

Abstract
Objective
Patatin-like phospholipase domain containing 3 (PNPLA3, adiponutrin) has been identified as a modifier of lipid metabolism. To better understand the physiological role of PNPLA3/adiponutrin, we have investigated its regulation in intact mice and human hepatocytes under various nutritional/metabolic conditions.
Material/Methods
PNPLA3 gene expression was determined by real-time PCR in liver of C57BL/6 mice after dietary treatments and in HepG2 cells exposed to various nutritional/metabolic stimuli. Intracellular lipid content was determined in HepG2 cells after siRNA-mediated knockdown of PNPLA3.
Results
In vivo, mice fed a high-carbohydrate (HC) liquid diet had elevated hepatic lipid content, and PNPLA3 mRNA and protein expression, compared to chow-fed mice. Elevated expression was completely abrogated by addition of unsaturated lipid emulsion to the HC diet. By contrast, in mice with high-fat diet-induced steatosis, Pnpla3 expression did not differ compared to low-fat fed mice. In HepG2 cells, Pnpla3 expression was reversibly suppressed by glucose depletion and increased by glucose refeeding, but unchanged by addition of insulin and glucagon. Several unsaturated fatty acids each significantly decreased Pnpla3 mRNA, similar to lipid emulsion in vivo. However, Pnpla3 knockdown in HepG2 cells did not alter total lipid content in high glucose- or oleic acid-treated cells.
Conclusions
Our results provide evidence that PNPLA3 expression is an early signal/signature of carbohydrate-induced lipogenesis, but its expression is not associated with steatosis per se. Under lipogenic conditions due to high-carbohydrate feeding, certain unsaturated fatty acids can effectively suppress both lipogenesis and PNPLA3 expression, both in vivo and in a hepatocyte cell line.
Keywords: Patatin-like phospholipase domain containing 3, Nonalcoholic fatty liver disease, Glucose, Lipid emulsion
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) currently affects about one-third of the population in Western countries (1, 2) and is an emerging public health problem worldwide. Patatin-like phospholipase domain-containing protein 3 (PNPLA3), also known as adiponutrin, first came to wide attention with regard to NAFLD when Romeo et al. (3) reported a strong association between the PNPLA3 polymorphism rs738409 C/G (I148M) and NAFLD. This association has been confirmed in a number of independent studies of fatty liver disease (4–6).
The regulation and function of PNPLA3 are still unclear. It was reported that PNPLA3 has both TG hydrolase (7, 8) and transacylase activity (9) in vitro. He et al. (8) demonstrated that the wild type PNPLA3 enzyme has TG hydrolase activity in vitro assays and that overexpression of PNPLA3-I148M increased the TG content of cultured Huh7 liver cells and mouse liver in vivo. Moreover, Huang et al. (10) showed that wild type PNPLA3 has a high hydrolytic activity for glycerolipids and that the I148M substitution dramatically decreased its enzymatic activity. Thus, these studies support that PNPLA3 is involved in lipid metabolism by functioning as a TG hydrolase. However, Qiao et al. (11) showed that overexpression of PNPLA3 increased TG content in primary mouse hepatocytes, suggesting that PNPLA3 is lipogenic. Consistent with this study, Kumari et al. (12) reported that PNPLA3 functions as lysophosphatidic acid (LPA) acyltransferase, which catalyzes the conversion of LPA into phosphatidic acid (PA) for phospholipid synthesis. Kumari et al. (12) concluded that the I148M variant of PNPLA3 is involved in development of NAFLD by a gain of lipogenic function. In addition, Kumashiro et al. (13) recently demonstrated that knockdown of PNPLA3 with specific antisense oligonucleotides prevented hepatic steatosis in high fat fed rats and reduced PA and the ratio of PA to LPA. However, two other studies failed to identify any metabolic changes including TG metabolism in PNPLA3 null mice, suggesting that PNPLA3 is not a major player in glucose/lipid metabolism or that other compensatory factors are activated in response to the loss of PNPLA3 (14, 15). Recently, it has been proposed that PNPLA3 plays roles in discriminating fatty acids and remodeling lipid droplets (16). Overall, the role of PNPLA3 in the pathogenesis of NAFLD appears to be complex, and remains an open question.
In the present study, we focused on the regulation of PNPLA3 expression by addressing three questions: Does the intake of a high-carbohydrate (HC) diet or a high-fat (HF) diet upregulate the expression of PNPLA3 in the mouse liver? Does inclusion of fat in the HC diet aggravate or attenuate the effect of the HC diet? Does glucose have a direct effect on PNPLA3 expression in human HepG2 cells as a hepatocyte model? Our data indicate that while both our HC and HF diets result in increased liver lipids, only the HC diet increased PNPLA3 gene expression. This effect was reduced by inclusion of fat in the HC diet or by addition of fatty acids to high glucose-treated HepG2 cells. Together our results show that PNPLA3 expression is tightly controlled by glucose and certain types of unsaturated fatty acids, and suggests that PNPLA3 likely reflects lipogenesis in the liver. However it is not necessarily associated with the development of steatosis.
2. Materials and Methods
2.1. Animals and dietary protocols
Animal protocols were approved by the Institutional Animal Use and Care Committee of Pennsylvania State University. To evaluate hepatic PNPLA3 expression in response to diet, two studies were conducted. In study 1, the diet was based on a liquid formula (Clinimix E®, Baxter, Abbott, Deerfield, IL) that was supplemented with vitamins (Pediatric Influvite®, Baxter, Abbott, Deerfield, IL) and minerals and trace elements (8 μg/L zinc chloride, 6.4 μg/L cupric sulfate, 1.2 μg/L manganous sulfate, 80 μg/L chromium chloride, 176 μg/L sodium selenite, and 1.13 g/L ferrous sulfate); this HC diet has been shown to induce hepatic steatosis in 4–5 weeks (17). In study 1, twenty 5-week-old male C57BL/6 mice were housed on a 12:12 h light/dark cycle and randomized into 4 groups which received either: 1) a regular rodent diet (normal chow, rodent diet 5001 containing 58% carbohydrate, 13.5% fat, and 28.5% protein. (The composition of all experimental diets is expressed in % energy.) 2) HC liquid diet containing 76.8% carbohydrate in the form of dextrose, 22.7% protein, and 0.5% fat, an amount which was added in the form of lipid emulsion (LE) (Intralipid 20%, Baxter, Abbott, Deerfield, IL), as a source of essential fatty acids; 3) the same formula with 4% LE (74% carbohydrate, 22% protein, and 4.0% fat), referred to as HC-4% LE; or 4) the same formula supplemented with 13.5% LE (66.8% carbohydrate, 19.7% protein, and 13.5% fat), referred to as HC-13.5% LE. For all mice fed liquid diets, a known volume of the appropriate diet was added daily to a feeding tube and the amount remaining the next day was determined to calculate total intake. The liquid diet was the only source of nutrition and hydration (17). In study 2, liver tissue was obtained from a study of high fat (HF) diet-induced metabolic syndrome (18). Male C57BL/6 mice were fed purified diets (Research Diet, Inc, New Brunswick, NJ) for 16 weeks which was either low fat (LF) (70% carbohydrate, 10% fat, and 20% protein); a HF diet (25% carbohydrate, 60% fat, and 15% protein) or they were treated with the same HF diet but allowed to voluntarily exercise through access to running wheels, see (18) for details. Mice in study 2 were housed on a 12-h light/dark cycle with free access to food and drinking water. Body weights (study 1 and 2) and liver TG levels (study 1) of mice have been published (17, 18). Briefly, final body weight among different groups were same in study 1, and exercise resulted in decreased final body weight (12%) compared to HF-fed mice.
At the end of each study (5 wk for study 1 and 16 wk for study 2), mice were either euthanized with carbon dioxide (study 1) or anesthetized and killed by exsanguination (study 2) and tissues were dissected. In study 1, portions of the left liver lobes were embedded in Optimal Cutting Temperature compound (Sakura Finetek, Torrance CA). In both experiments liver samples were immediately frozen in liquid nitrogen and stored at −80°C before use. All other tissues were stored at −80°C.
2.2. Cell culture and Oil Red O staining
To investigate whether glucose, fatty acids and other supplements affect PNPLA3 expression in liver cells, HepG2 cells were used. These human hepatocarcinoma cells represent a relatively well-differentiated cell model of hepatocyte function and possess a wide variety of liver-specific metabolic functions (19). Cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% (v/v) fetal bovine serum and 0.5% (v/v) penicillin-streptomycin at 37°C in a 5% CO2-air incubator. Cells were at approximately 80% confluence at the time of treatment.
For detection of lipid accumulation by Oil Red O staining, HepG2 cells were washed with PBS and fixed with 10% phosphate-buffered formalin for 1 hour. Then cells were washed with 60% isopropanol after fixation with formalin and incubated with Oil Red O working solution for 10 minutes. Cells were washed with warm tap water for 4 times. After photography, the dye was eluted by adding a precise volume of 100% isopropanol and the optical density at 510 nm was measured to quantify total lipid staining.
2.3 Liver TG quantification
Liver total lipid was extracted from 200 mg of fresh liver according to the method of Folch et al. (20). Then liver TG was assayed by a modification of the method of Sardesi and Manning (21). Briefly, the total lipid extract from liver was applied to a column of 5% water-deactivated aluminum and the TG fraction was eluted with 25% diethyl ether in hexane. Then the TG content in aliquots of the elute was determined spectrophotometrically.
2.4. Preparation of BSA-bound fatty acids
Albumin-bound fatty acids were prepared according to methods described elsewhere (22). Briefly, 0.1mM each fatty acid (oleic acid, linoleic acid, linolenic acid, eicosapentaenoic acid, and docosahexaenoic acid) was dissolved in 800 μL ethanol before adding 40 μL of 5 M NaOH. The ethanol was evaporated and 4 ml of pre-warmed sterile phosphate-buffered saline was added to suspend the sodium salts of fatty acids. Five ml of ice -cold BSA solution was added to final concentration of BSA-bound fatty acid of 10 mM with a molar ration of 5.5:1 (fatty acid: BSA) at pH ≈7.4. Aliquots of the fatty acids stock were covered with nitrogen gas and stored at −20°C.
2.5. Total RNA extraction and reverse transcription
Total RNA was extracted from mouse liver and adipose tissue using Trizol reagent (Life Technologies, Carlsbad, CA). Total RNA from HepG2 cells was isolated using QIAGEN RNeasy mini kit following the protocol provided by the manufacturer. The concentration of total RNA was determined by UV absorbance spectrophotometry (NanoDrop ND-1000). The reverse transcription reaction was performed in a total volume of 20 μl and consisted of 4 μl 5X M-MLV RT Buffer, 250 μM dNTP mix, 50 ng oligo-dT15 primer, 20 U of ribonuclease inhibitor and 200 U of M-MLV reverse transcriptase (Promega). The reaction was performed at 42 °C for 30 min, 37 °C for 30 min, and 94 °C for 5 min.
2.6. Quantitative Real Time PCR
The iQ SYBR Green Supermix (Bio-Rad) was used for RT-PCR. The reaction was conducted in a total volume of 20 μl in 96-well plates. The PCR reaction conditions for each cycle were as follows: 94°C for 5 min, followed by 40 cycles at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec. Each value for the mRNA of analyzed genes was normalized relative to the mRNA for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-actin analyzed on the same plate. The primers were made by the Nucleic Acid Facility, Penn State University, University Park, Gene primer sequences are listed in Supplementary Table 1.
2.7. Western Blotting
Whole cell or tissue lysates (30–50 μg protein) were separated by electrophoresis on a 10% SDS–PAGE gels and transferred to PVDF membranes. The blots were then incubated with anti-PNPLA3 polyclonal antibody (1:5000 dilution; Everest Biotech; EB08402), anti-PNPLA3 (1:2000; abcam, ab81874), anti-ERK2 polyoclonal antibody (1:5000; Santa Cruz; sc-154), and anti-β-actin (1:2000; Santa Cruz; sc-47778). For all blots, horseradish peroxidase-conjugated secondary antibody or IRDye (Li-cor) secondary antibody was used. The protein bands were visualized by ECL Western blotting analysis system (GE Healthcare Life Sciences) or Odyssey Imaging System (Li-cor).
2.8. RNA interference
PNPLA3 siRNA (Thermo Scientific, Cat# L-009564-02) was delivered into HepG2 cells using Dharma FECT transfection reagent (Thermo Scientific, Cat# T-2004-01) following the protocol provided by the manufacturer. Non-targeting siRNA (Thermo Scientific, Cat# D-001810-10-05) was used as a negative control.
2.9. Statistical analysis
Data are expressed as means ± standard error of the mean for the number of replicates indicated. Statistical analysis was performed using one-way ANOVA or Student’s t -test, as appropriate (Prism 5 software, GraphPad). Results from the NC and HC groups were compared only to each other, using Student’s t -test, since these diets are fundamentally different in composition. Results for the same type of diet, e.g., HC diet compared to modified HC diet (+4% LE or +13.5% LE), were analyzed by ANOVA. To establish correlations, Pearson’s regression analysis was performed. A significant difference was defined as P<0.05.
3. Results
3.1. High carbohydrate diet increases and lipid emulsion attenuates the expression of PNPLA3 in mouse liver
We first analyzed how a lipogenic diet affects the expression of PNPLA3 in liver of intact mice. Compared to the group fed a stock diet (normal chow, NC), PNPLA3 mRNA was increased 7-fold in the liver of mice fed HC diet (Fig. 1A). By contrast, the inclusion of LE in the HC diet, either as HC-4% LE diet or as HC-13.5% LE diet, prevented the induction of PNPLA3 mRNA dose-dependently (Fig. 1A). Results from western blotting showed that similar changes were evident for PNPLA3 protein (Fig. 1B). Liver total TG content was significantly correlated with PNPLA3 mRNA levels in mice fed HC diet, either with or without LE (Fig. 1C).
Fig. 1. Hepatic PNPLA3 gene expression is regulated by dietary carbohydrate-lipid fuel mixture in HC-fed mice.

Liver total RNA and protein were isolated from mice fed chow, HC diet, HC-4%LE diet, or HC-13.5%LE diet and subjected to real time PCR (A), and Western blotting (B) for PNPLA3. The correlation of liver total TG content and hepatic PNPLA3 expression for all groups was analyzed by regression analysis (C). For RT-PCR results, values were normalized to GAPDH mRNA for each sample and then expressed as the mean ± SEM (n=5), where the mean of the NC group was set to 1.00. * P < 0.001 vs. NC; † P < 0.05 vs. HC; ‡ P < 0.01 vs. HC.
3.2. PNPLA3 is correlated with SREBP1c and ACC-1 in mouse liver
To determine if a similar pattern existed for other genes that are important in the lipogenic pathway, we also examined expression of sterol regulatory element binding protein (SREBP1c), a key transcriptional factor that regulates many genes involved in lipid metabolism, and acetyl-CoA carboxylase (ACC-1), which is responsible for formation of malonyl-CoA in the process of de novo fatty acid synthesis. Both SREBP1c (Fig. 2A) and ACC-1 (Fig. 2B) showed a similar expression pattern to that of PNPLA3. We observed a correlation between PNPLA3 mRNA and SREBP1c mRNA (Fig. 2C; R2 =0.62; P<0.0001) and ACC-1 mRNA (Fig. 2D; R2 =0.84; P<0.0001). SREBP1c mRNA was also correlated with ACC-1 mRNA (Fig. 2E; R2 =0.85; P<0.0001). By contrast, PNPLA2 (ATGL), which can break down TG, did not differ among diets (Supplementary Fig. 1A). We also measured PNPLA3 gene expression in mouse epididymal tissue. PNPLA3 was not different among diet groups in this tissue (Supplementary Fig. 1B), although, as shown in Supplementary Fig. 1C, PNPLA3 expression is highly expressed in murine adipose tissue.
Fig. 2. PNPLA3 expression is correlated with several lipogenic genes in mouse liver.

Liver total RNA was subject to RT-PCR for SREBP1c (A), ACC1 (B), PNPLA2/ATGL (F), and epididymal adipose PNPLA3 gene expression was also measured (G). Regression analysis was performed for PNPLA3 with SREBP1c (C), PNPLA3 with ACC1 (D), and SREBP1c with ACC1 (E). Liver PNPLA2 (ATGL) (F) and epididymal adipose PNPLA3 (G) were also measured. For RT-PCR results, values were normalized to GAPDH mRNA for each sample and then expressed as the mean ± SEM (n=5), where the mean of the NC group was set to 1.00. * P < 0.001 vs. NC; † P< 0.05 vs. HC; ‡ P < 0.01 vs. HC; §P < 0.05 vs. HC+4%LE; ¶ P <0.001 vs. HC+4%LE.
3.3. Hepatic PNPLA3 expression is not elevated in the liver of mice fed HF diet but is suppressed by exercise
Next, we studied how high-fat feeding and physical activity may affect the expression of PNPLA3. In study 2, mice fed HF diet developed fatty liver indicated by the increased level of hepatic TG (Fig. 3A). This increase was attenuated by exercise (Fig. 3A). PNPLA3 expression was not elevated in mice with HF-induced fatty liver, compared to mice fed LF diet (Fig. 3B). Strikingly, voluntary exercise reduced PNPLA3 mRNA levels by 10-fold compared to the LF and HF diet groups (Fig. 3B). The mRNA levels of fatty acid synthase (FAS) (Fig. 3C) and SREPB1c (Fig. 3D) did not differ between LF and HF diet groups. By contrast, ACC-1 was lower in the HF compared to the LF diet group (Fig. 3E). Moreover, exercise significantly suppressed both SREBP1c and ACC-1 gene expression (Fig. 3D and E).
Fig. 3. Hepatic PNPLA3 was not changed in mice fed a HF diet but was suppressed by exercise in liver of HF-fed mice.

Liver TG content was measured spectrophotometrically (A). Liver total RNA from mice fed LF and HF diets, and HF+EXE, were subject to real time PCR for PNPLA3 (B), FAS (C), SREBP-1c (D), and ACC-1(E) expression. Values were normalized to GAPDH mRNA and are expressed as the mean ± SEM (n=5), where the mean for the LF group was set to 1.0. A, * P< 0.0001 vs. LF, † P < 0.01 vs. LF, ‡ P < 0.05 vs. HF; B, * P < 0.001 vs. HF; C, * P < 0.05 vs. HF; D, *P <0.01 vs HF and † P < 0.05 vs. LF; E *P < 0.05 vs. LF and † P < 0.01 vs. HF
3.4. PNPLA3 gene expression was regulated by glucose in HepG2 cells
We investigated PNPLA3 and carbohydrate response element binding protein (ChREBP) gene expression in HepG2 cells exposed to “depletion-re-feeding conditions” in which cells were cycled between no glucose (depletion), low, and high glucose media. PNPLA3 gene expression was reduced after 8 h of depletion (P < 0.05), while the level returned to base line 8 h after high glucose refeeding (Fig. 4A). After this, reduction in glucose resulted in a moderate reduction in expression, while high glucose for 8 to 24 h increased the level of expression, which, subsequently, was again completely abrogated after 16 h of starvation. ChREBP exhibited a similar pattern of expression to that of PNPLA3 (Fig. 4B), and correlation analysis showed that PNPLA3 mRNA levels were highly significantly correlated with ChREBP mRNA levels (Fig. 4C).
Fig. 4. PNPLA3 gene expression was regulated by glucose in HepG2 cells.
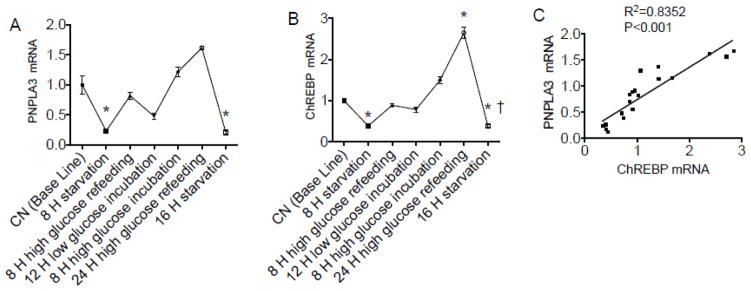
HepG2 cells were exposed to depletion-refeeding conditions in culture. Real time PCR was performed to determine mRNA levels of PNPLA3 (A) and ChREBP (B). The correlation of PNPLA3 and ChREBP was analyzed by regression analysis (C). The mean for the baseline group (A, B) was set to 1.0. A and B, * P < 0.05 vs. baseline; † P < 0.05 vs. 24 h high glucose refeeding.
3.5. PNPLA3 gene expression varies in response to glycolytic intermediates in HepG2 cells
A series of experiments were conducted to determine whether metabolism of glucose was essential for induction of PNPLA3, and whether a specific glycolytic intermediate could be responsible for regulating PNPLA3 expression. These results showed that treatment with the non-metabolizable glucose analog, 2-deoxy-D-glucose, did not induce PNPLA3 expression (Fig. 5A). Similarly, SREBP1c gene expression was induced two-fold by high glucose compared to low glucose, but suppressed by 2-deoxy-D-glucose (Fig 5B). Oil Red O staining showed that high glucose resulted in increased lipid accumulation, whereas 2-deoxy-D-glucose led to reduction of lipid content in cells (Supplementary Fig. 2A and B). Incubation with citrate, a Krebs cycle intermediate downstream of glucose metabolism, increased PNPLA3 levels, whereas pyruvate and acetate were ineffective (Fig. 5C). Expression patterns of FAS and ACC-1 were similar to that of PNPLA3 (Fig. 5D and E). In order to examine the specificity of PNPLA3 to citrate, cells were treated with the isomer isocitrate. The result showed that PNPLA3 was not regulated by isocitrate.
Fig. 5. PNPLA3 gene expression was regulated by glycolytic intermediates in HepG2 cells.

HepG2 cells were incubated in medium with high glucose (25 mM) or 2-deoxy-glucose (20 mM) for 24 h and the levels of PNPLA3 mRNA (A) and SREBP1c (B) were measured by RT-PCR. Glycolytic intermediates, citrate (7 mM), isocitrate (7mM), pyruvate (20 mM), and acetate (20 mM) were added to medium and cells were harvested after 24 h. Total RNA from HepG2 cells was subjected to RT-PCR analysis for PNPLA3 (C and F), FAS (D) and ACC-1 (E). Values were normalized to β-actin mRNA and are expressed as the mean ± SEM (n=3). A and B, * P < 0.05 vs. low glucose, † P< 0.05 vs. low glucose, ‡ P < 0.05 vs high glucose; C, D, and E,* P< 0.001 vs. all other groups.
3.6. PNPLA3 gene does not response to insulin and glucagon in HepG2 cells
Studies conducted with 3T3-L1 adipocytes (23) and human adipose tissue (24) have shown that PNPLA3 gene expression is regulated by insulin. In addition, one study showed that insulin increased promoter activity of PNPLA3 gene expressed in Chinese hamster ovary cells (25). Therefore, we examined whether hepatic PNPLA3 is regulated by insulin in HepG2 cells, which express the insulin receptor protein (26). Incubation of HepG2 cells with 10 nM insulin for 24 h did not increase PNPLA3 mRNA compare to control cells (Supplementary Fig. 3A). By contrast, the same treatment induced the expression of FAS, a known target of insulin action, by 2-fold (Supplementary Fig. 3A, B). Taken together, these data suggest that hepatic PNPLA3 does not response to insulin stimulation in HepG2 human hepatocytes. Because our in vivo data showed that exercise suppressed PNPLA3 expression, we hypothesized that exercise may stimulate glucagon secretion, and thereby decrease PNPLA3 gene expression. Treatment of HepG2 cells with 100 nM glucagon in low (5.5 mM) glucose medium for 24 hours had no effect on PNPLA3 mRNA expression (Supplementary Fig. 3C), whereas glucagon stimulated the expression of pyruvate carboxylase (PC), a known target of glucagon action (Supplementary Fig. 3D).
3.7. PNPLA3 gene expression is reduced in HepG2 cells by several unsaturated fatty acids
The major fatty acid components of the LE we employed, Intralipid®, are linoleic acid (LA), oleic acid (OA), and α-linolenic acid (ALA). Since the results of our studies demonstrated that Intralipid completely reversed the induction of PNPLA3 by HC-diet in mouse liver (Fig. 1), we investigated whether certain types of fatty acids affect PNPLA3 gene expression when added directly to HepG2 cells. Interestingly, treatment with either 400 μM OA (Fig. 6A) or LA (Fig. 6B) suppressed PNPLA3 gene expression in both low glucose and high glucose conditions. By contrast, ALA had no effect on PNPLA3 gene expression under either condition (Fig. 6C). We further explored the effects of two long-chain polyunsaturated fatty acids, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) on PNPLA3 gene expression. Both EPA and DHA suppressed PNPLA3 gene expression under both low and high glucose conditions (Fig. 6D). Moreover, the combination of OA, LA, and ALA (ratio: 6:3:1) significantly suppressed gene expression of PNPLA3 (Fig. 6E), SREBP1c (Fig. 6F), and FAS (Fig. 6G) in both low glucose and high glucose conditions. All of these fatty acids resulted in lipid accumulation as measured by Oil Red O staining (Supplementary Fig. 2A and C).
Fig. 6. PNPLA3 expression is regulated by unsaturated fatty acids in HepG2 cells cultured in low and high glucose media.
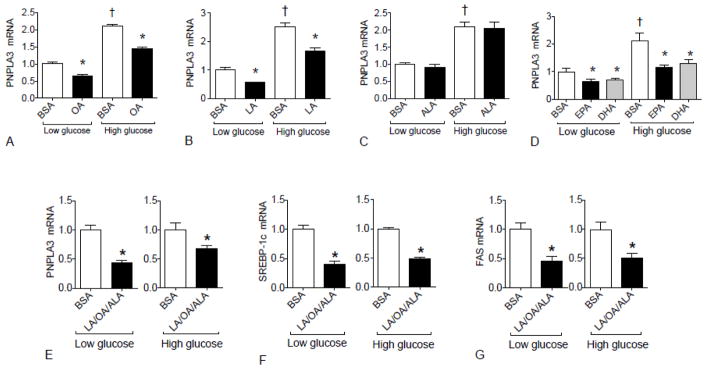
He pG2 cells were treated with 400 μM of the indicated individual fatty acids or the combination of LA, OA, and ALA (molar ratio: 6:3:1) or BSA alone either under low (5.5 mM) glucose or high (25 mM) glucose conditions for 24 h. RT-PCR analysis for PNPLA3 gene expression is shown for cells treated individually with OA (A), LA (B), ALA (C), or EPA or DHA (D), and with the mixture of LA, OA, and ALA in (E). SREBP1c (F) and FAS (G) were determined in cells treated with the mixture of LA, OA, and ALA. The mean for the albumin-treated control cells was set to 1.00. A, B, * P < 0.05 vs. BSA+Low glucose or BSA+High glucose; † P< 0.05 vs. BSA+Low glucose; C, † P < 0.05 vs. BSA+ Low glucose; D, * P < 0.05 vs. BSA+Low glucose or BSA+High glucose; † P < 0.05 vs. BSA+Low glucose; E, F and G * P < 0.05 vs. BSA.
3.8. PNPLA3 knockdown did not prevent lipid accumulation but increased FAS expression
Since PNPLA3 gene expression is regulated by nutritional signals and our data had implicated PNPLA3 as a lipogenic signal, we investigated whether the knockdown of PNPLA3 in HepG2 cells affects glucose or lipid metabolism. Cells transfected with scrambled siRNA as a non-targeting control were compared to cells transfected with PNPLA3-specific siRNA. Transfection with PNPLA3 siRNA decreased PNPLA3 mRNA and protein by ~65% and ~70%, respectively in HepG2 cells (Fig. 7A, B and C). However, Oil Red O staining showed no difference between cells with knocked down PNPLA3 and controls under either low or high glucose conditions (Fig. 7E). PNPLA3 knockdown also had no effect on lipid content when cells were incubated with exogenous fatty acid (OA) (Fig. 7E). Interestingly, we found that FAS mRNA was increased approximately 2 fold in PNPLA3 siRNA-treated cells compared to control siRNA-treated cells (Fig. 7D). By contrast, knockdown of FAS using a strategy similar to that used for PNPLA3 had no effect on the level of PNPLA3 expression (data not shown).
Fig. 7. PNPLA3 knockdown did not alter lipid accumulation from fatty acid but increased FAS expression in HepG2 cells.

The levels of PNPLA3 mRNA (A) and protein (B and C), and of FAS mRNA (D) were determined in HepG2 cells treated with control siRNA or PNPLA3-specific siRNA. The value for control siRNA cells was set to 1.0. In E, HepG2 cells that were pretreated with the indicated siRNA were subjected to either low (5.5 mM) glucose or high (25 mM) glucose for 48 h, or with either BSA or BSA-bound OA (400 μM) for 24 h and then stained with Oil Red O. The optical density of Oil Red O that was eluted from the stained and washed HepG2 cells was measured at 510 nm for quantification (F and G). The data are the mean ± SEM, n=3 wells/treatment. A, C and D, *P < 0.001 vs. control siRNA.
4. Discussion
In the present study, we investigated how dietary changes in carbohydrates and lipids affect PNPLA3 expression in mouse liver and HepG2 cells. The novel results of our in vivo studies include that PNPLA3 gene expression (mRNA) is increased ~7-fold in the liver of mice fed a HC diet with 0.5% fat. However, the addition of fat at either 4 or 13.5 % LE reduced PNPLA3 mRNA dose dependently in the liver of HC-fed mice. In our in vitro studies, we observed that glucose, citrate, and certain types of unsaturated fatty acids also modulated the level of PNPLA3 expression.
The exact role of PNPLA3 in the development of NAFLD remains poorly understood, although a number of studies have been performed (27). Our study revealed that PNPLA3 appears to serve as a reliable signal of the liver’s response to a high carbohydrate intake, and can potentially be used as a biomarker for NAFLD caused by carbohydrate overfeeding. In agreement with our finding, a human study showed that the interaction of PNPLA3 genotype and simple carbohydrate intake determines hepatic fat accumulation (28). The strong association of PNPLA3 with important lipogenic genes, SREBP1c and ACC-1, suggests that these genes are all part of a common program, which likely represents elevated lipogenesis under HC conditions and attenuated lipogenesis when the diet includes an appropriate amount of dietary fat. Our data are consistent with other studies that have shown that PNPLA3 is under the direct control of the transcription factor SREBP1c in response to glucose treatment (22, 29). By contrast, the expression level of PNPLA2 (also known as adipose triglyceride lipase, ATGL), which shares 70% similarity with PNPLA3, was not changed in HC-fed mice. The lack of regulation of PNPLA2, which is known to be an important TG hydrolase, and the lack of regulation of PNPLA3 in adipose tissue suggests that PNPLA3 in the liver is regulated independently of TG hydrolysis and in a tissue-specific manner. In addition, other studies have shown that nutritional regulation of PNPLA2 is opposite to that of PNPLA3 (14, 23). Notably, the expression pattern of PNPLA3 between human and rodent is different. Our present study showed that hepatic PNPLA3 mRNA is ~ 250-fold lower compared to PNPLA3 expression in adipose tissue. By contrast, human PNPLA3 expression is highest in liver and second highest in skin. The difference of tissue distribution of PNPLA3 between human and rodent are not yet understood. Nevertheless, the results of our studies in human liver cells and in mice in vivo were generally concordant.
The combined results of study 1 and study 2 strongly suggest that PNPLA3 expression is not responding to increases in body weight, or to accumulating liver fat content. In our HF diet-induced fatty liver model (study 2), we did not observe a significant change of PNPLA3 gene expression due to HF diet. These data indirectly support that the notion that PNPLA3 is related to de novo lipogenesis, since the hepatic steatosis in HF-fed mice is mainly due to overflow of fatty acids coming from lipolysis of adipose tissue and dietary fat, and de novo lipogenesis is less important (30,31). Therefore, hepatic steatosis per se does not alter PNPLA3 expression, but lipogenic stimuli do determine the level of hepatic PNPLA3 gene expression. Interestingly, exercise markedly lowered PNPLA3 expression, below that of LF-fed control mice in study 2. Moreover, exercise significantly suppressed both SREBP1c and ACC-1 expression, which indicates that exercise may be a mediator of hepatic lipogenesis. The fact that these genes are regulated in a similar manner by exercise further suggests they are all commonly related to hepatic lipogenesis. While we hypothesized that hormonal changes stimulated by exercise could be responsible for the reduction of PNPLA3, we did not observe any change in PNPLA3 gene expression in HepG2 cells treated directly with either glucagon or insulin. Together, these data suggest that PNPLA3 is positively regulated when there is a surfeit of carbohydrate relative to fat, and it is reduced when metabolism and energy utilization are increased, as in exercise.
Our studies in HepG2 cells further explored carbohydrate and fatty acids as potential regulators of PNPLA3 expression levels. PNPLA3 gene expression was highly sensitive to glucose concentration, and was rapidly and reversibly regulated under cellular depletion–refeeding conditions. The association of PNPLA3 with ChREBP suggests that this transcription factor may be a critical regulator. In agreement with our data, studies have shown that PNPLA3 is under the direct control of the transcription factor ChREBP in response to glucose treatment (22, 29). Our studies also reveal that the metabolism of glucose must be crucial, as PNPLA3 was reduced, by addition of 2-deoxy-D-glucose. The metabolic mediators that link glucose metabolism and lipogenic gene transcription have not been identified, but several candidates, such as pyruvate, acetate, and xylulose-5-phosphate, have been proposed as the key signaling compounds (32). Therefore, we explored the effects of citrate, pyruvate and acetate on PNPLA3 expression. The induction of PNPLA3 by citrate suggests that the oxidation of glucose, which generates adequate oxaloacetic acid for the production of citrate, or direct addition of citrate to cells, is required for the upregulation of PNPLA3 expression in this hepatocyte model.
The provision of fatty acids reduced PNPLA3 expression, both in vivo in the form of LE and in HepG2 cells in the form of added BSA-bound fatty acid. In HepG2 cells, the reduction in PNPLA3 expression by free fatty acid was observed regardless of glucose concentration, suggesting an independent regulatory effect of fatty acids. Suppression of PNPLA3 by OA or certain polyunsaturated fatty acids (linoleic, EPA and DHA, but not ALA) is surprising and unexplained, but interesting. As ALA, EPA and DHA are all n (omega)-3 fatty acids, while LA and OA are n-6 and n-9 fatty acids, respectively, the effect of fatty acid on PNPLA3 expression apparently cannot be attributed to any particular class of unsaturated fatty acid. Other studies have shown that polyunsaturated fatty acids inhibit lipogenic genes by reducing SREBP-1c nuclear abundance and inhibiting ChREBP nuclear translocation through transcriptional and posttranslational mechanisms, including suppression of SREBP-1c gene transcription, enhancement of SREBP1c mRNA decay, and inhibition of ChREBP phosphorylation (33, 34). Consistent with the above studies, our study revealed that the combination of OA and LA suppressed the main lipogenic genes, SREBP1c and FAS, and also PNPLA3. Further studies are needed to elucidate the mechanism by which fatty acids regulate PNPLA3 expression.
The knockdown of PNPLA3 did not affect total lipid content when HepG2 cells were incubated in normal medium or treated either with high glucose or OA. Consistent with our study, it has been show that knockdown or overexpression of PNPLA3 in vitro did not change cellular TG content (7, 23). It is also possible that because our PNPLA3 knockdown resulted in only a 70% reduction in PNPLA3 protein levels, this may not be sufficient to induce a phenotypic change. Alternatively, some compensatory mechanisms could be activated to maintain the homeostasis of glucose and lipid when PNPLA3 expression is reduced. Supporting the latter explanation, we observed an up-regulation of FAS mRNA in PNPLA3 knockdown cells. FAS is a critical enzyme in lipogenesis and thus the compensatory of up-regulation of FAS may contribute to unchanged lipid content in PNPLA3 deficient cells. The knockout of PNPLA3 in mice, which was without effect on phenotype (14, 15), supports this compensatory hypothesis.
In conclusion, our study provides strong evidence revealing that PNPLA3 is upregulated by a lipogenic diet, and suppressed by certain types of unsaturated fatty acids both in vivo and in vitro. These results suggest that PNPLA3 expression is an early marker of carbohydrate-induced lipogenesis, but its expression is not associated with steatosis per se. Thus, PNPLA3 is a marker of the pathway to the lipogenic condition rather than the outcome of increased hepatic lipid accumulation.
Supplementary Material
Acknowledgments
Funding: This work was supported by Graduate Program in Nutrition and NIH CA90214 (A.C. Ross), and NIH AT004678 (J.D. Lambert). We thank Abbott Laboratories (Baxter) for providing the liquid diet (Clinimix E®), vitamins, and lipid emulsion (Intralipid®) that were used in study 1, and for a postdoctoral research award to K.I.
Abbreviations
- ACC1
acetyl CoA-carboxylase
- ALA
α-linolenic acid
- BSA
bovine serum albumin
- ChREBP
carbohydrate response element binding protein
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- HC
high-carbohydrate
- HF
High fat
- LE
lipid emulsion
- LA
linoleic acid
- NAFLD
nonalcoholic fatty liver disease
- OA
oleic acid
- PNPLA3
Patatin-like phospholipase domain containing 3
- siRNA
silencing (short-interference) RNA
- SREBP1c
sterol regulatory element binding protein 1c
- TG
triglyceride
Footnotes
Conflict of interest/Financial Disclosure Statement: The authors have no conflicts of interest of financial disclosures.
Author contributions: LH, designed experiments (study 1 and in vitro experiments) and conducted assays, analyzed data, and wrote draft manuscript; KI, conducted in vivo experiments and reviewed manuscript; K-H H, conducted assays and reviewed manuscript; SS-t, conducted in vivo experiments (study 2), provided samples and reviewed manuscript; JDL, designed in vivo experiments (study 2), provided samples and reviewed manuscript; ACR, designed experiments (study 1 and in vitro), reviewed data, and edited final manuscript. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- 2.Li YY. Genetic and epigenetic variants influencing the development of nonalcoholic fatty liver disease. World J Gastroenterol. 2012;18:6546–6551. doi: 10.3748/wjg.v18.i45.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santoro N, Kursawe R, D’Adamo E, Dykas DJ, Zhang CK, Bale AE, Cali AM, et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology. 2010;52:1281–1290. doi: 10.1002/hep.23832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawaguchi T, Sumida Y, Umemura A, Matsuo K, Takahashi M, Takamura T, Yasui K, et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One. 2012;7:e38322. doi: 10.1371/journal.pone.0038322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Zhao Q, Wu K, Fan D. I148M variant of PNPLA3 confers increased risk for nonalcoholic fatty liver disease not only in European population, but also in Chinese population. Hepatology. 2011;54:2275. doi: 10.1002/hep.24567. [DOI] [PubMed] [Google Scholar]
- 7.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res. 2005;46:2477–2487. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, Cohen JC, Hobbs HH. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao A, Liang J, Ke Y, Li C, Cui Y, Shen L, Zhang H, et al. Mouse patatin-like phospholipase domain-containing 3 influences systemic lipid and glucose homeostasis. Hepatology. 2011;54:509–521. doi: 10.1002/hep.24402. [DOI] [PubMed] [Google Scholar]
- 12.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumashiro N, Yoshimura T, Cantley JL, Majumdar SK, Guebre-Egziabher F, Kursawe R, Vatner DF, et al. The role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology. 2012 doi: 10.1002/hep.26170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52:318–329. doi: 10.1194/jlr.M011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52:1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruhanen H, Perttilä J, Hölttä-Vuori M, Zhou Y, Yki-Järvinen H, Ikonen E, Käkelä R, Olkkonen VM. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55:739–46. doi: 10.1194/jlr.M046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito K, Hao L, Wray AE, Ross AC. Lipid emulsion administered intravenously or orally attenuates triglyceride accumulation and expression of inflammatory markers in the liver of nonobese mice fed parenteral nutrition formula. J Nutr. 2013;143:253–259. doi: 10.3945/jn.112.169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sae-tan S, Rogers CJ, Lambert JD. Voluntary exercise and green tea enhance the expression of genes related to energy utilization and attenuate metabolic syndrome in high fat-fed mice. Mol Nutr Food Res. 2013 Dec 27; doi: 10.1002/mnfr.201300621. [DOI] [PubMed] [Google Scholar]
- 19.Javitt NB. Hep G2 cells as a resource for metabolic studies: lipoprotein, cholesterol, and bile acids. FASEB J. 1990;4:161–168. doi: 10.1096/fasebj.4.2.2153592. [DOI] [PubMed] [Google Scholar]
- 20.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 21.Sardesi VM, Manning JA. The determination of triglycerides in plasma and tissues. Clin Chem. 1968;114:147–52. [Google Scholar]
- 22.Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, Hobbs HH. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes. 2006;55:148–157. [PMC free article] [PubMed] [Google Scholar]
- 24.Moldes M, Beauregard G, Faraj M, Peretti N, Ducluzeau PH, Laville M, Rabasa-Lhoret R, et al. Adiponutrin gene is regulated by insulin and glucose in human adipose tissue. Eur J Endocrinol. 2006;155:461–468. doi: 10.1530/eje.1.02229. [DOI] [PubMed] [Google Scholar]
- 25.Rae-Whitcombe SM, Kennedy D, Voyles M, Thompson MP. Regulation of the promoter region of the human adiponutrin/PNPLA3 gene by glucose and insulin. Biochem Biophys Res Commun. 2010;402:767–772. doi: 10.1016/j.bbrc.2010.10.106. [DOI] [PubMed] [Google Scholar]
- 26.Williams JF, Olefsky JM. Defective insulin receptor function in down-regulated HepG2 cells. Endocrinology. 1990;127:1706–1717. doi: 10.1210/endo-127-4-1706. [DOI] [PubMed] [Google Scholar]
- 27.Sookoian S, Pirola CJ. PNPLA3, the history of an orphan gene of the potate tuber PROTEIN family that found an organ: the Liver. Hepatology. 2013 doi: 10.1002/hep.26895. [DOI] [PubMed] [Google Scholar]
- 28.Davis JN, Le KA, Walker RW, Vikman S, Spruijt-Metz D, Weigensberg MJ, Allayee H, et al. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr. 2010;92:1522–1527. doi: 10.3945/ajcn.2010.30185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubuquoy C, Robichon C, Lasnier F, Langlois C, Dugail I, Foufelle F, Girard J, et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J Hepatol. 2011;55:145–153. doi: 10.1016/j.jhep.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 30.Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes Metab. 2010;12 (Suppl 2):83–92. doi: 10.1111/j.1463-1326.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 31.Ren LP, Chan SM, Zeng XY, Laybutt DR, Iseli TJ, Sun RQ, Kraegen EW, et al. Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS One. 2012;7:e30816. doi: 10.1371/journal.pone.0030816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Towle HC, Kaytor EN, Shih HM. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu Rev Nutr. 1997;17:405–433. doi: 10.1146/annurev.nutr.17.1.405. [DOI] [PubMed] [Google Scholar]
- 33.Jump DB, Tripathy S, Depner CM. Fatty acid-regulated transcription factors in the liver. Annu Rev Nutr. 2013;33:249–269. doi: 10.1146/annurev-nutr-071812-161139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab. 2013;24:257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.