHV-8 antibody negative by an indirect immunofluorescence microscopy assay (46). Blood plasma and serum and PBMC were derived from Pittsburgh MACS men who have sex with men (47), were Caucasian, of average age 36.6 years (range, 24 to 77 years) at the first visit, and were chosen based on HIV-1 and HHV-8 infection and development of KS. HHV-8 viral load was determined by PCR as described below. HIV-1 viral load was determined using the Roche Ultrasensitive RNA PCR assay (Hoffman-LaRoche). T cell numbers were determined using flow cytometry (48). The participants were classified into 3 groups of 7 healthy controls (HIV-1¯ HHV-8+), 13 HIV-1+ HHV-8+ KS negatives, and 13 HIV-1+ HHV-8+ KS positives. Blood samples were obtained within 1.5 years of KS diagnosis for KS positives and correspondingly for KS negatives.
B cells for in vitro studies.
PBMC were isolated by Ficoll-Hypaque density gradient separation. CD19+ B cells were collected by negative selection (B-Cell Isolation kit II; Miltenyi Biotec) and cultured in RPMI 1640 medium (Gibco) with 10% heat-inactivated fetal calf serum (FCS; GemCell). B cells were activated for 48 h at 37°C in 5% CO2 with 1 µg of trimeric Mega CD40L (Alexis) and 1,000 U recombinant human IL-4 per ml.
HHV-8 purification and infection of B cells in vitro.
HHV-8 was purified from a BCBL-1 cell line latently infected with HHV-8 (49). Prior to sucrose cushion ultracentrifugation, supernatants were treated with 1 U/100 µl DNase (Sigma). A total of 1×106 B cells/ml were left unexposed or exposed to 107 DNA copies of HHV-8 for 3 h at 37°C. Cells were washed and centrifuged twice. A total of 1×106 cells/ml were cultured in RPMI–10% FCS at 37°C.
Soluble cytokine and chemokine detection.
Supernatant samples were collected from unexposed and HHV-8-exposed B cells and screened for IL-1β, -2, -4, -6, -7, -8, and -10, IL-12p70, IFN-γ, TNF-α, IP-10, MIP-1α, MIP-1β, MCP-1, RANTES, and VEGF by CBA (BD) as per the manufacturer’s instructions. Samples were read on an LSR II flow cytometer (BD Immunocytometry Systems) and analyzed with FCAP Array software (BD). Sera were tested in duplicate for biomarkers by using an electrochemiluminescence Meso Scale Discovery (MSD) multiarray assay. Ultrasensitive kits for human IL-6, IL-8, TNF-α, MIP-1β, and MIP-1α and a human serum kit for VEGF (MSD) were used per the manufacturer’s instructions (Sector Imager 2400 electrochemiluminescence; MSD). These data were analyzed using the Discovery Workbench (version 3; MSD).
HHV-8 DNA quick real-time RT-PCR.
For the in vitro experiments, a total of 5×105 B cells and 500 µl of culture supernatant were collected post-HHV-8 exposure, pelleted, and assayed by PCR (50, 51). Samples were treated with 1 µl DNase in 10 µl buffer (Sigma) for 15 min and then lysed in easyMAG buffer (NucliSENS). DNA was extracted with an easyMAG automated extractor (bioMérieux). Phocine herpesvirus was added as an internal control (52). DNA was mixed with a primer set specific for HHV-8 K8.1 (51), and the real-time PCR was done using a 7000, 7500, or ViiA7 ABI system (Applied Biosystems). B cells isolated from PBMC obtained from KS cases and controls were treated and tested for HHV-8 DNA as described above.
TCID50 assay.
Supernatants collected at 3, 24, and 48 h post-B cell exposure were used in a TCID50 assay with T1H6-DC-SIGN cells, as described previously (12).
Microarray for B cell mRNA.
B cells were left unexposed or exposed to HHV-8 for 3 h, washed, and recultured. A total of 1×106 cells per treatment were collected after the wash (3 h) and at 4, 6, 9, 15, and 27 h postexposure. Genomic DNA was digested with RNase-free DNase, and RNA was extracted using an RNeasy minikit (Qiagen). Total concentrations (in ng/μl) were determined by using a NanoDrop 1000 spectrophotometer (Thermo Scientific). One microgram of RNA of each sample was labeled and directly hybridized to Illumina HT-12 v4 microchips by the University of Pittsburgh Genomics and Proteomics Core Laboratory. Samples were run in duplicate to determine RNA expression levels.
Intracellular staining and flow cytometry.
A total of 1×106 cells were resuspended in 100 µl phosphate-buffered saline (PBS) per well in a 96 V-bottom well plate. Staining for cytokines and chemokines was performed as previously described (53), except prior to intracellular staining, cells were treated with Super Blocking buffer (Pierce) for 30 min. Cells were analyzed on an LSR II or a Fortessa LSR flow cytometer. For HHV-8 viral proteins, cells were stained with anti-K8.1A/B or anti-ORF59 PF-8 MAb conjugated to Alexa Fluor 680 (AF680) by using the Zenon conjugation kit (Invitrogen). Purified mouse IgG1 and IgG2B (Sigma) were conjugated with AF680 and used as isotype gating controls (consistently gated at approximately 1% background positivity). For B cell subsets, cells were surface stained with 5 µl of CD19-brilliant violet, CD20-CF594, CD23-allophycocyanin (APC), CD27–APC-H7, CD38–peridinin chlorophyll protein (PerCP)-CY5.5, CD138-V450, 10 µl of CD209-fluorescein isothiocyanate (FITC; R&D Systems), 20 µl of IgM–phycoerythrin (PE)-Cy5, and IgD-PE. For combination panels of subsets and cytokines, cells were stained on the surface for CD19, CD20, and IgM as described above and 5 µl of CD27–PE-Cy7 and CD138–PerCP-CY5.5, as well as intracellularly with 5 µl of MIP-1β–APC-H7, IL-6–V450, TNF-α–APC, and MIP-1α–PE and 20 µl of IL-8–FITC. All antibodies were from BD unless otherwise noted.
cDNA synthesis and real-time RT-PCR.
A two-step RT-PCR assay was used to measure the levels of expression of host mRNAs as described elsewhere (54). Gene expression was normalized to the endogenous control mRNA, β-glucuronidase, and the values presented were calculated with the formula 2−ΔCT.
Statistics.
We used the paired Student’s t test to compare ORF59 PF-8pos and ORF59 PF-8neg cells within groups and an ANOVA for comparisons between groups. Flow cytometry data were analyzed using FloJo software (Tree Star). Polyfunctional cytokine and chemokine production levels were assessed by using SPICE permutation tests and Student’s t test (version 4.3; M. Roederer, Vaccine Research Center, NIAID, NIH).
SUPPLEMENTAL MATERIAL
Flow cytometry polyfunctional gating strategy. Singlet, live lymphocyte populations were determined and first gated (data not shown). An IgG2B control conjugated with AF-680 (not shown) was used to set the gate for ORF59 PF-8-positive and -negative cells. No differences in autofluorescence were seen between the 2 cell populations. (A) Unstained live, singlet CD19+ lymphocytes were used to set the gate for cytokine and chemokine markers. HHV-8-exposed ORF59 PF-8neg (B) and ORF59 PF-8pos (C) cells were gated against each cytokine and chemokine. Download
Flow cytometry B cell lineage subset gating strategy. Live, singlet populations were detected as previously described. (A) ORF59 PF-8neg/pos cells were selected and gated against CD20 and CD138 expression. CD20± CD138+ cells (dashed arrow) were further gated against CD38. Plasma cells were considered CD20± CD138+ CD38±. (B and C) CD20+ CD138¯ cells (solid arrow) were further gated against CD19 and CD27 expression. CD19+ CD27+ and CD19+ CD27¯cells were gated against IgD and IgM. Naive cells were CD19+ CD20+ IgM+ IgD+ CD27¯ CD138¯ (B), and IgM memory cells were considered CD19+ CD20+ IgM+ IgD± CD27+ CD138¯ (C). Download
ACKNOWLEDGMENTS
We thank Mariel Jais, Luann Borowski, Yue Chen, Kelly Gordon, and Robbie Mailliard for technical assistance and consultation, J. Margolick and J. Bream (Johns Hopkins University) for assistance in establishing the MSD assay, and Phalguni Gupta for reviewing the manuscript.
This work was supported by NIH grants R01-CA82053 and U01-AI35041 and NIH training grant fellowship T32 AI065380 to L.M.L.
Footnotes
Citation Knowlton ER, Rappocciolo G, Piazza P, Lepone LM, Nadgir SV, Bullotta A, Berendam SJ, Li J, Reinhart TA, Jenkins FJ, Rinaldo CR. 2014. Human herpesvirus 8 induces polyfunctional B lymphocytes that drive Kaposi’s sarcoma. mBio 5(5):e01277-14. doi:10.1128/mBio.01277-14.
REFERENCES
- 1. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 2. Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10:707–719. 10.1038/nrc2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gottwein E. 2012. Kaposi’s sarcoma-associated herpesvirus microRNAs. Front. Microbiol. 3:165. 10.3389/fmic.2012.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Polizzotto MN, Uldrick TS, Hu D, Yarchoan R. 2012. Clinical manifestations of Kaposi sarcoma herpesvirus lytic activation: multicentric Castleman disease (KSHV-MCD) and the KSHV inflammatory cytokine syndrome. Front. Microbiol. 3:73. 10.3389/fmicb.2012.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 156:743–749. 10.1016/S0002-9440(10)64940-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 113:124–136. 10.1172/JCI200417803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knowlton ER, Lepone LM, Li J, Rappocciolo G, Jenkins FJ, Rinaldo CR. 2012. Professional antigen presenting cells in human herpesvirus 8 infection. Front. Immunol. 3:427. 10.3389/fimmu.2012.00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uldrick TS, Polizzotto MN, Yarchoan R. 2012. Recent advances in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Curr. Opin. Oncol. 24:495–505. 10.1097/CCO.0b013e328355e0f3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rappocciolo G, Hensler HR, Jais M, Reinhart TA, Pegu A, Jenkins FJ, Rinaldo CR. 2008. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J. Virol. 82:4793–4806. 10.1128/JVI.01587-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Armitage RJ, Macduff BM, Spriggs MK, Fanslow WC. 1993. Human B cell proliferation and Ig secretion induced by recombinant CD40 ligand are modulated by soluble cytokines. J. Immunol. 150:3671–3680 [PubMed] [Google Scholar]
- 12. Nadgir SV, Hensler HR, Knowlton ER, Rinaldo CR, Rappocciolo G, Jenkins FJ. 2013. Fifty percent tissue culture infective dose assay for determining the titer of infectious human herpesvirus 8. J. Clin. Microbiol. 51:1931–1934. 10.1128/JCM.00761-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan SR, Chandran B. 2000. Characterization of human herpesvirus 8 ORF59 protein (PF-8) and mapping of the processivity and viral DNA polymerase-interacting domains. J. Virol. 74:10920–10929. 10.1128/JVI.74.23.10920-10929.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu L, Puri V, Chandran B. 1999. Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology 262:237–249. 10.1006/viro.1999.9900 [DOI] [PubMed] [Google Scholar]
- 15. Wang FZ, Akula SM, Pramod NP, Zeng L, Chandran B. 2001. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 75:7517–7527. 10.1128/JVI.75.16.7517-7527.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramanian R, Sehgal I, D’Auvergne O, Kousoulas KG. 2010. Kaposi’s sarcoma-associated herpesvirus glycoproteins B and K8.1 regulate virion egress and synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1 cells. J. Virol. 84:1704–1714. 10.1128/JVI.01889-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. 2012. Advances in human B cell phenotypic profiling. Front. Immunol. 3:302. 10.3389/fimmu.2012.00302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackson SM, Wilson PC, James JA, Capra JD. 2008. Human B cell subsets. Adv. Immunol. 98:151–224. 10.1016/S0065-2776(08)00405-7 [DOI] [PubMed] [Google Scholar]
- 19. Moore PS, Kingsley LA, Holmberg SD, Spira T, Gupta P, Hoover DR, Parry JP, Conley LJ, Jaffe HW, Chang Y. 1996. Kaposi’s sarcoma-associated herpesvirus infection prior to onset of Kaposi’s sarcoma. AIDS 10:175–180. 10.1097/00002030-199602000-00007 [DOI] [PubMed] [Google Scholar]
- 20. Jacobson LP, Jenkins FJ, Springer G, Munoz A, Shah KV, Phair J, Zhang Z, Armenian H, et al. 2000. Interaction of human immunodeficiency virus type 1 and human herpesvirus type 8 infections on the incidence of Kaposi’s sarcoma. J. Infect. Dis. 181:1940–1949. 10.1086/315503 [DOI] [PubMed] [Google Scholar]
- 21. Della Bella S, Taddeo A, Colombo E, Brambilla L, Bellinvia M, Pregliasco F, Cappelletti M, Calabrò ML, Villa ML. 2010. Human herpesvirus-8 infection leads to expansion of the preimmune/natural effector B cell compartment. PLoS One 5:e15029. 10.1371/journal.pone.0015029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Engels EA, Biggar RJ, Marshall VA, Walters MA, Gamache CJ, Whitby D, Goedert JJ. 2003. Detection and quantification of Kaposi’s sarcoma-associated herpesvirus to predict AIDS-associated Kaposi’s sarcoma. AIDS 17:1847–1851. 10.1097/00002030-200308150-00015 [DOI] [PubMed] [Google Scholar]
- 23. Boivin G, Côté S, Cloutier N, Abed Y, Maguigad M, Routy JP. 2002. Quantification of human herpesvirus 8 by real-time PCR in blood fractions of AIDS patients with Kaposi’s sarcoma and multicentric Castleman’s disease. J. Med. Virol. 68:399–403. 10.1002/jmv.10217 [DOI] [PubMed] [Google Scholar]
- 24. Martró E, Cannon MJ, Dollard SC, Spira TJ, Laney AS, Ou CY, Pellett PE. 2004. Evidence for both lytic replication and tightly regulated human herpesvirus 8 latency in circulating mononuclear cells, with virus loads frequently below common thresholds of detection. J. Virol. 78:11707–11714. 10.1128/JVI.78.21.11707-11714.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jenner RG, Maillard K, Cattini N, Weiss RA, Boshoff C, Wooster R, Kellam P. 2003. Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. U. S. A. 100:10399–10404. 10.1073/pnas.1630810100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klein U, Gloghini A, Gaidano G, Chadburn A, Cesarman E, Dalla-Favera R, Carbone A. 2003. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 101:4115–4121. 10.1182/blood-2002-10-3090 [DOI] [PubMed] [Google Scholar]
- 27. Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS Lett. 581:3485–3488. 10.1016/j.febslet.2007.06.056 [DOI] [PubMed] [Google Scholar]
- 28. Moir S, Fauci AS. 2009. B cells in HIV infection and disease. Nat. Rev. Immunol. 9:235–245. 10.1038/nri2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hassman LM, Ellison TJ, Kedes DH. 2011. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Invest. 121:752–768. 10.1172/JCI44185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boivin G, Gaudreau A, Routy JP. 2000. Evaluation of the human herpesvirus 8 DNA load in blood and Kaposi’s sarcoma skin lesions from AIDS patients on highly active antiretroviral therapy. AIDS 14:1907–1910. 10.1097/00002030-200009080-00004 [DOI] [PubMed] [Google Scholar]
- 31. Laichalk LL, Hochberg D, Babcock GJ, Freeman RB, Thorley-Lawson DA. 2002. The dispersal of mucosal memory B cells: evidence from persistent EBV infection. Immunity 16:745–754. 10.1016/S1074-7613(02)00318-7 [DOI] [PubMed] [Google Scholar]
- 32. Dorner M, Zucol F, Alessi D, Haerle SK, Bossart W, Weber M, Byland R, Bernasconi M, Berger C, Tugizov S, Speck RF, Nadal D. 2010. β1 integrin expression increases susceptibility of memory B cells to Epstein-Barr virus infection. J. Virol. 84:6667–6677. 10.1128/JVI.02675-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hensler HR, Rappocciolo G, Rinaldo CR, Jenkins FJ. 2009. Cytokine production by human herpesvirus 8-infected dendritic cells. J. Gen. Virol. 90:79–83. 10.1099/vir.0.006239-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. West JA, Gregory SM, Sivaraman V, Su L, Damania B. 2011. Activation of plasmacytoid dendritic cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 85:895–904. 10.1128/JVI.01007-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Santarelli R, Gonnella R, Di Giovenale G, Cuomo L, Capobianchi A, Granato M, Gentile G, Faggioni A, Cirone M. 2014. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci. Rep. 4:4241. 10.1038/srep04241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, Kim HJ, Bar-Or A. 2007. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J. Immunol. 178:6092–6099. 10.4049/jimmunol.178.10.6092 [DOI] [PubMed] [Google Scholar]
- 37. Agrawal S, Gupta S. 2011. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J. Clin. Immunol. 31:89–98. 10.1007/s10875-010-9456-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim CH, Pelus LM, White JR, Applebaum E, Johanson K, Broxmeyer HE. 1998. CK beta-11/macrophage inflammatory protein-3 beta/EBI1-ligand chemokine is an efficacious chemoattractant for T and B cells. J. Immunol. 160:2418–2424 [PubMed] [Google Scholar]
- 39. Teague RM, Harlan LM, Benedict SH, Chan MA. 2004. MIP-1alpha induces differential MAP kinase activation and IκB gene expression in human B lymphocytes. J. Interferon Cytokine Res. 24:403–410. 10.1089/1079990041535656 [DOI] [PubMed] [Google Scholar]
- 40. Schulte KM, Talat N. 2010. Castleman’s disease: a two compartment model of HHV8 infection. Nat. Rev. Clin. Oncol. 7:533–543. 10.1038/nrclinonc.2010.103 [DOI] [PubMed] [Google Scholar]
- 41. Miles SA, Rezai AR, Salazar-González JF, Vander Meyden M, Stevens RH, Logan DM, Mitsuyasu RT, Taga T, Hirano T, Kishimoto T, Martfnez-Maza O. 1990. AIDS Kaposi sarcoma-derived cells produce and respond to interleukin 6. Proc. Natl. Acad. Sci. U. S. A. 87:4068–4072. 10.1073/pnas.87.11.4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dourado I, Martínez-Maza O, Kishimoto T, Suzuki H, Detels R. 1997. Interleukin 6 and AIDS-associated Kaposi’s sarcoma: a nested case control study within the Multicenter AIDS Cohort Study. AIDS Res. Hum. Retroviruses 13:781–788. 10.1089/aid.1997.13.781 [DOI] [PubMed] [Google Scholar]
- 43. Choi YB, Nicholas J. 2010. Induction of angiogenic chemokine CCL2 by human herpesvirus 8 chemokine receptor. Virology 397:369–378. 10.1016/j.virol.2009.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu Y, Ganem D. 2007. Induction of chemokine production by latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J. Gen. Virol. 88:46–50. 10.1099/vir.0.82375-0 [DOI] [PubMed] [Google Scholar]
- 45. Bottero V, Sharma-Walia N, Kerur N, Paul AG, Sadagopan S, Cannon M, Chandran B. 2009. Kaposi sarcoma-associated herpes virus (KSHV) G protein-coupled receptor (vGPCR) activates the ORF50 lytic switch promoter: a potential positive feedback loop for sustained ORF50 gene expression. Virology 392:34–51. 10.1016/j.virol.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang FZ, Akula SM, Sharma-Walia N, Zeng L, Chandran B. 2003. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J. Virol. 77:3131–3147. 10.1128/JVI.77.5.3131-3147.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaslow RA, Ostrow DG, Detels R, Phair JP, Polk BF, Rinaldo CR., Jr. 1987. The multicenter AIDS cohort study: rationale, organization, and selected characteristics of the participants. Am. J. Epidemiol. 126:310–318. 10.1093/aje/126.2.310 [DOI] [PubMed] [Google Scholar]
- 48. Schenker EL, Hultin LE, Bauer KD, Ferbas J, Margolick JB, Giorgi JV. 1993. Evaluation of a dual-color flow cytometry immunophenotyping panel in a multicenter quality assurance program. Cytometry 14:307–317. 10.1002/cyto.990140311 [DOI] [PubMed] [Google Scholar]
- 49. Cerimele F, Curreli F, Ely S, Friedman-Kien AE, Cesarman E, Flore O. 2001. Kaposi’s sarcoma-associated herpesvirus can productively infect primary human keratinocytes and alter their growth properties. J. Virol. 75:2435–2443. 10.1128/JVI.75.5.2435-2443.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stamey FR, Patel MM, Holloway BP, Pellett PE. 2001. Quantitative, fluorogenic probe PCR assay for detection of human herpesvirus 8 DNA in clinical specimens. J. Clin. Microbiol. 39:3537–3540. 10.1128/JCM.39.10.3537-3540.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Qu L, Triulzi DJ, Rowe DT, Jenkins FJ. 2011. Detection of HHV-8 (human herpesvirus-8) genomes in induced peripheral blood mononuclear cells (PBMCs) from US blood donors. Vox Sang. 100:267–271. 10.1111/j.1423-0410.2010.01404.x [DOI] [PubMed] [Google Scholar]
- 52. Niesters HG. 2002. Clinical virology in real time. J. Clin. Virol. Off. Publ. Pan American Society For Clinical Virology 25(Suppl 3):S3–12. 10.1016/S1386-6532(02)00026-4 [DOI] [PubMed] [Google Scholar]
- 53. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789. 10.1182/blood-2005-12-4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanghavi SK, Reinhart TA. 2005. Increased expression of TLR3 in lymph nodes during simian immunodeficiency virus infection: implications for inflammation and immunodeficiency. J. Immunol. 175:5314–5323. 10.4049/jimmunol.175.8.5314 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flow cytometry polyfunctional gating strategy. Singlet, live lymphocyte populations were determined and first gated (data not shown). An IgG2B control conjugated with AF-680 (not shown) was used to set the gate for ORF59 PF-8-positive and -negative cells. No differences in autofluorescence were seen between the 2 cell populations. (A) Unstained live, singlet CD19+ lymphocytes were used to set the gate for cytokine and chemokine markers. HHV-8-exposed ORF59 PF-8neg (B) and ORF59 PF-8pos (C) cells were gated against each cytokine and chemokine. Download
Flow cytometry B cell lineage subset gating strategy. Live, singlet populations were detected as previously described. (A) ORF59 PF-8neg/pos cells were selected and gated against CD20 and CD138 expression. CD20± CD138+ cells (dashed arrow) were further gated against CD38. Plasma cells were considered CD20± CD138+ CD38±. (B and C) CD20+ CD138¯ cells (solid arrow) were further gated against CD19 and CD27 expression. CD19+ CD27+ and CD19+ CD27¯cells were gated against IgD and IgM. Naive cells were CD19+ CD20+ IgM+ IgD+ CD27¯ CD138¯ (B), and IgM memory cells were considered CD19+ CD20+ IgM+ IgD± CD27+ CD138¯ (C). Download