

Abstract
How intestinal epithelial cells recognize pathogens and activate inflammasomes at intestinal surfaces is poorly understood. We hypothesized that intestinal epithelial cells utilize integrin receptors to recognize pathogens and initiate inflammation within the intestinal tract. We find that intestinal epithelial cells infected with Yersinia enterocolitica, an enteric pathogen, use β1 integrins as pathogen recognition receptors detecting the bacterial adhesin invasin. The invasin-integrin interaction provides the first signal for NLRP3 inflammasome activation with the type three secretion system translocon providing the second signal for inflammasome activation resulting in release of IL-18. During infection, Yersinia employs two virulence factors, YopE and YopH, to counteract invasin-mediated integrin-dependent inflammasome activation. Further, NLRP3 inflammasome activation in epithelial cells requires components of the focal adhesion complex signaling pathway, focal adhesion kinase and rac 1. The binding of invasin to β1 integrins rapidly induces IL-18 mRNA expression suggesting integrins provide a first signal for NLRP3 inflammasome activation. These data suggest integrins function as pathogen recognition receptors on intestinal epithelial cells to rapidly induce inflammasome-derived IL-18-mediated responses.
Introduction
Mucosal surfaces are exquisitely sensitive to inflammation-mediated immune pathologies, necessitating strict regulation of inflammatory responses (1, 2). To minimize unwanted inflammation, epithelial cells in environments with high microbial burdens such as the intestine, selectively express pathogen-associated molecular pattern receptors (PAMPR) on the basolateral surface or in endosomal compartments (3). Additionally, production of inflammatory cytokines interleukin-1β (IL-1βand IL-18 are tightly regulated transcriptionally by PAMPRs and post-translationally by the inflammasome (4–7).
Inflammasomes are macromolecular machines that promote activation of caspase-1 in response to PAMP molecules, cellular stress, or cellular damage (8). Ultimately, inflammasome activation leads to the initiation of inflammatory signaling through cleavage of pro IL-1β and IL-18 to their active forms and secretion of these cytokines from the cell. Inflammasomes are usually composed of a sensor protein from the nucleotide-binding domain and leucine-rich repeat protein family (NLR), an adaptor protein such as apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1. The activation of inflammasomes are thought to involve two signals; the first signal results from PAMP recognition and leads to the increased expression of the inflammasome components and substrate cytokines; various cellular insults such as pore formation can provide the second signal for the NLRP3 inflammasome resulting in caspase-1 cleavage and cytokine release. Inflammasome activation is best studied in macrophages; however, several cell types can produce IL-1β and IL-18. Intestinal epithelial cells (IECs) do not express IL-1β but they do express pro-IL-18 and IL-18 derived from IECs and hematopoietic cells is known to protect against colitis and colorectal cancer in mouse models (9–11).
The intestinal epithelium resides in a unique immunological environment where it is potentially in contact with microorganisms constituting the normal flora. Additionally, the intestine is a major portal for infectious diseases, suggesting IECs evolved mechanisms to distinguish between innocuous flora and dangerous pathogens (1). The mechanisms underlying inflammasome activation in response to infection of IECs are not understood.
However, a critical step in the pathogenesis of food borne bacterial pathogens is attachment or attachment and invasion of IECs (12). Some of the best-characterized invasive pathogens of the intestine are Yersinia enterocolitica and the closely related Y. pseudotuberculosis.
Y. enterocolitica is a zoonotic bacterial food borne pathogen of humans causing terminal ileitis, entero-colitis, and mesenteric lymphadenitis (13, 14). To attach to and invade intestinal tissues, Yersinia expresses an adhesin called invasin (Inv) (15, 16). Inv binds to β1 -integrins predominantly expressed on micro-fold epithelial cells (M-cells) overlying Peyer’s Patches (PP) (17). Inv-mediated integrin binding facilitates invasion of the intestinal epithelium and PP colonization. Once Y. enterocolitica establishes infection of the PP, it is an extracellular pathogen that utilizes numerous virulence factors to modulate host responses to infection (18, 19).
Y. enterocolitica encodes virulence factors on both the chromosome and the 70kb virulence plasmid, pYV (14, 19). pYV encodes several effector toxins and a type three secretion system (TTSS) that provides a conduit to secrete these effectors from the bacterial cytosol directly into the host cell cytoplasm (19, 20). The TTSS translocon proteins form a pore in the host cell membrane serving as a signal for NLRP3 inflammasome activation and inflammatory cytokine secretion from macrophages (21–24). The effectors known as Yersinia outer proteins (Yops) are mostly enzymes; for example, YopH is a protein tyrosine phosphatase that dephosphorylates focal adhesion kinase (FAK), p130 cas, and other components of the focal adhesion complex to disrupt the actin cytoskeleton in epithelial cells (25–30). YopE, a GTPase activating protein (GAP), targets signaling of the small G-proteins Rac, Rho, and Cdc42 (31). Both YopH and YopE act together to inhibit β1 -integrin signaling. YopP/J a protease and protein acetylase has been implicated in modulation of the inflammasome in macrophages through inhibition of NF-κB. Yops also inhibit cytokine expression as a means of immune evasion (23, 24, 32–35). The molecular mechanisms utilized by other Yops, such as YopM, have yet to be completely defined. Interestingly, YopM in the closely related pathogens Yersinia pseudotuberculosis and Y. pestis does not appear to be an enzyme but is a protein that binds and sequesters host-signaling proteins including caspase-1. In activated macrophages, YopM can be a potent inhibitor of the inflammasome through its interaction with caspase-1 (24). However, mechanisms of Y. enterocolitca-derived inflammasome signaling in IECs and the role of the Yops in this process are completely unknown.
In the current study, we identify the mechanism of NLRP3 inflammasome-activation in IECs. We also describe a novel first-signal signaling pathway for NLRP3 inflammasome activation in IECs requiring signaling through the focal adhesion-signaling complex. We determine Yops E and H as the virulence factors counteracting integrin-mediated first signals and inflammasome activation in IECs. These data represent a novel pathway providing the first signal for inflammasome activation in non-myeloid cells.
Materials and Methods
In-frame Yop deletions of Y. enterocolitica
All Y. enterocolitica strains used in this project were derived from JB580V (Table S1). In frame deletions of YopE, YopH, and YopQ were generated through homologous recombination by using the 500bp upstream and downstream sequences of each gene. The up and downstream sequences were first amplified by PCR from the virulence plasmid pYVe8081 with pre-designed primers (Table SII). These DNA fragments were cloned into pCR2.1-TOPO vector by TOPO TA cloning (K4500) from Invitrogen according to manufacturer’s instructions. Primers were made with 5′ overhangs of SalI and PacI restriction site sequences (underlined), which allowed for the 500bp up and downstream fragments to be cloned into a single pCR2.1-TOPO vector. Subsequently, the ~1kb DNA fragment was cloned into the SalI and NotI sites of the suicide vector pSR47s and introduced into E. coli s17λpir mating strain. Mating between JB580V and s17λpir conjugated the suicide plasmid into JB580V. Kanamycin and nalidixic acid resistance selected for recombination of the suicide plasmid into the virulence pYV plasmid. The loss of the integrated pSR47s from the kanamycin and Nalidixic acid resistant JB580V clones was achieved by selecting for sucrose resistance. PCR and SDS-PAGE coomassie blue staining of all secreted Yops verified the successful deletion of each Yop.
Invasin single amino acid substitutions
Single amino acid substitutions to generate E. coli expressing Inv D760A and Inv D809A were performed through the QuikChange Site-Directed Mutagenesis Kit (Strategene) according to the manufacturer’s instructions. The mutagenic oligonucleotide primers used to generate the substitution are listed on (Table SII).
RT-PCR
Caco-2 cells were pretreated for 1h with 5 μM PF-431396 and 100 μM NSC 23766 to inhibit FAK and rac1 respectively. Control cells were treated with DMSO (solvent). Cells were infected with the indicated strain of Y. enterocolitica for 30 mins and then total RNA was extracted using the trizol method. Two micrograms of total RNA was reverse transcribed to cDNA. Following cDNA synthesis, RT-PCR was performed for caspase-1, nlrp3, and GAPDH with the resulting product fractionated by agarose gel electrophoresis.
Cells and infection
human enterocyte cell line Caco-2 were cultured at 37°C with 5% CO2 in 20% FBS MEM medium. For infections, cells were grown to confluence (four days) in 12- or 6-well dishes. Yersinia (Table SII) were grown overnight in LB medium containing 20μg/ml Nalidixic acid at 28°C. The cultures were then diluted 1:100 into fresh media and grown 37°C for 2 hrs. Salmonella were grown overnight in LB medium 37°C, diluted into fresh LB medium, and grown standing for 3.5 hr at 37°C. All bacteria were washed with PBS and diluted into OPTI-MEM at an OD of 0.2 before infecting Caco-2 cells at an MOI of 10 or as otherwise specified. After 1hr of infection, 20μg/ml of gentamycin was added to kill extracellular bacteria and maintained throughout the course of the experiment. E. coli HB101 strains (Table SII) were also grown at 37°C overnight in LB medium under 100μg/ml ampicillin selection then diluted and grown at 37°C for an additional 1 hr. Caco-2 cells were infected with E. coli strains for 2 hr and subsequently treated with 25μM nigericin (Sigma Aldrich) and 20μg/ml of gentamycin for the reminder of the infection time course.
Chemical inhibition studies
Caco-2 cells cultured on 12 or 6-well plates were pre-treated for 1h with 100μM of the irreversible caspase-1 inhibitor Ac-YVAD-cmk from Bachem. Potassium efflux was blocked with either 50μM of the ion-channel blocker glibenclamide or 130mM KCl both from Sigma Aldrich. For Rac 1 and FAK inhibition, 50 or 100μM of the Rac 1 inhibitor NSC23766 (EMD Millipore) and the indicated concentrations of the FAK inhibitor PF-431396 (Sigma Aldrich) were used. Solvents used were water for NSC23766 and DMSO for the remaining inhibitors; solvents also served as the negative (vehicle) controls. After pre-treatment with the inhibitors, cells were infected as indicated for 8hr with inhibitors maintained in culture throughout the infection.
IL-18 ELISA
assayed cell culture supernatants as we have described before (32). Briefly, anti-human IL-18 antibody (D044-3) was used as the capture antibody and its paired clone biotin-labeled anti-human IL-18 antibody (D045-6) as the secondary antibody, all purchased from MBL International (Woburn, MA). Each sample was assayed in triplicate and the experiment repeated independently two-four times.
IL-18 Bioassay
we assessed the production of IFN-γ from KG-1 cells, a human myelomonocytic cell line, stimulated by conditioned media from Caco-2 cells. KG-1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS in 96-well plates seeded at 2×105 cells/well. Cell culture supernatants from Caco-2 cells infected with Y. enterocolitica strains or S. typhimuirum were added onto the KG-1 cells. Recombinant mature IL-18 (1ng/ml) was also added as a positive control. After 24 hr incubation, media was analyzed for the concentration of IFN-γ using the Human IFN-γ ELISA from BD Bioscience.
Immunoblots
of cell lysates or supernatants was used to monitor protein expression and the cleavage products of IL-18 and capase-1. Cells were lysed in RIPA buffer (150mM NaCl, 50mM Tris (pH 8), 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS) supplemented with protease inhibitor cocktail (RPI corp.) and 2 mM phenylmethanesulfonyl fluoride. Supernatant proteins were precipitated by the methanol-chloroform extraction method as described (36). Protein concentration in the cell lysate was determined using bicinchoninic acid (BCA) assay kit. Equal concentrations of protein were separated through 10–13.5% denaturing SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were incubated overnight at 4°C with primary antibodies diluted (1:1000) against: human activated caspase-1 p20, β1 integrin, FAK, and phospho-FAK all from Cell Signaling Technology. Other antibodies used were against human IL-18 from Santa Cruz Biotechnology; against Rac1 from Epitomics, a loading control antibody against Hsp70 from Santa Cruz Biotechnology. Blots were then washed as before and protein bands visualized by chemiluminescence. Relative caspase-1 protein levels were determined by densitometric analysis of Western blot bands using a Molecular Imager Gel Doc XR System (BioRad, Hercules, CA).
β1 integrin blocking
Confluent Caco-2 cells were treated with 0.5mM of inhibitory peptide cyclo-GRGDSP or a non-blocking control peptide cyclo-GRGESP, from Anaspec. Integrins were blocked with the indicated concentrations of anti-human integrin α5β1 mAb, JBS5, from EMD Millipore, a control antibody against integrin α5 chain clone V5 (BD Bioscience) or mIgG from Sigma Aldrich. Prior to infection, Caco-2 cells were pre-treated with antibodies for 30 minutes at room temperature, then infected and incubated at 37°C for the remaining time-course.
RNA Interference
Caco-2 cells cultured to 50% confluency, then transfected with siRNAs using Lipofectamine RNAiMAX (Invitrogen) according to manufacturer’s instructions. siRNAs included 50nM cryopyrin NLRP3 ((sc-45469) Santa Cruz Biotechnology); 100nM FAK 1 siRNA and control siRNA (Cell Signaling Technology); ON-TARGETplus SMARTpool siRNA (100nM) against β1-integrin (L-004506-00-0005), and control siRNA (D-001810–10-05) from (Dharmacon, Thermo Scientific, USA). Rac1 knockdown was achieved by transfecting 0.5ug/well of Rac 1 shRNA vector (pRNAT.mCherry shRac1) or control (pRNAT.mCherry shScr) for 48–72hrs before infection with bacteria. The knockdown of transcription was verified by qRT-PCR or immunoblot analysis.
Quantitative RT-PCR
qRT-PCR reactions were performed with TaqMan gene expression assays according to the manufacturer’s instructions and the reaction performed using the StepOnePlus Real-Time PCR System (Applied Biosystems). Probes from Life Technologies were used: NLRP3: Hs00918082 m1, IL-18: Hs01038788 m1, and GAPDH: Hs99999905-m1. Expression levels of the target transcripts in each sample were calculated by the comparative Ct method after normalization to GAPDH expression. Each sample was analyzed in triplicate and repeated in three independent experiments.
Statistical Analysis
ANOVA was performed using the Prism 4 software package (GraphPad, La Jolla, CA). Data are presented as means ± the standard deviation with p values ≤ 0.05 considered significant.
Results
Y. enterocolitica YopE and YopH inhibit secretion of active IL-18 from IECs
In mouse models of yersiniosis, IL-18 is critical in the host response to Y. enterocolitica infection (37). To investigate if IL-18 secretion is modulated during Y. enterocolitica infection of IECs, we infected the human enterocyte cell line Caco-2 with Y. enterocolitica or S. typhimurium as a positive control. Wild-type bacteria failed to induce the secretion of IL-18 (Figure 1A), but we hypothesized that Y. enterocolitica employs one or more Yop(s) to disrupt the production and secretion of active IL-18. IECs were infected with Yop mutants and ELISA determined the concentrations of IL-18 secreted into the culture supernatants. IL-18 secretion was detected from cells infected with the yopE mutant. In contrast to the yopE mutant, infection of cells with the yopH or other Yop mutants did not result in detectable IL-18 secretion. However, a yopEH mutant induced robust secretion of IL-18 (Figs. 1A and 1B). These findings indicate YopH and YopE act synergistically to inhibit secretion of IL-18 from IECs. In the closely related pathogen, Y. pseudotuberculosis, the YopK protein was reported to inhibit activation of caspase-1 and IL-1β secretion from bone marrow-derived macrophages (23). We found the Y. enterocolitica yopQ mutant (homologous to YopK) did not alter secretion of IL-18 suggesting differences in inflammasome modulation between IECs and macrophages (Fig. 1). Notably, Y. enterocolitica JB580c, a strain lacking the pYV virulence plasmid encoding the Yops and TTSS machinery, is unable to stimulate IL-18 (Fig. 1). This finding suggests that other properties of the TTSS such as the translocon are required for IL-18 secretion during Y. enterocolitica infection of IECs (22).
Figure 1. Secretion of mature IL-18 from intestinal epithelial cells during Y. enterocolitica infection is synergistically inhibited by YopE and YopH.

(A) Caco-2 cells were infected with the indicated Yop mutants, WT (JB580V) and pYV-plasmid cured (JB580C) strains of Y. enterocolitica or with S. typhimurium (STm) as a positive control. ELISA determined the concentration of IL-18 in the culture supernatants. (B) Caco-2 cell supernatants and lysates subjected to immunoblotting with anti-IL-18 antibody or Hsp70 antibody as loading control. (C) The bioactivity of IL-18 was assessed by exposing KG-1 cells, which produce IFN-γ in the presence of mature IL-18, to conditioned media of infected Caco-2 cells or 1ng/ml of recombinant mature IL-18. IFN-γ secreted by KG-1 cells was analyzed by ELISA. (D) Caco-2 cells were infected with the indicated bacteria and then ELISA determined IL-1β concentrations in cell supernatants. In all samples IL-1β concentrations were below the level of detection for the assay as indicated by the solid line (4 pg/ml). Data are presented as mean ± SD of three independent experiments. **p<0.001; ***p<0.0001
IL-18 bioactivity was tested by assaying the ability of conditioned media from cells infected with the Y. enterocolitica mutants to induce the production of interferon-γ (IFN- γ) from KG-1 cells (38). As shown in Figure 1C, IFN- γ production was induced by the supernatants of cells infected with the yopEH mutant but not with supernatants from the other Yersinia strains tested. These results indicate that in the absence of Yops E and H, Y. enterocolitica induces the secretion of mature IL-18.
Activation of the inflammasome in macrophages leads to the maturation of IL-1β However, literature suggests that IECs produce IL-18 but do not produce IL-1β (39). To test the ability of IECs to produce IL-1β in response to Yersinia infection, we infected Caco-2 cells with the various strains of Yersinia or Salmonella as a positive control. As shown in Figure 1D, Caco-2 cells do not secrete appreciable levels of IL-1β under our infection conditions with all samples having IL-1β concentrations below the limit of detection for the assay.
IL-18 secretion is dependent on caspase-1 and the NLRP3 inflammasome
Maturation of IL-18 requires caspase-1 proteolytic activity. To test if IL-18 secreted during Y. enterocolitica infection was dependent on caspase-1, we treated cells with an irreversible caspase-1 inhibitor (Ac-YVAD-cmk) prior to infection and then monitored maturation of IL-18. IL-18 maturation was dramatically reduced in the presence of inhibitor indicating caspase-1 activity was required for IL-18 maturation (Figs. 2A and B). The reduction in mature IL-18 corresponded with a reduction in the active form of caspase-1 p20 from IECs infected with Yersinia strains or Salmonella. Corresponding to reductions in cleaved IL-18 in the supernatant following caspase-1 inhibitor treatment, inhibitor treatment reduced caspase-1 band intensity from 0.19, 0.40, and 0.48 arbritary units (au) in the solvent treated cells to 0.12, 0.02, and 0.17 au in the inhibitor treated cells for JB580v, yopEH and Salmonella respectively (Fig. 2B). Altogether these data suggests that production of mature IL-18 from IECs requires active caspase-1.
Figure 2. IL-18 secretion requires active caspase-1 and the NLRP3 inflammasome.

(A) Caco-2 cells were pretreated for 1hr with 100μM of the caspase-1 inhibitor prior to infection with the indicated Y. enterocolitica strains or S. typhimurium (STm). Cell supernatants were evaluated for IL-18 secretion by ELISA eight hours post infection. (B) Levels of pro caspase-1, IL-18, and Hsp70 in the cell lysates and mature IL-18 and caspase-1 p20 in cell supernatants were detected by immunoblotting. (C, D) Caco-2 cells were transfected with NLRP3 siRNA or control non-targeting siRNA for 72hrs. (C) The knockdown of the Nlrp3 transcript was confirmed by quantitative RT-PCR. (D) siRNA-transfected cells were infected with wild-type (JB580V) and yopEH mutant strains of Y. enterocolitica and supernatants were evaluated for IL-18 secretion by ELISA. Data are presented as mean ± SD of two-three independent experiments. *p<0.05; ***p<0.0001. See also Fig. S1.
The NLRP3 inflammasome is activated in macrophages infected with Yersinia (23, 24, 35). To investigate NLRP3 inflammasome activation in IECs, we utilized siRNA-targeting nlrp3 and verified significant siRNA-mediated knockdown of nlrp3 transcript (Fig. 2C). When Caco-2 cells were transfected with NLRP3-specific siRNA followed by infection with the yopEH mutant, the secretion of IL-18 was markedly decreased relative to cells transfected with non-targeting siRNA (Fig. 2D).
Bacterial pore-forming toxins can trigger NLRP3 inflammasome activation (40) through efflux of intracellular potassium. To examine K+ efflux in Y. enterocolitica-induced inflammasome activation, we infected Caco-2 cells in the presence of 130mM extracellular KCl which led to a 50% reduction in secretion of IL-18 from cells infected with the yopEH mutant (Fig. S1). To further test the role of K+, we pretreated cells with glibenclamide to block potassium efflux. In agreement with the KCl results, infection of Caco-2 cells with the yopEH mutant in the presence of 50μM glibenclamide resulted in a two-fold decrease in secreted IL-18 (Fig. S1). These findings suggest Y. enterocolitica infection of Caco-2 cells causes K+ efflux.
Invasin binding to α5β1 integrins promotes IL-18 secretion
The major adhesin of Yersinia, Inv, binds β1 integrins aiding in contact-dependent TTSS and facilitates phagocytosis (41). The injection of Yops E and H by the TTSS disrupts signaling downstream of the β1 integrin inhibiting phagocytosis (19). We hypothesized that Inv-activated β1 integrin signaling acts as a primary trigger for inflammasome activation in IECs. Therefore, we infected Caco-2 cells with a Y. enterocolitica yopEH inv triple mutant and found that the absence of Inv resulted in 40-fold decrease in IL-18 secretion compared to cells infected with the yopEH mutant expressing Inv (Fig. 3A). There was a minute but detectable level of IL-18 produced by cells infected by the yopEH inv triple mutant but IL-18 was undetectable with the YopEH sufficient inv mutant.
Figure 3. Invasin binding to α5β1 integrins activates caspase-1 and induces IL-18 secretion.

(A) Caco-2 cells were infected with wild-type (JB580V), yop mutants and inv mutant strains or were left uninfected (NI). Eight hours post infection; ELISA determined IL-18 concentrations. (B) Caco-2 cells were treated with nigericin or vehicle after 2hrs of infection with JB580C or the JB580C inv mutant. Cell supernatants were assessed for IL-18 levels. (C) Caco-2 cells transfected with β1 integrin siRNA for 72hrs were infected with wild type and yopEH mutant strains of Yersinia or S. typhimurium (STm). β1 integrin, caspase-1, IL-18 and Hsp70 in the cell lysates and caspase-1 (p20) in the cell supernatants were assessed by immunoblotting. (D–E) Caco-2 cells were pretreated with (D) a blocking α5β1 integrin antibody (JBS5) or a non-blocking α5β1 integrin antibody at the indicated concentrations or E) 0.5mM of cyclo-GRGDSP and cyclo-GRGESP peptides for 30 minutes prior to infection. ELISA measured IL-18 concentrations in cell supernatants. Data are presented as mean ± SD of two-three independent experiments. *p<0.01; **p<0.001; ***p<0.0001.
We hypothesized that if pore-formation were present as a second signal, then the pYV plasmid-cured strain (JB580c) would trigger inflammasome activity. Thus, we infected Caco-2 cells with JB580c in the presence of a pore-forming toxin, nigericin (40). Consistent with our hypothesis, JB580c infection coupled with nigericin treatment induced dose-dependent IL-18 secretion (Fig. 3B). Nigericin or JB580c alone failed to induce significant IL-18 secretion, even at a high multiplicity of infection (MOI), suggesting that individually they are insufficient to trigger inflammasome activity (Figs. 3B and 4A). To further test the necessity of Inv and pore formation, we deleted inv in the JB580c background and assessed its ability to induce IL-18 secretion from Caco-2 cells in the presence of nigericin. Infection with the JB580c inv mutant failed to induce IL-18 secretion in either the presence or absence of nigericin or with increasing bacterial MOI (Figs. 3B and 4A). These data suggests that Y. enterocolitica-mediated induction of IL-18 secretion from IECs is dependent on at least two properties: Inv activity and pore-formation.
Figure 4. Invasin-mediated dose dependent changes in IL-18 secretion.

A) Caco-2 cells were infected for 2 hours with JB580C or the JB580C inv mutant at the indicated MOI. After 2hrs, cells were treated with 25μM of the pore-forming toxin nigericin, and after 30 minutes, 20μg/ml of gentamycin to kill extracellular bacteria. ELISA determined the concentration of IL-18 in the conditioned media. Data represents mean ± SD of triplicate samples from two independent experiments. *p < 0.01; ** p < 0.001; ***p <0.0001. B) Caco-2 cells were infected with E. coli carrying an empty vector or expressing WT Y. enterocolitica invasin at the indicated MOI for 2hrs and then cells were treated with 25μM of nigericin. ELISA determined the concentration of IL-18 in the conditioned media. Data represents mean ± SD of three independent experiments. *p < 0.05; **p<0.01.
Since our data suggests that Inv provides the first signal for activation of the inflammasome, we tested if β1 integrin receptors are a component of the pathogen detection and inflammasome-signaling pathways in epithelial cells. We silenced expression of β1 integrins using siRNA and tested if integrins stimulate caspase-1 cleavage and IL-18 secretion. Caco-2 cells transfected with β1 integrin siRNA and then infected with the yopEH mutant, showed a five-fold decrease in the concentration of secreted IL-18 and a decrease in cleaved forms of caspase-1 (p20 subunit) (Fig. 3C). When caspase-1 p20 band intensity on the immunoblot was quantified, cells transfected with control siRNA had 0.008, 0.13, and 0.09 au whereas β1 integrin siRNA treated cells had 0.007, 0.003, and 0.11 au for JB580v, yopEH and Salmonella respectively. Altogether these data suggests that β1 integrins are important for caspase-1 activation and IL-18 production from IECs infected with Yersinia following yopEH mutant infection but have little impact on IECs infected with JB-580v or Salmonella.
Although Inv binds to several β1 integrins, Inv preferentially binds α5β1 integrins in the intestinal tract, which are predominantly expressed on IECs (42, 43). To test if Inv binding to α5β1 integrins triggers inflammasome activity, we neutralized α5β1 integrins on Caco-2 cells with an anti- α5β1 integrin-blocking antibody. When cells were infected with the yopEH mutant, the antibody inhibited IL-18 secretion in a dose-dependent manner with a 75% reduction in secreted IL-18 at the highest antibody concentration (Fig. 3D). Treatment of Caco-2 cells with a non-neutralizing α5β1 integrin antibody did not alter secretion of IL-18 during infection (Fig. 3D). Further, RGD-containing peptide (cyclo-GRGDSP), a synthetic α5β1 integrin ligand, competitively inhibits Inv binding to α5β1 integrins expressed on Caco-2 cells by two-fold whereas an RGE-containing control peptide did not affect IL-18 secretion (Fig. 3E).
IECs secrete IL-18 when treated with nigericin and infected with E. coli expressing Y. enterocolitica Inv on their surface
We wanted to rule out other significant contributions from PAMPs present in and on Yersinia. Therefore, we ectopically expressed inv in a nonpathogenic strain of E. coli and tested the ability of Inv expressed on E. coli to mediate IL-18 secretion from IECs. Importantly, using nonpathogenic E. coli expressing Inv allowed us to test the ability of Caco-2 cells to respond to common PAMPs present on other common Gram-negative bacteria that are part of the normal flora.
Caco-2 cells were infected with E. coli-expressing Y. enterocolitica inv (E. coli +Inv), E. coli strain carrying an empty vector (E. coli vector), and JB580c, in the presence or absence of nigericin. E. coli +Inv and JB580c induced IL-18 secretion 25 and 4-fold, respectively, when compared with uninfected cells and controls (Fig. 5A). IL-18 secretion was MOI-dependent when cells were infected with E. coli +Inv and JB580c, but not with E. coli +vector (Figs. 4A and B respectively). IL-18 secretion during E. coli +Inv infection was abrogated by the α5β1 integrin neutralizing antibody and reduced by over two-fold by cyclo-GRGDSP (Figs. 5B and 5D). These data suggest that Inv, when coupled with pore-formation, is sufficient to stimulate IL-18 secretion in IECs. Importantly, these data also suggest that IECs are not responding to PAMPs other than Inv as a means to detect the presence of pathogenic bacteria.
Figure 5. Invasin-mediated α5β1 integrin-dependent IL-18 secretion from nigericin treated IECs.

(A–B, D) Caco-2 cells were infected for 2 hours with E. coli carrying an empty vector (E. coli vector) or expressing Y. enterocolitica Inv (E. coli + inv) at an MOI of 200 and JB580C at an MOI of 10 or (C) E. coli expressing inv with single point mutations D760A or D809A. (B, D) Caco-2 cells were pretreated with: (B) 7.5μg/ml of blocking α5β1 integrin antibody (JBS5) or a non-blocking α5β1 integrin antibody or (D) 0.5mM of cyclo-GRGDSP and cyclo-GRGESP peptides for 30 minutes. (A–D) After infection for 2hrs, cells were then treated with 25μM of nigericin. The conditioned medias were subjected to IL-18 ELISA. Data represents mean ± SD of two-three independent experiments. *p < 0.01; **p < 0.001; ***p < 0.0001.
Inv from Y. pseudotuberculosis contains an aspartic acid at position 911 that is essential for integrin binding (44, 45). This critical amino acid is at position 760 in Y. enterocolitica Inv. We tested if D760 was necessary for IL-18 production and secretion by infecting cells with an Inv-D760A point mutant. We also infected IECs with a control Inv mutant, D809A, an aspartic acid predicted to be on the same face of Inv as D760, but not involved in integrin binding (45). In comparison to IECs infected with E. coli expressing wild type Inv, IL-18 secretion decreased by over 2-fold when cells were infected with E. coli Inv D760A (Fig. 5C). The Inv D809A mutant was still capable of inducing IL-18 to similar levels as E. coli expressing wild type Inv. Altogether, these data suggest that Inv binding to integrins through the critical amino acid D760 is necessary for the induction of IL-18 secretion.
FAK and Rac 1 are involved in inflammasome signaling and IL-18 production in IECs
Our data suggests Y. enterocolitica utilizes Yops H and E to disrupt integrin signaling to attenuate caspase-1 activity and IL-18 secretion from IECs. β1 integrin binding by Inv causes receptor clustering, triggering the assembly of focal adhesion complexes and tyrosine kinase activity (41). FAK is a major target of YopH (27, 29). We hypothesized FAK contributes to the integrin signal activating the inflammasome in IECs. To test FAK, we assessed IL-18 secretion from IECs treated with increasing concentrations of PF-431396, an inhibitor that blocks FAK phosphorylation. Following infection with the yopEH mutant, we found that phosphorylation of FAK was decreased with inhibitor concentrations starting at 1 μM corresponding to a dose-dependent decrease in IL-18 secretion (Fig. S2). Cells were pretreated with increasing concentrations of PF-431396 and infected with wild type Y. enterocolitica, the yopEH mutant, and as a positive control S. typhimurium. Each incremental increase in inhibitor concentration decreased IL-18 secretion by about 2-fold from cells infected with the yopEH mutant whereas IL-18 secretion from cells infected with S. typhimurium or wild type Yersinia was not impacted (Fig. 6A). To further test FAK, we used RNAi to deplete FAK prior to infection. Caco-2 cells were transfected with siRNA targeting FAK as well as control siRNA. Following FAK depletion, cells were infected with wild type Y. enterocolitica, the yopEH mutant, or S. typhimurium. Analysis of the culture media indicated that knock-down of FAK decreased IL-18 concentrations by 3-fold in comparison to control siRNA-transfected cells, but had no significant effect on IL-18 secreted by cells infected with S. typhimurium (Fig. 6B). These data suggest FAK signaling is involved in inducing the secretion of IL-18 during Y. enterocolitica infection with the yopEH mutant.
Figure 6. FAK and Rac 1 are involved in the secretion of IL-18.

Caco-2 cells were pretreated for 1hr with increasing concentrations of: (A) the FAK inhibitor PF-431396 or C) the Rac 1 inhibitor NSC23766 or (E) with 5μM of PF-431396, 100μM of NSC23766, a combination of either inhibitors or the vehicle (DMSO). (A, C, E) Cells were then infected with indicated bacterial strains at an MOI of 10. ELISA determined the concentration of IL-18 in the culture media. (B) Caco-2 cells were transfected with 100nM of FAK siRNA or control siRNA for 72 hours prior to infection. Cell supernatants were analyzed for IL-18 and lysates subjected to immunoblot analysis of phospho-FAK, total-FAK and the loading control Hsp70. (D) Caco-2 cells were transfected with 0.5ug of Rac1 shRNA vector or a control vector expressing scrambled shRNA for 48 hours. Cells were infected and processed as in (B) and Rac 1 was assessed by immunoblotting. Data represents mean ± SD of three independent experiments. *p < 0.01; ***p<0.0001. See also Fig. S2.
Inv-mediated integrin signaling activates Rho GTPases such as Rac 1 to regulate actin cytoskeleton rearrangements. YopE is an inhibitor of Rac1, Cdc42, and RhoA activity (31, 33). To test Rac1 in inflammasome activation, we used the Rac 1 inhibitor NSC23766, which does not affect the activity of Cdc42 and RhoA (46). Caco-2 cells were treated with increasing concentrations of NSC23766 prior to infection. Concentrations of secreted IL-18 following infection with the yopEH mutant decreased in a dose-dependent manner resulting in a 2-fold decrease at the highest inhibitor concentration (Fig. 6C). Additionally, Caco-2 cells were transfected with shRNA plasmid targeting rac1. As shown in Figure 6D, yopEH mutant infection of cells after rac 1 knockdown reduced IL-18 by 2-fold relative to controls. Rac1 inhibition or shRNA knockdown did not significantly inhibit IL-18 secretion during S. typhimurium infection (Figs. 6C and D). Since we did not observe complete inhibition of IL-18 secretion in the presence of Rac1 inhibitor or shRNA, our data does not rule out other YopE targets in IL-18 secretion including RhoA (47).
Yops E and H synergistically inhibit inflammasome activity. We tested if this synergism could be chemically complimented by inhibiting FAK and Rac 1 activity prior to yopEH mutant infection. Caco-2 cells were pre-treated with either FAK inhibitor (5μM) or Rac 1 inhibitor (100μM) or a combination of the two inhibitors. In comparison to vehicle treated cells, FAK inhibitor alone dramatically reduced secreted IL-18 4-fold relative to the rac 1 inhibitor, which reduced IL-18 concentrations 2-fold (Fig. 6E). The combination of the two inhibitors reduced IL-18 secretion to levels just above the limit of detection corresponding to a 50-fold decrease in IL-18 concentrations relative to controls (Fig. 6E). These data suggest FAK and Rac1 act synergistically to transduce first signals from integrins to the inflammasome.
IL-18 expression is regulated by integrin signaling in IECs
We investigated if integrin signaling provides a first signal for inflammasome activation in IECs. Caco-2 cells infected with wild type or the yopEH mutant were evaluated for changes in expression of casp1, nlrp3 and IL-18 mRNA. Expression of casp1 mRNA was not modulated at the early time points tested and remained similar to uninfected cells (data not shown). Expression of nlrp3 was inhibited by FAK and rac1 inhibitors in mock-treated IECs, wild type Yersinia infected cells, and yopEH mutant infected IECs relative to untreated cells demonstrating decreased basal nlrp3 mRNA expression but mRNA levels were similarly decreased in all treatment groups (data not shown). These data suggest that integrin binding to matrix likely leads to basal nlrp3 expression but that infection does not modulate nlrp3 expression at the early time points tested. However, there was an early induction (0.5hpi) in IL-18 mRNA in cells infected with the yopEH mutant. This induction resulted in a 2-fold increase in IL-18 mRNA relative to cells infected with wild type Yersinia and uninfected cells. Wild type Y. enterocolitica failed to augment the expression of IL-18 mRNA resulting in IL-18 transcript levels similar to uninfected cells (Fig. 7A). To test if integrin signaling was responsible for up-regulating IL-18 mRNA expression, we pretreated cells with either JBS5 α5β1 neutralizing antibody or α5β1 non-neutralizing antibody. Caco-2 cells pretreated with JBS5 antibody and infected with the yopEH mutant showed a 2-fold decrease in IL-18 expression relative to the non-neutralizing antibody. In the presence of JBS5 antibody, IL-18 mRNA levels from infected cells remained the same as from uninfected cells (Fig. 7B). We tested FAK and rac1 in the expression of IL-18 mRNA by pretreating cells with the FAK and rac1 inhibitors and then the levels of IL-18 mRNA were evaluated after 30 minutes of infection. IL-18 transcript levels during infection with the yopEH mutant decreased to basal levels similar to uninfected cells and cells infected with wild type Y. enterocolitica (Fig. 7C). These data suggests that integrin signaling involving the activities of both FAK and Rac1 can influence inflammasome activity by up-regulating IL-18 mRNA. Altogether, these data suggests that integrins can provide the first signal for inflammasome activation in epithelial cells.
Figure 7. Invasin-integrin binding provides a first signal for inflammasome activation.
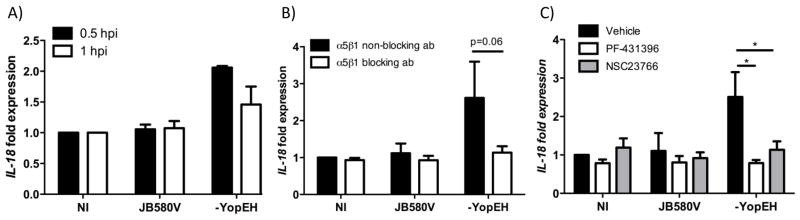
(A) Caco-2 cells infected with the indicated Yersinia strains for 30 min or 1hr. (B) Cells were pretreated with 7.5μg/ml of blocking α5β1 integrin antibody (JBS5) or a non-blocking α5β1 integrin antibody for 30 minutes or (C) with 5μM of PF-431396, 100μM of NSC23766 or DMSO for 1hr. (B–C) Cells were then infected for 30 min. (A–C) After infection, cells were processed to isolate RNA and IL-18 transcript levels were measured by qRT-PCR; data was normalized to GAPDH expression. Data represents triplicate samples with the mean ± SD of two-three independent experiments *p < 0.01.
Discussion
IECs serve as central regulators of gut homeostasis responding to insults by secreting cytokines (IL-18) to initiate both innate immunity and epithelial repair. In this study, we demonstrate for the first time that α5β1 integrins on IECs when bound to Y. enterocolitica Inv trigger IL-18 secretion through activation of the NLRP3 inflammasome. As part of integrin-mediated signaling to the inflammasome, we report that the activities of FAK and Rac 1 are involved in signal transduction (Fig. 8). However, the full inflammasome signal requires both invasin-dependent integrin binding and pore-formation. We further show Y. enterocolitica utilizes at least two TTSS effectors, YopE and YopH, to block the integrin-mediated inflammasome signal (Fig. 8).
Figure 8. A model depicting the proposed mechanism for invasin-mediated first signals and induction of inflammasome activity.
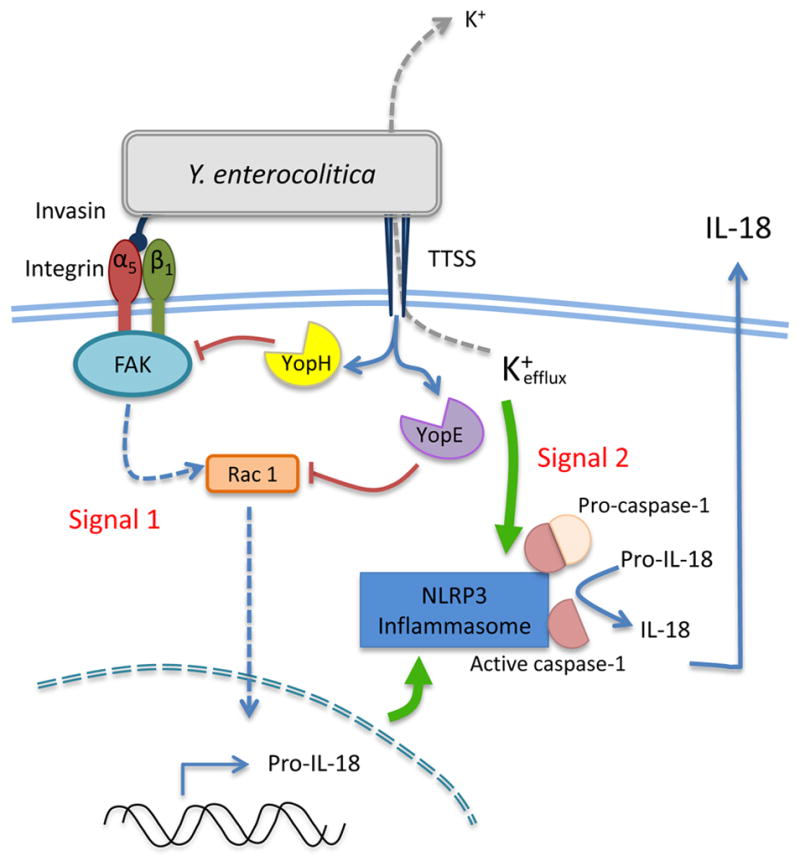
Invasin binding to β1 integrins on epithelial cells triggers a signaling cascade involving focal adhesion proteins including FAK followed by the activation of the Rho GTPase Rac 1. Activation of the focal adhesion complex ultimately leads to IL-18 gene expression. This integrin signaling pathway coupled with TTSS translocon-mediated pore-formation leads to the activation of the NLRP3 inflammasome and the secretion of IL-18. This signaling cascade is potently inhibited by the activity of the virulence factors YopH and YopE during infection with wild type Yersinia enterocolitica.
In the intestine, the response of IECs to microbes requires an acute ability to discriminate pathogenic bacteria from commensal flora. Because both normal flora and pathogenic microbes share conserved PAMPs sensed by host PAMPRs, we hypothesized that intestinal IECs must sense and respond to other PAMPs on enteric pathogens. Adhesins such as Inv are common among invasive bacteria and often bind their receptors with affinities significantly higher than natural ligands (48). A recent report demonstrated that T. denticola surface protein Td92 binds to α5β1 integrins leading to the full activation of the NLRP3 inflammasome in THP-1 monocytes (49). Although our findings similarly show that Inv-mediated integrin signaling is important for inflammasome activation in response to Y. enterocolitica infection, we found that in IECs, unlike macrophages that express other PAMPRs, integrins provide the inflammasome-priming signal by upregulating IL-18 transcription. These data strongly suggests the Inv-integrin interaction provides the first signal in a two-signal model of inflammasome activation. Full activation of the inflammasome requires an additional signal triggered by Yersinia TTSS or in some of our experimental systems by treating cells with the pore-forming toxin nigericin (50).
Pore formation by the Yersinia TTSS translocon in LPS primed macrophages, activates the NLRP3 inflammasome (21, 23, 35, 51, 52). However, our work is the first to demonstrate that Inv binding to α5β1 integrins can facilitate the priming step often called the first signal. This is of particular importance during intestinal innate immunity because IECs have muted responses to LPS due to low expression of TLR4, MD-2, and CD14 (3, 53). The relatively low expression of TLRs is critical for immune tolerance to commensal bacteria during homeostasis, and further implies that IECs utilize other mechanisms to rapidly detect pathogenic bacteria.
A tenet of our hypothesis is that IECs discriminate between normal flora and pathogens by utilizing novel PAMPRs. We directly tested this hypothesis in experiments utilizing non-pathogenic E. coli, a component of the normal flora. E. coli ectopically expressing Inv could induce IL-18 expression in the presence of nigericin. However even at high MOIs, E. coli with an empty vector failed to stimulate significant amounts of IL-18 in the presence of nigericin. Further, the IEC response to Inv as a PAMP required the Inv-integrin interaction as antibodies or peptides that block the receptor-ligand interaction dramatically reduced IL-18 secretion. A point mutation in Inv, D760A, which disrupts active site binding (44, 45), abolished IL-18 secretion strongly suggesting that IECs are sensing Inv as a PAMP through interactions with integrins. These data also demonstrate that IECs did not respond to common PAMPs present on both E. coli and Yersinia. Altogether, these data strongly support the hypothesis that integrins expressed on IECs can function as PAMPRs discriminating between pathogens and normal flora. Pathogens are capable of exploiting integrins for attachment and uptake because they often bind with much higher affinity than natural ligands; for example, Inv binds with a 100-fold higher affinity than fibronectin (48). This property of pathogen-associated adhesins could potentially provide a mechanism for how IECs accomplish integrin-dependent pathogen detection.
Many pathogens gain access to host cells and tissues by binding to integrins. Interestingly, vaccinia virus uptake is mediated by β1 integrins and like Yersinia, vaccinia has numerous virulence factors targeting inflammasome signaling (54). Considering the large number of pathogens exploiting integrins for invasion, our findings potentially highlight a common mechanism evolved to facilitate rapid recognition and inflammasome responses to invasive pathogens at mucosal surfaces.
Analysis of the impact of Yops on inflammasome activity in macrophages identified an inhibitory activity for YopK/YopQ, YopE, and YopM (23, 24, 33). However, the inhibitory activity of YopE is not yet clear because over expression studies found that Y. enterocolitica YopE disrupted inflammasome complex assembly, while Y. pseudotuberculosis YopE did not exhibit inflammasome inhibitory activity in mouse macrophages (23, 33). Our data demonstrates that YopE alone does not fully account for the potent inhibitory capability of Y. enterocolitica on inflammasome activity, but acts in concert with YopH. However, we did not test over expression of YopE in the IEC model and it is possible that it would be sufficient to disrupt inflammasome assembly. Prior to our work, YopH had not been identified as an inflammasome inhibitor. Previous work showed that Y. pseudotuberculosis YopK masks the Yersinia TTSS translocon from detection by the inflammasome in macrophages (Brodsky et al., 2010). Contrary to these findings, our data found no demonstrable role for Y. enterocolitica YopQ, the homologue of YopK, in counteracting the activation of the inflammasome in IECs. In fact, the yopEH mutant is YopQ and YopM sufficient and induces significant IL-18 suggesting that these Yops are not major inhibitors of inflammasome activity in IECs. The reason for this discrepancy might be due to Yersinia species-specific differences in YopQ/YopK activity. However, it is reasonable to speculate that the differences observed in Yops regulating inflammasome activity are due to cell type differences. In support of this hypothesis, recent work investigating the targets of YopH in neutrophils isolated from infected animals clearly showed cell-type-specific targets of YopH in vivo (30).
Collectively, these data highlight the importance of integrin signaling for inflammasome-mediated inflammatory responses in IECs and show that Y. enterocolitica escapes these responses by utilizing YopE and YopH to interfere with integrin signaling. These data illustrate how pathogens can control inflammasome-mediated inflammatory signaling by blocking signaling pathways required for the first signal. Elucidating the potential of integrin signaling as a common inflammasome activation mechanism in epithelial cells will provide important insight into host-pathogen interactions dictating early inflammatory responses at mucosal sites.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants AI067716 and AI060789 to PHD, AI083387 to SB, 5T32 AI007271 to JAS, and a Translational Science Training Fellowship to JT.
The authors wish to thank Dr. Virginia Miller for bacterial strains and reagents. The authors would also like to thank Ms. Cecilia Hinojosa for assistance with immunoblot quantification.
Footnotes
Author contributions: JT, SB, and PD designed the experiments; JT, JAS, SB and PD performed the experiments and wrote the manuscript.
Conflict of interest: The authors declare they have no conflicts of interest.
References
- 1.Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 2.Winter SE, Keestra AM, Tsolis RM, Baumler AJ. The blessings and curses of intestinal inflammation. Cell Host Microbe. 2010;8:36–43. doi: 10.1016/j.chom.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 2001;167:1609–1616. doi: 10.4049/jimmunol.167.3.1609. [DOI] [PubMed] [Google Scholar]
- 4.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 7.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 8.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- 9.Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, McCafferty DM, Rioux KP, Ghosh S, Xavier RJ, Colgan SP, Tschopp J, Muruve D, MacDonald JA, Beck PL. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359–1372. doi: 10.1002/ibd.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isberg RR. Uptake of enteropathogenic Yersinia by mammalian cells. Curr Top Microbiol Immunol. 1996;209:1–24. doi: 10.1007/978-3-642-85216-9_1. [DOI] [PubMed] [Google Scholar]
- 13.Bottone EJ. Yersinia enterocolitica: The Charisma Continues. Clinical Microbiol Rev. 1997;10:257–276. doi: 10.1128/cmr.10.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dube P. Interaction of Yersinia with the gut: mechanisms of pathogenesis and immune evasion. Curr Top Microbiol Immunol. 2009;337:61–91. doi: 10.1007/978-3-642-01846-6_3. [DOI] [PubMed] [Google Scholar]
- 15.Isberg RR, Falkow S. A single genetic locus encoded by Yersinia pseudotuberculosis permits invasion of cultured animal cells by Escherichia coli K-12. Nature. 1985;317:262–264. doi: 10.1038/317262a0. [DOI] [PubMed] [Google Scholar]
- 16.Miller VL, Falkow S. Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect Immun. 1988;56:1242–1248. doi: 10.1128/iai.56.5.1242-1248.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark MA, Hirst BH, Jepson MA. M-cell surface beta1 integrin expression and invasin-mediated targeting of Yersinia pseudotuberculosis to mouse Peyer’s patch M cells. Infect Immun. 1998;66:1237–1243. doi: 10.1128/iai.66.3.1237-1243.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelis GR. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat Rev Mol Cell Biol. 2002;3:742–752. doi: 10.1038/nrm932. [DOI] [PubMed] [Google Scholar]
- 19.Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. 2005;59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 20.Buttner D. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol Mol Biol Rev. 2012;76:262–310. doi: 10.1128/MMBR.05017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auerbuch V, Golenbock DT, Isberg RR. Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog. 2009;5:e1000686. doi: 10.1371/journal.ppat.1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bliska JB, Wang X, Viboud GI, Brodsky IE. Modulation of innate immune responses by Yersinia type III secretion system translocators and effectors. Cell Microbiol. 2013;15:1622–1631. doi: 10.1111/cmi.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe. 2012;12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Black DS, Bliska JB. Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J. 1997;16:2730–2744. doi: 10.1093/emboj/16.10.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruce-Staskal PJ, Weidow CL, Gibson JJ, Bouton AH. Cas, Fak and Pyk2 function in diverse signaling cascades to promote Yersinia uptake. J Cell Sci. 2002;115:2689–2700. doi: 10.1242/jcs.115.13.2689. [DOI] [PubMed] [Google Scholar]
- 27.Persson C, Carballeira N, Wolf-Watz H, Fallman M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 1997;16:2307–2318. doi: 10.1093/emboj/16.9.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weidow CL, Black DS, Bliska JB, Bouton AH. CAS/Crk signalling mediates uptake of Yersinia into human epithelial cells. Cell Microbiol. 2000;2:549–560. doi: 10.1046/j.1462-5822.2000.00079.x. [DOI] [PubMed] [Google Scholar]
- 29.Alrutz MA, Isberg RR. Involvement of focal adhesion kinase in invasin-mediated uptake. Proc Natl Acad Sci U S A. 1998;95:13658–13663. doi: 10.1073/pnas.95.23.13658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rolan HG, Durand EA, Mecsas J. Identifying Yersinia YopH-targeted signal transduction pathways that impair neutrophil responses during in vivo murine infection. Cell Host Microbe. 2013;14:306–317. doi: 10.1016/j.chom.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Black DS, Bliska JB. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol. 2000;37:515–527. doi: 10.1046/j.1365-2958.2000.02021.x. [DOI] [PubMed] [Google Scholar]
- 32.Bose R, Thinwa J, Chaparro P, Zhong Y, Bose S, Zhong G, Dube PH. Mitogen-activated protein kinase-dependent interleukin-1alpha intracrine signaling is modulated by YopP during Yersinia enterocolitica infection. Infect Immun. 2012;80:289–297. doi: 10.1128/IAI.05742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schotte P, Denecker G, Van Den Broeke A, Vandenabeele P, Cornelis GR, Beyaert R. Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1beta. J Biol Chem. 2004;279:25134–25142. doi: 10.1074/jbc.M401245200. [DOI] [PubMed] [Google Scholar]
- 34.Shin H, Cornelis GR. Type III secretion translocation pores of Yersinia enterocolitica trigger maturation and release of pro-inflammatory IL-1beta. Cell Microbiol. 2007;9:2893–2902. doi: 10.1111/j.1462-5822.2007.01004.x. [DOI] [PubMed] [Google Scholar]
- 35.Zheng Y, Lilo S, Brodsky IE, Zhang Y, Medzhitov R, Marcu KB, Bliska JB. A Yersinia effector with enhanced inhibitory activity on the NF-kappaB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog. 2011;7:e1002026. doi: 10.1371/journal.ppat.1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bohn E, Sing A, Zumbihl R, Bielfeldt C, Okamura H, Kurimoto M, Heesemann J, Autenrieth IB. IL-18 (IFN-gamma-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J Immunol. 1998;160:299–307. [PubMed] [Google Scholar]
- 38.Meng X, Leman M, Xiang Y. Variola virus IL-18 binding protein interacts with three human IL-18 residues that are part of a binding site for human IL-18 receptor alpha subunit. Virology. 2007;358:211–220. doi: 10.1016/j.virol.2006.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. The Journal of clinical investigation. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 41.Grassl GA, Bohn E, Muller Y, Buhler OT, Autenrieth IB. Interaction of Yersinia enterocolitica with epithelial cells: invasin beyond invasion. Int J Med Microbiol. 2003;293:41–54. doi: 10.1078/1438-4221-00243. [DOI] [PubMed] [Google Scholar]
- 42.Kuhn K, Eble J. The structural bases of integrin-ligand interactions. Trends Cell Biol. 1994;4:256–261. doi: 10.1016/0962-8924(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 43.Isberg RR, Leong JM. Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell. 1990;60:861–871. doi: 10.1016/0092-8674(90)90099-z. [DOI] [PubMed] [Google Scholar]
- 44.Leong JM, Morrissey PE, Marra A, Isberg RR. An aspartate residue of the Yersinia pseudotuberculosis invasin protein that is critical for integrin binding. EMBO J. 1995;14:422–431. doi: 10.1002/j.1460-2075.1995.tb07018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamburger ZA, Brown MS, Isberg RR, Bjorkman PJ. Crystal structure of invasin: a bacterial integrin-binding protein. Science. 1999;286:291–295. doi: 10.1126/science.286.5438.291. [DOI] [PubMed] [Google Scholar]
- 46.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mejia E, Bliska JB, Viboud GI. Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog. 2008;4:e3. doi: 10.1371/journal.ppat.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saltman LH, Lu Y, Zaharias EM, Isberg RR. A region of the Yersinia pseudotuberculosis invasin protein that contributes to high affinity binding to integrin receptors. J Biol Chem. 1996;271:23438–23444. doi: 10.1074/jbc.271.38.23438. [DOI] [PubMed] [Google Scholar]
- 49.Jun HK, Lee SH, Lee HR, Choi BK. Integrin alpha5beta1 activates the NLRP3 inflammasome by direct interaction with a bacterial surface protein. Immunity. 2012;36:755–768. doi: 10.1016/j.immuni.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin HCG. Type III secretion translocation pores of Yersinia enterocolitica trigger maturation and release of pro-inflammatory IL-1beta. Cell Microbiol. 2007;9:2893–2902. doi: 10.1111/j.1462-5822.2007.01004.x. [DOI] [PubMed] [Google Scholar]
- 52.Bergsbaken T, Cookson BT. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 2007;3:e161. doi: 10.1371/journal.ppat.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Funda DP, Tuckova L, Farre MA, Iwase T, Moro I, Tlaskalova-Hogenova H. CD14 is expressed and released as soluble CD14 by human intestinal epithelial cells in vitro: lipopolysaccharide activation of epithelial cells revisited. Infect Immun. 2001;69:3772–3781. doi: 10.1128/IAI.69.6.3772-3781.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PW, Liao CL, Chang W. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J Virol. 2012;86:6677–6687. doi: 10.1128/JVI.06860-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.