

Abstract
Background and Purpose
Follistatin-like 1 (FSTL1), an extracellular glycoprotein, has been reported to decrease apoptosis in ischemic cardiac diseases but its effect in ischemic stroke has not been examined. We hypothesized that recombinant FSTL1 attenuates neuronal apoptosis through its receptor DIP2A and the Akt pathway after middle cerebral artery occlusion (MCAO) in rats.
Methods
One hundred and forty male Sprague-Dawley rats were subjected to 2 hours of MCAO followed by reperfusion. In a subset of animals, the time course and location of FSTL1 and DIP2A were detected by western blot and immunofluorescence double staining. Another set of animals were intracerebroventricularly given either recombinant FSTL1 1 hour after reperfusion or FSTL1-siRNA 48 hours before reperfusion. Additionally, DIP2A was knockdown by siRNA in some animals. Infarction volume and neurological deficits were measured, and the expression of FSTL1, DIP2A, phosphorylated Akt, cleaved caspase-3 and TUNEL were quantified using Western blot.
Results
The expression of FSTL1 and DIP2A were increased in neurons and peaked 24 hours after MCAO. Recombinant FSTL1 reduced brain infarction and improved neurologic deficits 24 and 72 hours after MCAO via activation of its receptor DIP2A and downstream phosphorylation of Akt. These effects were reversed by DIP2A-siRNA and FSTL1-siRNA.
Conclusion
Recombinant FSTL1 decreases neuronal apoptosis and improves neurological deficits through phosphorylation of Akt by activation of its receptor DIP2A after MCAO in rats. Thus, FSTL1 may have potentials as a treatment for ischemic stroke patients.
Keywords: FSTL1, DIP2A, AKT, apoptosis, MCAO
Introduction
Stroke is one of the leading causes of morbidity and mortality worldwide, of which more than 80% are ischemic caused by obstruction of the cerebral arteries1. Currently, tissue plasminogen activator (tPA) is the only Food and Drug Administration-approved treatment for acute ischemic stroke and is given to about 2-5% of stroke patients2. However, tPA has a few limitations which fuel the interest in the development of new neuroprotective therapies. Recent studies have revealed that stroke-induced neuron apoptosis, predominantly in the ischemic penumbra or peri-infarct zone, may be caspase-dependent. Thus neuronal apoptosis is potentially recoverable in the early stage after stroke onset3 and a treatment manipulating apoptotic pathways remains an appealing strategy.
Follistatin-like 1 (FSTL1) is an extracellular glycoprotein that belongs to the follistatin family of proteins. Accumulating evidence indicates that FSTL1 has protective effects on ischemia-reperfusion injury in muscle and heart tissue associated with anti-apoptosis4, 5. The plasma protein levels of FSTL1 were increased after ischemia injury or under hypoxia/reoxygenation stress in the heart or hind-limb5, 6. Elevated levels of circulating FSTL1 were also observed in heart failure patients7. These findings suggest that FSTL1 may have broad cardiovascular-protective activities in heart ischemia. The protective effects of FSTL1 against apoptosis in cardiac ischemia were mediated by activation of the receptor Disco-interacting protein 2 homolog A (DIP2A) and phosphorylation of Akt8. However, the therapeutic impact of FSTL1 and its receptor DIP2A on brain ischemia has not been previously investigated nor the molecular mechanisms underlying the effects of FSTL1 on neuron apoptosis. The present study aims to investigate the neuroprotective effect and potential molecular mechanisms of FSTL1 in a rat MCAO model.
Materials and Methods
Animal Model and Experimental Protocol
All experiments were approved by the Institutional Animal Care and Use Committee of Loma Linda University.
Sprague-Dawley male rats weighting 260 to 300g were subjected to middle cerebral artery occlusion (MCAO) as previously described9, with some modifications. Briefly, anesthesia was induced intraperitoneally with ketamine (80mg/kg) and xylazine (10mg/kg) followed by atropine at a dose of 0.1mg/kg. The right common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA) were surgically exposed. The ECA was coagulated and 4-0 nylon suture with silicon was inserted into the ICA through the ECA stump to occlude the MCA. After 2 hours of MCA occlusion, the suture was carefully removed to induce reperfusion. Sham rats underwent the same procedures except that the MCA was not occluded. After closing the skin incision, rats were kept at approximately 37°C on an electric heating blanket and were housed separately until completely recovered from anesthetic.
To test whether delivery of FSTL1 protein affects acute brain ischemic injury in rat, male SD rats were injected intracerebroventricularly with either one of two dosages of recombinant FSTL1 protein (R&D Systems, 100mg/kg or 300mg/kg) or vehicle (0.1M Phosphate buffer solution (PBS)) at 1 hours after reperfusion.
siRNA injection
Three different formats of FSTL1-siRNA (OriGene Technologies) or DIP2A-siRNA (Santa Cruz Biotechnology) were applied 48 hours before MCAO by intraventricular injection (I.C.V) as previously described10. A scalp incision was made along the midline and a burr hole (1mm) was drilled into the skull above the right hemisphere (1.0 mm lateral of the bregma). The FSTL1-siRNA or DIP2A-siRNA mixture or Scramble-siRNA (100 pmol/2μl) was delivered into the ipsilateral ventricle with a Hamilton syringe and administered over 2 minutes. The needle was left in place for an additional 5 minutes after injection to prevent possible leakage and was then slowly withdrawn over 4 minutes. After the needle was removed, the burr hole was sealed with bone wax. The incision was closed with sutures and the rat was allowed to recover.
Neurological Scores
Twenty-four or 72 hours after MCAO, the Garcia test was performed by a blinded investigator as previously described with modifications11. The scores given to each rat at completion of the evaluation was the summation of 7 individual test scores (spontaneous activity, symmetry in the movement of four limbs, forepaw outstretching, climbing, body proprioception, response to vibrissae touch, and beam walking). The neurological scoring ranged from 2 (most severe deficit) to 21 (maximum).
2, 3, 5-triphenyltetraolium chloride staining
Infarction volume was determined by staining with 2, 3, 5-triphenyltetraolium chloride (TTC, Sigma) at 24 and 72 hours after MCAO as previously described with some modification11. Briefly, general anesthesia was reintroduced with a mixture of ketamine and xylazine and decapitated; the brains were rapidly removed and sectioned coronally into 2-mm thick slices starting from the frontal pole. Slices were stained with TTC for 15min at 37°C. The infarct and total hemispheric areas of each section were traced and analyzed using ImageJ (ImageJ, NIH). The possible interference of brain edema with infarct volume was corrected by standard methods (whole contralateral hemisphere volume − nonischemic ipsilateral hemisphere volume) and the infracted volume was expressed as a ratio of the corrected infarct to the whole contralateral hemisphere.
Immunofluorescence Staining
Rats were euthanized 24 hours after MCAO for double immunofluorescence staining as previously described11, using the neuronal marker of neuronal nuclei (NeuN) (Millipore), FSTL1 (sc-12542, Santa Cruz Biotechnology) and DIP2A (sc-6755, Santa Cruz Biotechnology). Brain sections were incubated with a mixture of either NeuN and FSTL1 or NeuN and DIP2A primary antibodies over night at 4°C, followed by a mixture of secondary antibodies for 1 hour at room temperature. Microphotographs were analyzed with a fluorescent microscope (Olympus OX51, Japan).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
Twenty-four hours after MCAO, TUNEL staining was performed with Insitu apoptosis detection kit (Roche) according to the manufacturer's instruction. Four images were taken from the ischemic border of each section using a 40x objective lens and then the number of TUNEL positive neurons and the number of TUNEL positive non-neurons were counted. The total number of neurons and non-neurons in the ischemic hemisphere at 40x magnification were counted in three arbitrary fields. The number of TUNEL-positive neurons or non-neurons was expressed as a ratio to that in MCAO+Vehicle group, respectively.
Western blots
Brain samples were collected 24 hours after MCAO. Proteins of the ipsilateral hemisphere were extracted by homogenizing in RIPA lysis buffer (sc-24948, Santa Cruz Biotechnology). Western blotting was performed as described previously12. Primary antibodies used were: FSTL1 (sc-12542, Santa Cruz Biotechnology), DIP2A (sc-67556, Santa Cruz Biotechnology), phosphorylated Akt (p-Akt, Cell Signaling Technology), cleaved caspase-3 (CC3, Cell Signaling Technology) and goat polyclonal β-actin (sc-1616, Santa Cruz Biotechnology).
Statistical Analysis
Data from different groups were compared using one-way analysis of variance (ANOVA) followed by post-hoc Tukey tests. The data are presented as means ± SEM. A value of p<0.05 was considered statistically significant.
Results
Expressions of FSTL1 and DIP2A were increased in neurons and peaked 24 hours after MCAO
The protein level of FSTL1 and DIP2A at 6, 12, 24, and 48 hours in the ischemic penumbra after MCAO were measured by Western blot. Analysis showed that FSTL1 began increasing as early as 6 hours after ischemia, peaking at 12 hours, and was sustained to 24 hours (both 12 and 24 hours were statistically significant from Sham, p<0.05). FSTL1 returned to a level indistinguishable from Sham by 48 hours (Figures 1A and 1B). A similar trend was observed for the expression of DIP2A, the FSTL1 receptor; DIP2A significantly increased at 12 and 24 hours after MCAO, and returned backed to Sham levels at 48 hours (Figure 1C). Double immunofluorescence staining of FSTL1 and NeuN, or DIP2A and NeuN showed that both FSTL1 and DIP2A were up-regulated in neurons in the penumbra 24 hours after MCAO. FSTL1 was primarily expressed in the cytoplasm and DIP2A is located in the membranes of neurons (Figure 1D).
Figure 1.

Time course and location of FSTL1 and DIP2A in rat brain after MCAO. (A) Representative Western blots of FSTL1 and DIP2A. (B) FSTL1 expression increases significantly 12 and 24 hours after MCAO, and declines 48 hours after MCAO. (C) DIP2A expression increases significantly 12 and 24 hours after MCAO, and declines 48 hours after MCAO. (D) Immunofluorescence double staining of FSTL1 (red), DIP2A (red), and Neuronal Nuclei (NeuN, green) showed that the expression of FSTL1 and DIP2A were increased and localized in neurons of the penumbra 24 hours after ischemia. n=5 per group for Western blots and n=1 per group for immunohistochemistry. * p<0.05 vs. Sham. Scale bars=100 μm.
Recombinant FSTL1 reduced brain infarction and improved neurological function 24 and 72 hours after MCAO
Analysis of the infarct stained with TTC showed that the ratio of the infarcted tissue volume to the volume of the contralateral tissue 24 hours after MCAO was 0.334±0.1102. Administering the Vehicle did not significantly reduce the infarct ratio (0.324±0.1302, p>0.05 vs. MCAO). Treatment with the low dose of FSTL1 (100mg/kg) had an infarct ratio of 0.2194±0.0395 (p>0.05 vs. MCAO). However, treatment with the high dose of FSTL1 (300mg/kg) significantly decreased the infarct ratio (Figures 2A and 2B, 0.190±0.0397, p<0.05 vs. MCAO).
Figure 2.

FSTL1 decreased infarct ratio and improved neurological function 24 hours after MCAO. (A) Representative TTC staining images of coronal sections. (B) Quantified Infarct ratio and (C) neurological scores showed that high dosage of FSTL1 decreases infarction and neurological deficits 24 hours after MCAO. * p<0.05 vs. Sham, & p<0.05 vs. MCAO, n=6 for each group.
MCAO resulted in neurological deficits 24 hours after MCAO (p<0.05 vs. Sham). Administering the Vehicle or low dose of FSTL1 did not have any significant effect on neurological deficits. Yet, the high dose of FSTL1 significantly improved the neurological function of rats 24 hours after MCAO (Figure 2C, p<0.05 vs. MCAO).
Seventy-two hours after MCAO, the infarct ratio was 0.3193±0.0775 in untreated group (MCAO) and 0.3222±0.0536 in the Vehicle treated group (MCAO+Vehicle). FSTL1 at a dose of 300mg/kg significantly decreased the infarct ratio compared to MCAO (0.151±0.0382, p<0.05 vs. MCAO) (Figures 3A and 3B). Furthermore, FSTL1 siginificantly improved the performance of MCAO rats on the neurobehavioral tests (Figure 3C, p<0.05 vs. MCAO).
Figure 3.

FSTL1 decreased infarct ratio and improved neurological function 72 hours after MCAO. (A) Representative TTC staining images of coronal sections. (B) Quantified Infarct ratio and (C) neurological scores showed that high dose of FSTL1 decreases infarction and neurological deficits 24 hours after MCAO. * p<0.05 vs. Sham, & p<0.05 vs. MCAO, n=6 for each group.
FSTL1 decreased neuronal apoptosis through phosphorylation of Akt
Twenty-four hours after MCAO, there were abundant TUNEL positive neurons in the ischemic penumbra, and treatment with 300mg/kg FSTL1 decreased the number of TUNEL positive neurons (Figures 4A and 4B, p<0.05 vs. MCAO+Vehicle). Knocked down of FSTL1 by intracerebroventricular injection of FSTL1-siRNA 48 hours before MCAO significantly increased the number of TUNEL positive neurons when compared to either the MCAO+Vehicle or the scramble-siRNA group (Figures 4A and 4B, p<0.05 vs. MCAO+Vehicle and MCAO+Scramble-siRNA). Neurological function was exacerbated by FSTL1-siRNA (Figure 4B, p<0.05 vs. MCAO+Vehicle and MCAO+Scramble-siRNA). Additionally, administration of FSTL1 reduced the number of TUNEL positive non-neuronal cells, FSTL1-siRNA increased the number of TUNEL positive non-neuronal cells, and FSTL1+DIP2A-siRNA increased the number of TUNEL positive non-neuronal cells (Figure I in the online-only Data Supplement).
Figure 4.
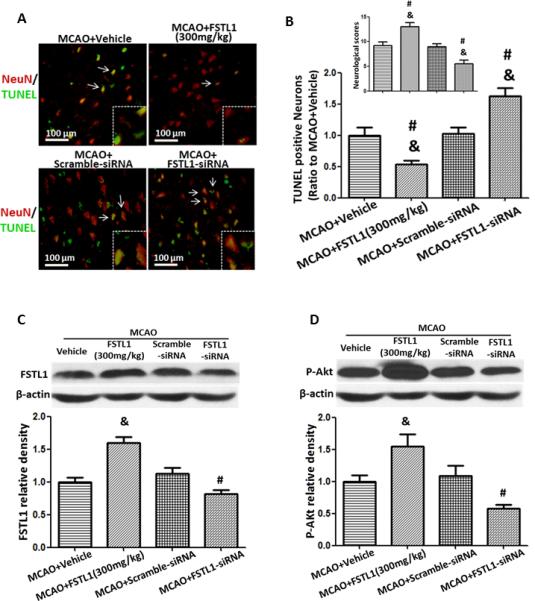
FSTL1 decreased apoptosis by increasing Akt phosphorylation 24 hours after MCAO. (A) Immunofluorescence double staining of TUNEL positive cells (green) and neurons (NeuN, red). (B) TUNEL positive neurons are decreased by FSTL1 and increased by FSTL1-siRNA.Neurological score is improved by FSTL1 and reduced by FSTL1-siRNA. (C). Western blot analysis of FSTL1 expression. (D) Western blot analysis of phosphorylated Akt. Akt phosphorylation is increased by FSTL1 and reversed by FSTL1-siRNA. DIP2A (96 kDa), FSTL1 (35 kDa) and β-actin (42 kDa) were run on the same gel. n=6 for each group in Western blot and immunohistochemistry. & p<0.05 vs. MCAO+Vehicle, # p<0.05 vs. MCAO+Scramble-siRNA, Scale bars=100 μm.
Western blot analysis showed that administration of FSTL1 significantly increased the expression of FSTL1 and enhanced the phosphorylation of Akt (Figures 4C and 4D, p<0.05 vs. MCAO+Vehicle). FSTL1-siRNA effectively suppressed the expression of FSTL1 and reduced the level of phosphorylated Akt 24 hours after MCAO (Figures 4C and 4D, p<0.05 vs. MCAO+Vehicle, MCAO+FSTL1, and MCAO+Scramble-siRNA).
FSTL1 phosphorylation of Akt is dependent on its receptor DIP2A
DIP2A-siRNA was injected intracerebroventricularly 48 hours before MCAO and rats were treated with FSTL1 1 hour after MCAO. DIP2A-siRNA abolished the effects of FSTL1 by increasing neuron apoptosis (Figures 5A and 5B, increased TUNEL positive neurons, p<0.05 vs. MCAO+FSTL1+scramble-siRNA group) and increasing the neurological deficits (Figure 5C, p<0.05 vs. MCAO+FSTL1+scramble- siRNA group). Western blot analysis showed that DIP2A-siRNA reduced the expression of DIP2A, decreased the level of phosphorylation of Akt, and raised the level of CC3 (Figure 6, p<0.05 vs. MCAO+FSTL1+scramble-siRNA group).
Figure 5.
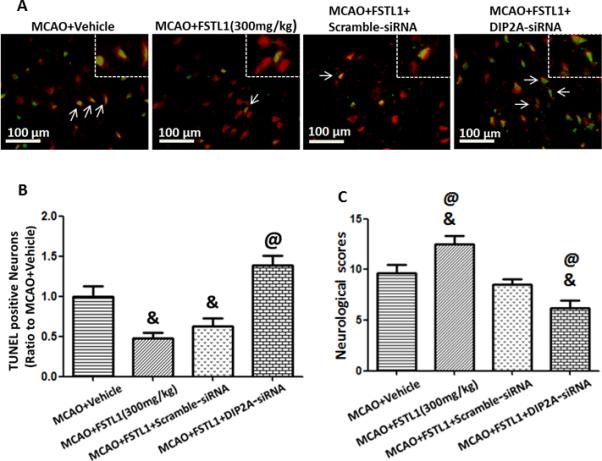
FSTL1 decreased neuron apoptosis after MCAO was dependent on its receptor, DIP2A. DIP2A-siRNA increased neuron apoptosis (A, B) and increased neurological deficits (C) in FSTL1 treated rats after MCAO. n=6 for each group. & p<0.05 vs. MCAO+Vehicle, @ p<0.05 vs. MCAO+FSTL1+Scramble-siRNA. Scale bars=100 μm.
Figure 6.
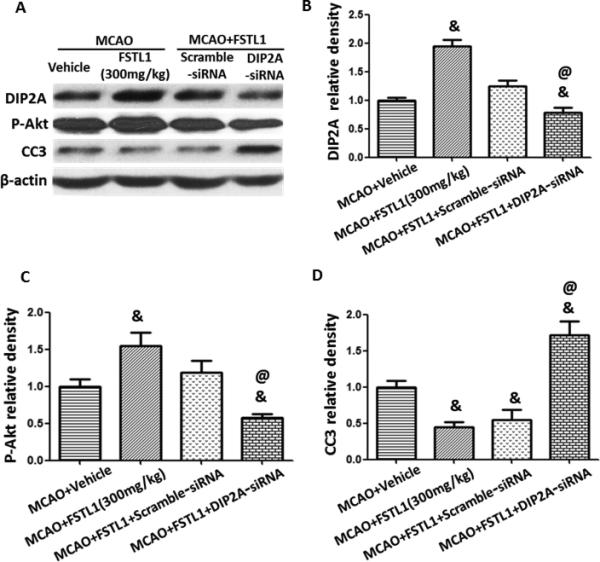
FSTL1 prevented apoptosis though the DIP2A/Akt pathway after MCAO in rats. Western blots for DIP2A, Akt and CC3 (A) showed DIP2A-siRNA prevented the expression of DIP2A after FSTL1 treatment (B), reduced Akt phosphorylation (C), and increased the expression of CC3 (D). n=6 for each group. & p<0.05 vs. MCAO+Vehicle, @ p<0.05 vs. MCAO+FSTL1+Scramble-siRNA.
Discussion
Apoptosis has been suggested to play a role in neuronal death after stroke in patients as well as in animal models of stroke. Cerebral ischemia triggers two general pathways of apoptosis: the intrinsic pathway which originates from mitochondrial release of cytochrome c and associated stimulation of caspase-3, and the extrinsic pathway which originates from the activation of cell surface death receptors and subsequent stimulation of caspase-83. Neurons in the ischemic penumbra or peri-infarct zone undergo apoptosis within hours after stroke onset. Targeting and preventing apoptosis in the penumbra would seem to be a logical therapeutic goal for limiting cerebral infarct volume after stroke. In the present study we showed that administration of recombinant FSTL1 can reduce infarction volume and improve neurological function by suppressing neuron apoptosis after MCAO in rats. We also found that the anti-apoptotic mechanism of FSTL1 treatment is via decreased caspase-3 cleavage through phosphorylation of Akt by activation of the FSTL1 receptor, DIP2A. These observations suggested that treatment with FSTL1 might be a potential therapeutic intervention for reducing infarction in ischemic stroke patients.
FSTL1 was originally identified from a transforming growth factor (TGF)-responsive gene in osteoblast cells that have a follistatin-like domain. Although developmental studies have suggested that FSTL1 regulates organ tissue formation in embryos13, its functions in fully developed tissue has only been partially elucidated to. FSTL1 has been reported to suppress cancer14 and modulate inflammation15 in animals. Recently, accumulating evidence indicates FSTL1 is cardiac-protective after ischemia stress. Up-regulation of FSTL1 transcription was observed in the hearts of Akt1 transgene-induced hypertrophy mice16, 17, and FSTL1 levels are increased in patients with acute coronary syndrome or heart failure 7, 18, 19. In experimental cardiac ischemia, systemic administration of FSTL1 was protective and suggested to be anti-apoptotic6. Additionally, administration of FSTL1 accelerated revascularization in the hind-limb ischemia model, and in vitro studies demonstrated an anti-apoptotic effect of FSTL1 on primary endothelial cells5. The results of these studies suggest that FSTL1 functions as an anti-apoptotic protein after ischemia. However, little is known about its function in cerebrovascular disease. We found that FSTL1 was endogenously expressed in brain and was robustly increased 12 hours after MCAO. FSTL1 was observed in the cytoplasm of neurons. This study also provides additional evidence of the anti-apoptotic function of FSTL1 after ischemia. Administration of recombinant FSTL1 decreased infarction volume and improved neurological function by reducing apoptosis, and knockdown of FSTL1 exacerbates these outcomes. Furthermore, FSTL1 was found to decrease apoptosis both in neurons and non-neuronal cells, suggesting that FSTL1 may play a role in other cell types after cerebral ischemia.
The serine/threonine protein kinase Akt (also known protein kinase B) is a key regulator of cell growth and survival, and is essential for cellular adaptation to stress. Therefore, Akt plays a role in several critical pathways making it a compelling target for neuroprotection after brain ischemia. FSTL1 has been implicated in the protection of cardiovascular cells from stress via the activation of phosphatidylinositol 3-kinase (PI3K) and Akt signaling6. In endothelial cells, FSTL1 activation of PI3K/Akt signaling led to the activation of endothelial nitric oxide synthase and subsequent nitric oxide production5. In this study, it was found that recombinant FSTL1 increases the phosphorylation of Akt 24 hours after MCAO, and knockdown of FSTL1 has the reverse effect.
Although FSTL1 is categorized as a follistatin-like protein, there is relatively little functional similarity with other follistatin family proteins which are binding partners of the TGF-β family proteins. Recently, DIP2A was found to function as an FSTL1 receptor. In endothelial cells, DIP2A functions in the anti-apoptotic effects of FSTL1 and mediates FSTL1 activation of Akt8. DIP2A has been observed on the surface of endothelial cells, and knockdown of DIP2A by siRNA reduced the binding of FSTL1. Furthermore, in endothelial cell cultures, DIP2A knockdown diminishes FSTL1-stimulated survival, migration, and differentiation in network structures and inhibited FSTL1-induced Akt phosphorylation. Finally, DIP2A was also identified as a candidate receptor molecule for FSTL1 by molecular interaction methods20. To determine the mechanism of FSTL1 in Akt phosphorylation after focal cerebral ischemia, we first examined the localization of DIP2A and FSTL1. We found that both DIP2A and FSTL1 were primarily expressed by neurons and increased 24 hours after MCAO, indicating that locally produced FSTL1 may activate signaling pathways through the DIP2A receptor. Knockdown of DIP2A by siRNA removed the anti-apoptosis effects of FSTL1 via decreased the Akt phosphorylation after MCAO in rats.
In conclusion, our findings indicate that FSTL1 may contribute to neuronal survival after brain ischemia. FSTL1 inhibited apoptosis through phosphorylation of Akt via activation its receptor, DIP2A. This study provides new information on the function of FSTL1 following MCAO, and makes FSTL1 a potential therapeutic candidate for ischemic stroke patients.
Supplementary Material
Acknowledgments
Sources of Funding
This study was supported partially by a grant from NIH NS043338 JHZ.
Footnotes
Disclosure
None.
References
- 1.O'Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the interstroke study): A case-control study. Lancet. 2010;376:112–123. doi: 10.1016/S0140-6736(10)60834-3. [DOI] [PubMed] [Google Scholar]
- 2.Brainin M, Teuschl Y, Kalra L. Acute treatment and long-term management of stroke in developing countries. Lancet Neurol. 2007;6:553–561. doi: 10.1016/S1474-4422(07)70005-4. [DOI] [PubMed] [Google Scholar]
- 3.Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 4.Ogura Y, Ouchi N, Ohashi K, Shibata R, Kataoka Y, Kambara T, et al. Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models. Circulation. 2012;126:1728–1738. doi: 10.1161/CIRCULATIONAHA.112.115089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, et al. Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J Biol Chem. 2008;283:32802–32811. doi: 10.1074/jbc.M803440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oshima Y, Ouchi N, Sato K, Izumiya Y, Pimentel DR, Walsh K. Follistatin-like 1 is an akt-regulated cardioprotective factor that is secreted by the heart. Circulation. 2008;117:3099–3108. doi: 10.1161/CIRCULATIONAHA.108.767673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Armouche A, Ouchi N, Tanaka K, Doros G, Wittkopper K, Schulze T, et al. Follistatin-like 1 in chronic systolic heart failure: A marker of left ventricular remodeling. Circ Heart Fail. 2011;4:621–627. doi: 10.1161/CIRCHEARTFAILURE.110.960625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouchi N, Asaumi Y, Ohashi K, Higuchi A, Sono-Romanelli S, Oshima Y, et al. Dip2a functions as a fstl1 receptor. J Biol Chem. 2010;285:7127–7134. doi: 10.1074/jbc.M109.069468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Q, Ma Q, Zhan Y, He Z, Tang J, Zhou C, et al. Isoflurane enhanced hemorrhagic transformation by impairing antioxidant enzymes in hyperglycemic rats with middle cerebral artery occlusion. Stroke. 2011;42:1750–1756. doi: 10.1161/STROKEAHA.110.603142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. Nlrp3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol. 2013;75:209–219. doi: 10.1002/ana.24070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Q, Chen C, Yan J, Yang X, Shi X, Zhao J, et al. Therapeutic application of gene silencing mmp-9 in a middle cerebral artery occlusion-induced focal ischemia rat model. Exp Neurol. 2009;216:35–46. doi: 10.1016/j.expneurol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Kristian T, Balan I, Schuh R, Onken M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J Neurosci Res. 2011;89:1946–1955. doi: 10.1002/jnr.22626. [DOI] [PubMed] [Google Scholar]
- 13.Sylva M, Moorman AF, van den Hoff MJ. Follistatin-like 1 in vertebrate development. Birth Defects Res C Embryo Today. 2013;99:61–69. doi: 10.1002/bdrc.21030. [DOI] [PubMed] [Google Scholar]
- 14.Chan QK, Ngan HY, Ip PP, Liu VW, Xue WC, Cheung AN. Tumor suppressor effect of follistatin-like 1 in ovarian and endometrial carcinogenesis: A differential expression and functional analysis. Carcinogenesis. 2009;30:114–121. doi: 10.1093/carcin/bgn215. [DOI] [PubMed] [Google Scholar]
- 15.Clutter SD, Wilson DC, Marinov AD, Hirsch R. Follistatin-like protein 1 promotes arthritis by up-regulating ifn-gamma. J Immunol. 2009;182:234–239. doi: 10.4049/jimmunol.182.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiekofer S, Shiojima I, Sato K, Galasso G, Oshima Y, Walsh K. Microarray analysis of akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure. Physiol Genomics. 2006;27:156–170. doi: 10.1152/physiolgenomics.00234.2005. [DOI] [PubMed] [Google Scholar]
- 18.Lara-Pezzi E, Felkin LE, Birks EJ, Sarathchandra P, Panse KD, George R, et al. Expression of follistatin-related genes is altered in heart failure. Endocrinology. 2008;149:5822–5827. doi: 10.1210/en.2008-0151. [DOI] [PubMed] [Google Scholar]
- 19.Widera C, Horn-Wichmann R, Kempf T, Bethmann K, Fiedler B, Sharma S, et al. Circulating concentrations of follistatin-like 1 in healthy individuals and patients with acute coronary syndrome as assessed by an immunoluminometric sandwich assay. Clin Chem. 2009;55:1794–1800. doi: 10.1373/clinchem.2009.129411. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka M, Murakami K, Ozaki S, Imura Y, Tong XP, Watanabe T, et al. Dip2 disco-interacting protein 2 homolog a (drosophila) is a candidate receptor for follistatin-related protein/follistatin-like 1--analysis of their binding with tgf-beta superfamily proteins. FEBS J. 2010;277:4278–4289. doi: 10.1111/j.1742-4658.2010.07816.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.