

Background: Tumor necrosis factor α (TNFα) impairs insulin signaling in the retina.
Results: Pioglitazone reduced TNFα- and SOCS3-activated insulin resistance pathways in retinal cells as well as in lysates from whole rat retina.
Conclusion: PPARγ regulates insulin signaling in retina.
Significance: Increased understanding of retinal insulin signaling may lead to new therapies for type 2 diabetes.
Keywords: Insulin Receptor, Insulin Receptor Substrate 1 (IRS-1), Insulin Resistance, Suppressor of Cytokine Signaling 3 (SOCS3), Tumor Necrosis Factor (TNF), Type 2 Diabetes, Diabetic Retinopathy
Abstract
Dysfunctional insulin signaling is a key component of type 2 diabetes. Little is understood of the effects of systemic diabetes on retinal insulin signaling. A number of agents are used to treat patients with type 2 diabetes to normalize glucose levels and improve insulin signaling; however, little has been done to investigate the effects of these agents on retinal insulin signal transduction. We hypothesized that pioglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) agonist, would normalize retinal insulin signal transduction through reduced tumor necrosis factor α (TNFα) and suppressor of cytokine signaling 3 (SOCS3) activities in whole retina and retinal endothelial cells (REC) and Müller cells. To test this hypothesis, we used the BBZDR/Wor type 2 diabetic rat model, as well as REC and Müller cells cultured in normoglycemia and hyperglycemic conditions, to investigate the effects of pioglitazone on TNFα, SOCS3, and downstream insulin signal transduction proteins. We also evaluated pioglitazone's effects on retinal function using electroretinogram and markers of apoptosis. Data demonstrate that 2 months of pioglitazone significantly increased electroretinogram amplitudes in type 2 diabetic obese rats, which was associated with improved insulin receptor activation. These changes occurred in both REC and Müller cells treated with pioglitazone, suggesting that these two cell types are key to insulin resistance in the retina. Taken together, these data provide evidence of impaired insulin signaling in type 2 diabetes rats, which was improved by increasing PPARγ activity. Further investigations of PPARγ actions in the retina may provide improved treatment options.
Introduction
Rates of type 2 diabetes are expected to continually increase due to increased obesity worldwide (1). In order to best treat these patients, we need to understand both systemic and organ-specific changes related to diabetes and the mechanisms by which therapeutics offer potential treatment. Because diabetic retinopathy is a debilitating complication of both type 1 and type 2 diabetes, it is particularly important to understand how treatments for controlling systemic glucose might affect retinal function. Peroxisome proliferator-activated receptor γ (PPARγ)2 agonists, such as rosiglitazone or pioglitazone, have been commonly prescribed for systemic glucose control, yet their effects on retinal function have yet to be established. Systemic effects of these drugs show promise for treatment of diabetes based on their ability to increase insulin sensitivity, modify lipid profiles, decrease blood pressure, and decrease inflammatory mediators (2–4).
Cell-specific effects of the PPARγ agonist pioglitazone have been reported in adipocytes, indicating that treatment eliminates TNFα-induced insulin resistance (5). It has also been shown that pioglitazone increases IRS-1 levels in muscle and liver lysates from streptozotocin-treated rats or rats fed a high sucrose diet (6). An important but unresolved question is whether PPARγ agonists alter interactions between TNFα in two retinal cell types, retinal endothelial cells (REC) and Müller cells, known to be pivotal in the development of diabetic retinopathy.
We hypothesize that pioglitazone inhibition of TNFα actions may be key in its potential protective effects in diabetic retinopathy, because TNFα has been linked to insulin resistance in other tissues. Results from our laboratory and others have indicated that diabetes increases TNFα levels, which in turn triggers apoptosis of retinal cells and leads to diabetic retinopathy (7, 8). Use of TNFα receptor knock-out mice to investigate whether diabetes-induced increase in TNFα in the retina is key to diabetic retinopathy showed that loss of TNFα protected the retina against the effects of high glucose on formation of degenerate capillaries and pericyte ghosts, two key markers of vascular damage in early diabetic retinopathy (7). Also, we have recently shown that TNFα can induce phosphorylation of IRS-1 on serine 307 to block Akt activity, leading to apoptosis in both REC (9) and Müller cells (10). Increased understanding of pioglitazone actions on TNFα may provide novel insight into mechanisms by which TNFα can induce insulin resistance in retinal cells.
In addition to directly regulating insulin resistance through phosphorylation of IRS-1, TNFα also increases SOCS1/SOCS3 levels (11), which in turn blocks IRS-1, resulting in increased REC apoptosis (9). SOCS3 is also reported to inhibit insulin signaling by other potential mechanisms as well, including increased phosphorylation of insulin receptor on tyrosine 960 (IRTyr-960), which inhibits the interaction between insulin receptor and IRS-1 (12). We recently have shown that SOCS3 can also directly regulate TNFα,3 suggesting a cross-talk between TNFα and SOCS3 pathways that may induce insulin resistance. Of particular interest is the report that PPARγ agonists decrease both SOCS3 and TNFα in hepatic tissues in rats fed a high fat diet (13).
The goal of this study was to investigate whether pioglitazone could restore insulin signal transduction through reduction of TNFα/SOCS3 pathways in REC and Müller cells in vitro and in a type 2 diabetic rat model, BBZDR/Wor, in vivo. The data demonstrate that pioglitazone normalized glucose blood levels in obese diabetic rats and improved retinal function measured by electroretinogram amplitudes. Additionally, pioglitazone reduced TNFα and SOCS3-activated insulin resistance pathways, including phosphorylation of IRS-1 on serine 307 and insulin receptor on tyrosine 960, in REC and Müller cells, as well as in lysates from whole rat retina. This reduction in TNFα/SOCS3 was associated with reduced apoptotic proteins and increased levels of Akt and Bcl-xL, two key antiapoptotic proteins.
MATERIALS AND METHODS
Animals
BBZDR/Wor lean (10 male) and obese rats (10 male) were purchased from Biomedical Research Models (Worcester, MA). Glucose measurements were taken weekly, and once obese rats reached a glucose of >250 mg/dl, an initial electroretinogram (ERG) was taken prior to treatment initiation. Five lean and five obese rats were placed into treatment groups and received a daily intraperitoneal injection of 25 mg/kg pioglitazone for 2 months. After 2 months of treatment, animals in all four groups (lean, lean + pioglitazone, obese, obese + pioglitazone) underwent an additional ERG prior to sacrifice.
The BBZDR/Wor rat model of type 2 diabetes has been used for studies of autonomic neuropathy (14, 15), myogenic tone of the ophthalmic artery (16), and vascular damage repair using endothelial cell progenitors (17). The BBZDR/Wor rat was produced using the BBZDP/Wor (BB rat with an Iddm2 type 1 genetic locus) and mating with lean BBZ/Wor rats (BB rat with Zucker diabetic gene) to remove the Iddm2 locus. The obese male BBZDR/Wor rat spontaneously develops type 2 diabetes at 10 weeks of age (100%) when fed standard rat chow (18, 19). The obese BBZDR/Wor rat is lymphopenic, hyperinsulinemic with peripheral insulin resistance and develops spontaneous autoimmune non-insulin-dependent diabetes mellitus at an age of 70 days (18). Results in the oxygen-induced retinopathy model and the STZ model using endothelial cell progenitor cells suggest that the BBZDR/Wor rat is a good model of type 2 diabetic retinal changes (17).
ERG
Prior to sacrifice for protein analyses, animals were subjected to ERG analyses to evaluate the changes in the electrical activity of the retina as we have described previously (20, 21). Briefly, rats were dark-adapted overnight. ERG responses were recorded from both eyes together using platinum wire corneal electrodes, a forehead reference electrode, and a ground electrode in the tail. Pupils were fully dilated using 1% tropicamide solution (Alcon). Methylcellulose (Celluvise, Allergan, Irvine, CA) drops were applied as well to maintain a good electrical connection, and body temperature was maintained at 37 °C by a water-based heating pad. All ERG experiments were approved by the University of Tennessee Institutional Animal Care and Use Committee on Protocol 1992. ERG waveforms were recorded with a bandwidth of 0.3–100 Hz and samples were recorded at 2 kHz by a digital acquisition system and analyzed by a custom-built program (MatLab). Statistical analysis was applied to the mean ± S.D. amplitudes of the A- and B-wave of each treatment group. Comparisons were made of ERG amplitudes, but implicit times were not measured.
Retinal Endothelial Cells
Primary human REC were obtained from Cell Systems Corp. (Kirkland, WA). Cells were grown in M131 medium containing microvascular growth supplements, 10 μg/ml gentamycin, and 0.25 μg/ml amphotericin B (Invitrogen). Before the experiments, cells were transferred to high glucose (25 mm) medium or maintained in normal glucose (5 mm) medium and grown to 80% confluence. Primary cells (passages 2–4) were used. Cells were quiesced by incubating in high or normal glucose medium without growth factors for 18–24 h and then treated with 25 μm pioglitazone for 24 h.
Müller Cells
Rat retinal Müller cells (rMC-1; courtesy of Vijay Sarthy, Northwestern University) were grown in 5 or 25 mm glucose DMEM (HyClone Laboratories, Logan, UT) supplemented with 10% FBS and antibiotics. Cells were cultured to 80% confluence (2–4 days), and then cells were starved for 18–24 h by reduction to 2% FBS in the growth medium to eliminate any residual growth factors in the serum. We chose to reduce serum to 2% rather than remove it completely, in order to eliminate activation of apoptotic pathways. We have used this method in the past for measurements of TNFα and insulin pathways (22). In preliminary studies, we initiated work in Müller cells using 25 μm pioglitazone because this was effective in increasing PPARγ activity in REC experiments; however, this dose did not significantly increase PPARγ activity in Müller cells. Therefore, we increased the dose to 50 μm pioglitazone in Müller cells and found significantly increased PPARγ at this dose (Fig. 7), so 50 μm pioglitazone was used for all further work.
FIGURE 7.
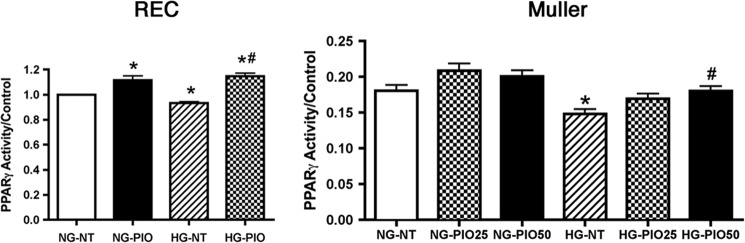
PPARγ activity is increased in REC and Müller cells after pioglitazone. For both REC and Müller cells, cells were grown in normal glucose (NG) or high glucose (HG). For REC, cells were treated with 25 μm pioglitazone (PIO), whereas Müller cells were untreated (NT) or treated with 25 or 50 μm pioglitazone. *, p < 0.05 versus normal glucose untreated; #, p < 0.05 versus high glucose untreated. Data are mean ± S.E. n = 4 for all treatments.
PPARγ Antagonist
Both REC and Müller cells were treated with T0070907 prior to pioglitazone treatment to verify that pioglitazone actions on the cells were specific to PPARγ actions. T0070907 is a highly selective PPARγ antagonist. Cells were treated at 50 nm (23) for 30 min prior to pioglitazone administration, as described above. The dose used to inhibit PPARγ activity in our system is much lower than reported in other studies (ranging from 1 to 50 μm).
Western Blotting
After appropriate treatments and rinsing with cold phosphate-buffered saline, whole retinal lysates or cell lysates were collected in lysis buffer containing protease and phosphatase inhibitors and scraped into the tubes. Protein extracts were prepared by sonication. Equal amounts of protein from the cell or tissue extracts were separated on the precast Tris-glycine gel (Invitrogen), blotted onto a nitrocellulose membrane. After blocking in TBST (10 mm Tris-HCl buffer, pH 8.0, 150 mm NaCl, 0.1% Tween 20) and 5% (w/v) BSA, the membrane was treated with appropriate primary antibodies followed by incubation with secondary antibodies labeled with horseradish peroxidase. Antigen-antibody complexes were detected by a chemiluminescence reagent kit (Thermo Scientific). Primary antibodies used were SOCS3, phosphorylated IRS-1 (Ser-307), total IRS-1, phosphorylated Akt (serine 473), Akt, cytochrome c, Bax, Bcl-xL, phosphorylated insulin receptor (tyrosine 1150/1151), and insulin receptor (all purchased from Cell Signaling, Danvers, MA). Insulin receptor phosphorylated on tyrosine 960 was purchased from Cell Applications (San Diego, CA). PPARγ antibody was purchased from R&D Systems (Minneapolis, MN). β-Actin was purchased from Santa Cruz Biotechnology, Inc.
PPARγ Transcription Factor Assay
PPARγ activity was determined with the ELISA-based PPARγ transcription factor assay kit (Abcam, Cambridge, MA) following the manufacturer's protocol. In brief, 100 μg of nuclear extract from retinal endothelial cells and Müller cells was added to each well of a 96-well plate precoated with a specific double-stranded DNA (dsDNA) sequence containing the peroxisome proliferator response element (PPRE). The wells were incubated overnight at 4 °C without agitation. Specific primary antibody directed against PPARγ was added, followed by HRP-conjugated secondary antibody. PPARγ was detected with sensitive colorimetric readout at 450 nm after the addition of developing and stop solution.
ELISA Analyses
A TNFα ELISA (Fisher) and IGFBP-3 ELISA (R&D Systems) were used according to the manufacturer's instructions. Fifty μg of protein was used to ensure equal protein amounts in all wells. A cleaved caspase 3 ELISA was purchased from Cell Signaling (Danvers, MA) with equal protein loaded, so optical density measurements can be used for analyses.
Statistics
For all experiments, excluding ERG studies, data are presented as means ± S.E. Five animals were used in each group for all in vivo experiments. At least four separate dishes of each treatment were used for all cell culture experiments. Data were analyzed by a Kruskal-Wallis test, followed by a post hoc Dunn's test. A representative blot is shown for Western blot results.
RESULTS
Pioglitazone Treatment Improved the Glucose Levels of Obese Rats, with No Change in Intraocular Pressure or Body Weight
Within 1 week of daily treatment with 25 mg/kg pioglitazone, the BBZDR/Wor obese rats had glucose levels similar to those of lean animals, which were maintained throughout the remainder of the 2-month study (Table 1). Treatment of lean and obese rats with pioglitazone did not alter intraocular pressure levels. Pioglitoazone increased the weight of the obese animals by 36% (Table 1). We measured intraocular pressure to ensure that pioglitazone did not produce deleterious effects in the eye. This suggests that although pioglitazone did not decrease the obesity of the rats, it was still highly effective in lowering blood glucose levels.
TABLE 1.
Summary data for lean (control), lean + pioglitazone (25 mg/kg intraperitoneally; Ctrl+pio) for 2 months, obese (Diab), and obese + pioglitazone (Diab+pio)
IOP, intraocular pressure.
| Groups | n | Body weight | Glucose | IOP |
|---|---|---|---|---|
| g | mg/dl | mm Hg | ||
| Ctrl | 5 | 475.5 ± 21.8 | 96.7 ± 20.1 | 10.1 ± 1.8 |
| Ctrl+pio | 5 | 470.3 ± 27.5 | 82.3 ± 15.2 | 9.8 ± 1.2 |
| Diab | 5 | 661 ± 16.5a | 524.8 ± 34.1a | 9.1 ± 1.1 |
| Diab+pio | 5 | 900 ± 38.1b | 89 ± 22.9b | 9.5 ± 1.6 |
a p < 0.05 versus lean.
b p < 0.05 versus obese.
Pioglitazone Treatment Significantly Increased PPARγ Activity in Whole Retinal Lysates of BBZDR/Wor Obese Rats
To ensure that pioglitazone was working through expected pathways in the retina, we measured PPARγ levels in the retina of all animals. We had to use a Western blot for this assay because we only had whole retinal lysates from the animals and could not isolate the nuclear extract as we did for cell culture work. PPARγ levels were decreased in retinas from untreated type 2 diabetic BBZDR/Wor obese rats but were significantly increased in retinas from both lean and type 2 obese rats treated with pioglitazone (Fig. 1), demonstrating that systemic pioglitazone reaches the retina to elicit its effects through activation of the PPARγ pathway.
FIGURE 1.
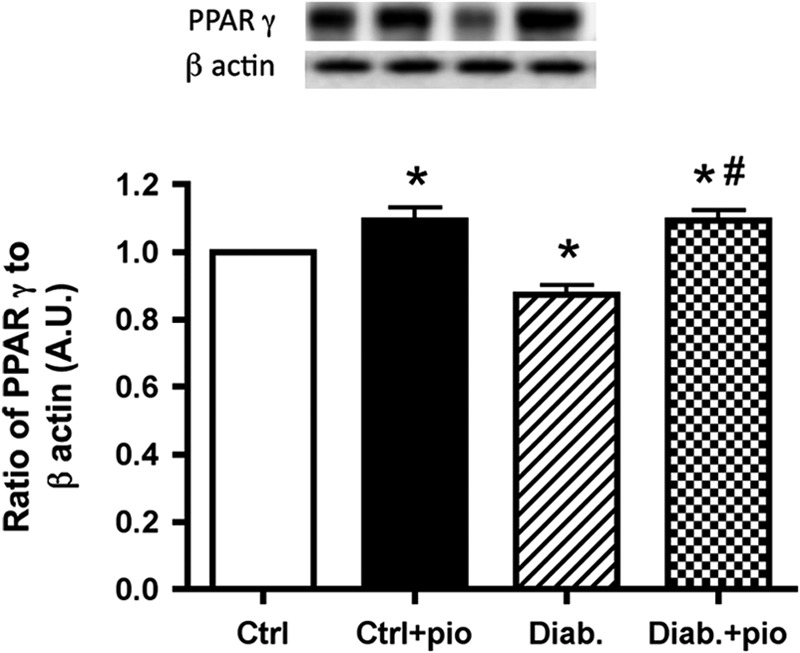
Pioglitazone increased PPARγ activity in whole retinal lysates of type 2 diabetic rats. Western blot results from whole retina of lean (Ctrl), lean + pioglitazone (Ctrl+pio), BBZDR/Wor obese (Diab), and BBZDR/Wor obese + pioglitazone (Diab+pio) mice. *, p < 0.05 versus control; #, p < 0.05 versus diabetic. Data are mean ± S.E. (error bars); n = 5 for each group. A.U., arbitrary units.
Pioglitazone Treatment Protected against the Loss of ERG Amplitudes in Type 2 Diabetes Obese Rats within 2 Months
Although the ultimate goal of these studies was to evaluate whether pioglitazone could restore normal insulin signaling in type 2 diabetic BBZDR/Wor obese rats, we also wanted to evaluate whether effects of pioglitazone were linked to improved retinal function. To test this, we performed ERG analyses on both lean and type 2 diabetic obese rats prior to initiation of pioglitazone treatment and just prior to sacrifice after 2 months of treatment. Fig. 2 demonstrates that diabetic animals had reduced A-wave, B-wave, and oscillatory potentials prior to treatment. Treatment with pioglitazone significantly improved all wave amplitudes but did not restore amplitudes to the same level observed in lean animals (Fig. 2). This suggests that pioglitazone did improve retinal function, probably through improved glucose tolerance.
FIGURE 2.

B-wave amplitudes are improved in diabetic rats treated with pioglitazone. Top, ERG amplitudes for A-wave (left), B-wave (middle), and oscillatory potentials (right) before treatment was started. Bottom, effects of 2 months of pioglitazone treatment. *, p < 0.05 versus diabetic. Data are mean ± S.D. (error bars); n = 5 in all groups.
Pioglitazone Significantly Reduced TNFα Levels in the Retinas of Type 2 Diabetic Obese Rats, Leading to Reduced IRS-1Ser-307 Phosphorylation
Studies in adipocytes (24) and myeloid progenitor cells (25) suggest that TNFα induces insulin resistance through phosphorylation of IRS-1 on serine 307. We wanted to determine whether pioglitazone could inhibit this insulin resistance pathway. Fig. 3 demonstrates that pioglitazone given to type 2 diabetic BBZDR/Wor obese rats significantly decreased TNFα levels, leading to decreased IRS-1Ser-307 phosphorylation, which should improve insulin signal transduction. These data provide in vivo evidence of PPARγ regulation of TNFα in the retina in a type 2 diabetic rat model.
FIGURE 3.
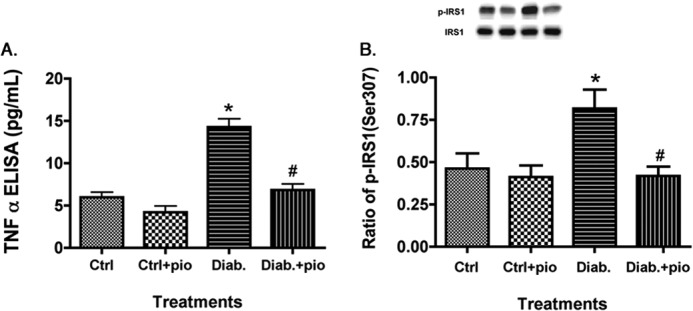
Pioglitazone reduced TNFα and IRS-1Ser-307 in type 2 diabetic rats. For both A and B, data presented include whole retinal analyses of lean (Ctrl), lean + pioglitazone (Ctrl+pio), BBZDR/Wor obese (Diab), and BBZDR/Wor obese + pioglitazone (Diab+pio) animals. A, ELISA results for TNFα; B, Western blot results for IRS-1Ser-307. *, p < 0.05 versus control; #, p < 0.05 versus diabetic. Data are mean ± S.E. (error bars); n = 5 in all groups.
SOCS3 and IRTyr-960 Were Decreased Significantly after Pioglitazone Treatment in the Retinas of Type 2 Diabetic Obese Rats
In addition to TNFα enhancement of phosphorylation of IRS-1Ser-307 and resulting inhibition of insulin signaling, increased TNFα levels can also activate SOCS3 (11) to promote insulin resistance (26). SOCS3 can inhibit the insulin pathway through phosphorylation of the insulin receptor on tyrosine 960, which blocks the interaction between insulin receptor and IRS-1 (26). Because pioglitazone was able to significantly reduce TNFα levels, we also wanted to investigate whether SOCS3 and IRTyr-960 levels were attenuated with pioglitazone treatment. Our results show that pioglitazone significantly decreased SOCS3 levels in the diabetic rats, which led to a decrease in the phosphorylation of IRTyr-960 (Fig. 4). This suggests that pioglitazone restores insulin signal transduction, probably through inhibition of both IRS-1Ser-307 and IRTyr-960.
FIGURE 4.
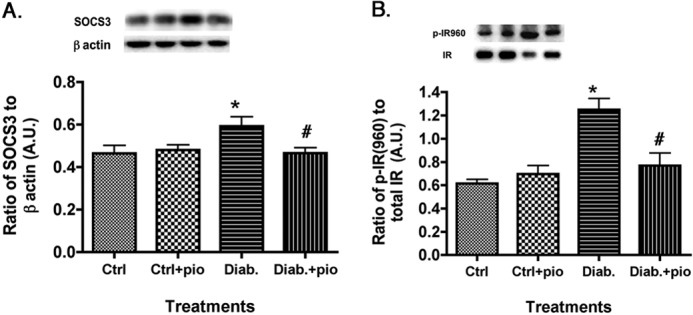
SOCS3 and IRTyr-960 are reduced in pioglitazone-treated type 2 diabetic rats. For both A and B, data presented include whole retinal analyses of lean (Ctrl), lean + pioglitazone (Ctrl+pio), BBZDR/Wor obese (Diab), and BBZDR/Wor obese + pioglitazone (Diab+pio) animals. A, Western blot results for SOCS3; B, Western blot results for IRTyr-960. *, p < 0.05 versus control; #, p < 0.05 versus diabetic. Data are mean ± S.E. (error bars); n = 5 in all groups. A.U., arbitrary units.
Pioglitazone Significantly Increased IGFBP-3 Protein Levels in the Retinas of Type 2 Obese BBZDR/Wor Rats, Probably Associated with Increased Insulin Receptor Activity
Although pioglitazone clearly decreased TNFα and SOCS3 actions, we also wanted to measure whether pioglitazone could increase insulin receptor phosphorylation. Additionally, because we recently showed that IGFBP-3 could induce insulin receptor phosphorylation in the retina of diabetic rats, we measured IGFBP-3 levels (27). Fig. 5 demonstrates that obese animals have reduced IGFBP-3 levels, which are restored with pioglitazone treatment. This is the first demonstration that pioglitazone can regulate IGFBP-3 levels in diabetic rats. Additionally, pioglitazone significantly restored insulin receptor phosphorylation in the type 2 diabetic rats (Fig. 5B). These results show that pioglitazone probably works in multiple pathways to restore normal glucose levels, through both inhibition of TNFα and SOCS3 inhibitory pathways and restoration of insulin-activating pathways.
FIGURE 5.
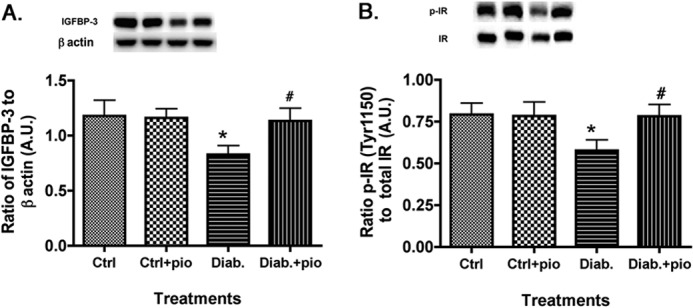
IGFBP-3 and phosphorylation of insulin receptor increased after pioglitazone treatment. For both A and B, data presented include whole retinal analyses of lean (Ctrl), lean + pioglitazone (Ctrl+pio), BBZDR/Wor obese (Diab), and BBZDR/Wor obese + pioglitazone (Diab+pio) animals. A, Western blot results for IGFBP-3; B, Western blot results for IRTyr-1150/1151. *, p < 0.05 versus control; #, p < 0.05 versus diabetic. Data are mean ± S.E.; n = 5 in all groups.A.U., arbitrary units.
Apoptotic Markers Were Decreased after Pioglitazone Treatment in the Retinas of Type 2 Diabetic Obese Rats
The key outcome of normal insulin signal transduction is increased phosphorylation of Akt and inhibition of apoptosis. Data in Fig. 6 demonstrate that type 2 diabetic rats have increased apoptotic markers (Fig. 6, C–E), which were significantly reduced by pioglitazone treatment. Retinal lysates from the BBZDR/Wor obese rats had decreased antiapoptotic markers, which were increased after pioglitazone therapy (Fig. 6, A and B). Taken with the other animal findings, these data demonstrate that pioglitazone is highly effective in reducing insulin resistance in whole retina of type 2 diabetic rats, leading to increased survival of retinal cells and improved function.
FIGURE 6.

Apoptotic markers are reduced in type 2 diabetic rats treated with pioglitazone. For both A and B, data presented include whole retinal analyses of lean (Ctrl), lean + pioglitazone (Ctrl+pio), BBZDR/Wor obese (Diab), and BBZDR/Wor obese + pioglitazone (Diab+pio) animals. A and B show Western blot results for the anti-apoptotic proteins (Akt and Bcl-xL). C–E, Western blot results for pro-apoptotic proteins (Bax, cytochrome c, and cleaved caspase 3). *, p < 0.05 versus control; #, p < 0.05 versus diabetic. Data are mean ± S.E. n = 5 in all groups. A.U., arbitrary units.
Pioglitazone Increased PPARγ Activity in Both REC and Müller Cells
Pioglitazone was highly effective in protecting whole retinal function and insulin signaling. In order to determine which cell types might be most responsive to pioglitazone actions, we investigated the effects of pioglitazone on insulin signal transduction in both REC and Müller cells cultured in normal and high glucose to mimic diabetic-like conditions. We chose to focus on these cell types because we have previously reported that TNFα is key to impaired insulin signaling in both REC (9) and Müller cells (22). Before we could initiate these studies, we first needed to determine the optimal dose of pioglitazone to increase PPARγ activity in each cell type. Because we used 25 mg/kg in vivo, we started cell work in both REC and Müller cells at 25 μm pioglitazone. This dose was effective in significantly increasing PPARγ activity in REC cultured in high glucose (Fig. 7A); however, it did not significantly increase activity in Müller cells. Therefore, we increased the dose of pioglitazone to 50 μm pioglitazone and found that this dose was able to significantly increase PPARγ activity (Fig. 7B). In the remaining cell culture studies, we treated REC with 25 μm pioglitazone and Müller cells with 50 μm pioglitazone to maximize PPARγ activation. It is unclear why a higher concentration of pioglitazone is required to significantly increase PPARγ activity in rat Müller cells versus human REC. This will be a focus of further studies.
The Pioglitazone-induced Increase in PPARγ Activity Led to a Significant Reduction in Apoptotic Markers in Both REC and Müller Cells
Because the key outcome of restoration of normal insulin signal transduction is prevention of apoptosis, the first question we addressed in both REC and Müller cells was whether pioglitazone reduced apoptotic markers (Bax, cytochrome c, and caspase 3) while increasing antiapoptotic markers (Akt and Bcl-xL). We found that in both REC (Fig. 8, A–E) and Müller cells (Fig. 8, F–J), pioglitazone increased levels of key antiapoptotic factors (Fig. 8, A, B, F, and G). As would be expected with increased antiapoptotic levels, pioglitazone also decreased proapoptotic proteins in both cell types (Fig. 8, C–E and H–J). Data suggest that pioglitazone probably protects the retina through reduction of apoptosis in both REC and Müller cells.
FIGURE 8.

Proapoptotic proteins are reduced in REC and Müller cells treated with pioglitazone. All work was done in REC and Müller cells left untreated (NT) or treated with pioglitazone. A–E, data in REC; F–J, Müller cell data. Data show that pioglitazone (PIO) reduced proapoptotic proteins (C–E and H–J) in both cell types. *, p < 0.05 versus normal glucose (NG) untreated; #, p < 0.05 versus high glucose (HG) untreated. Data are mean ± S.E. (error bars); n = 4 for all treatments. A.U., arbitrary units.
Pioglitazone Treatment Reduced TNFα and IRS-1Ser-307 Phosphorylation in Both REC and Müller Cells
We have previously reported that high glucose significantly increased TNFα and IRS-1Ser-307 in both REC (9) and Müller cells (10), which was associated with increased apoptosis. In this study, we wanted to ascertain whether pioglitazone could reduce the glucose-dependent increase in TNFα and IRS-1Ser-307 in REC and Müller cells. Fig. 9 showed that high glucose increased TNFα and IRS-1 in both REC and Müller cells, which matches previously published work. Pioglitazone significantly reduced both TNFα (Fig. 9. A and C) and IRS-1Ser-307 (Fig. 9, B–D) in both REC (Fig. 9, A and B) and Müller cells (Fig. 9, C and D). These data suggest that pioglitazone works to protect retinal cells by blocking TNFα-induced insulin resistance.
FIGURE 9.

TNFα and IRS-1Ser-307 are reduced in both REC and Müller cells after pioglitazone treatment. All work was done in REC and Müller cells left untreated (NT) or treated with pioglitazone (PIO). A and B, data in REC; C and D, Müller cell data. Data show that pioglitazone reduced TNFα and IRS-1Ser-307 in both cell types. *, p < 0.05 versus normal glucose (NG) untreated; #, p < 0.05 versus high glucose (HG) untreated. Data are mean ± S.E. (error bars); n = 4 for all treatments. A.U., arbitrary units.
Pioglitazone Treatment Also Significantly Decreased SOCS3 and IRTyr-960 in REC and Müller Cells
To ensure that pioglitazone works similarly in both REC and Müller cells as it did in whole retinal lysates, we also measured SOCS3 and IRTyr-960 in both cell types after high glucose exposure. High glucose significantly increased both SOCS3 and IRTyr-960, as we have reported previously (9, 10). Based on the reduced TNFα levels, it was expected that SOCS3 and IRTyr-960 would be reduced in both cell types after pioglitazone. Indeed, Fig. 10 demonstrates that pioglitazone significantly reduced SOCS3 (Fig. 10, A and C) as well as IRTyr-960 phosphorylation (Fig. 10, B and D). Combining all of the data presented, pioglitazone restored insulin signal transduction in the retina through a reduction in TNFα- and SOCS3-induced insulin resistance in both REC and Müller cells. This restoration of normal insulin signaling inhibited apoptosis in retinal cells as well as improved retinal function, as shown by increased ERG amplitudes in treated diabetic rats.
FIGURE 10.

SOCS3 and IRTyr-960 are reduced by pioglitazone. All work was done in REC and Müller cells left untreated (NT) or treated with pioglitazone. A and B, data in REC; C and D, Müller cell data. Data show that pioglitazone (PIO) reduced SOCS3 and IRTyr-960 in both cell types. *, p < 0.05 versus normal glucose (NG) untreated; #, p < 0.05 versus high glucose (HG) untreated. Data are mean ± S.E. (error bars); n = 4 for all treatments. A.U., arbitrary units.
PPARγ Antagonist T0070907 Antagonized Pioglitazone Effects on TNFα, SOCS3, and IRTyr-1150/1151 on REC and Müller Cells
To verify that actions on insulin signaling occur through pioglitazone activity and not through an alternative pathway, we treated both REC and Müller cells with both pioglitazone as we did above, with some cells pretreated with a highly selective PPARγ antagonist, T0070907, at 50 nm for 30 min prior to pioglitazone administration. As a control for these studies to verify that T0070907 is an effective inhibitor of pioglitazone, the PPARγ activity assay was completed for REC (Fig. 11A) and Müller cells (Fig. 11E), demonstrating that 50 nm T0070907 was effective in reducing pioglitazone actions in the cells. Data demonstrate that pioglitazone decreased TNFα (Fig. 11, B and F) and SOCS3 (Fig. 11, C and G) levels while increasing IRTyr-1150/1151 (Fig. 11, D and H), as we reported above. TNFα levels were much higher in the rMC-1 cells, probably due to the new modification of the TNFα ELISA from Pierce; however, the PPAR inhibitor had the same actions in both REC and Müller cells. Use of the PPARγ inhibitor showed that pioglitazone actions on TNFα, SOCS3, and IRTyr-1150/1151 occur through PPARγ activities because all pioglitazone actions were inhibited by T0070907. Taken together, these data suggest that pioglitazone regulates insulin signaling through PPARγ actions.
FIGURE 11.

PPARγ antagonist T0070907 (50 nm) blocks pioglitazone actions on TNFα, SOCS3, and IRTyr-1150/1151. All work was done in REC and Müller cells left untreated (NT), treated with pioglitazone (PIO), treated with T0070907 only, or treated with pioglitazone + T0070907. A–D, REC results; E–H, Müller cell results. A and E show that T0070907 inhibited pioglitazone actions using the PPARγ activity assay. B and F demonstrate that T00709707 antagonized pioglitazone actions on TNFα, whereas C and G show T0070907 actions on SOCS3. D and H show that pioglitazone's ability to increase IRTyr-1150/1151 requires PPARγ because it was blocked by T0070907. *, p < 0.05 versus normal glucose (NG) untreated; #, p < 0.05 versus high glucose untreated; $, p < 0.05 versus high glucose (HG) + pioglitazone. Data are mean ± S.E.; n = 4 for all treatments. A.U., arbitrary units.
DISCUSSION
The major finding of our study is that pioglitazone treatment in vitro of retinal cells cultured in high glucose as well as treatment in vivo of type 2 diabetic BBZDR/Wor rats led to a direct increase in endogenous PPARγ and a concomitant decrease in a variety of cell death markers as well as improvement in retinal function. Pioglitazone is known to increase PPARγ in other tissues; pioglitazone treatment has been reported to improve insulin sensitivity in patients with type 2 diabetes. The intent of our study was to utilize the BBZDR/Wor rat model and retinal cell culture to establish the mechanisms of action of PPARγ in protection of retinal tissue against glucose-induced retinopathy. Our results reported here show that the BBZDR/Wor model exhibits all of the expected features of Type 2 diabetes, as reported by previous work from our laboratory and others, including diabetes-induced changes in TNFα/SOCS3 pathways. Thus, the BBZDR/Wor model provided a valid platform for full evaluation of the mechanisms of action of the target drug, pioglitazone.
Our data demonstrate that pioglitazone is effective in increasing retinal PPARγ levels in vivo, which in turn protects against cell death associated with diabetic retinopathy. This is in agreement with reports that pioglitazone enhancement of PPARγ protects against retinal cell death in a variety of other retinal degeneration models, including the streptozotocin-induced diabetic rat model (2), the ischemia/reperfusion damage model in rats (28), and the rat model of optic nerve crush damage (29). The pioglitazone-induced increase in PPARγ activity in whole retina is probably responsible for the improvement in the ERG because others have reported that pioglitazone improved the ERG in the ischemia/reperfusion model in rats (28).
Our results suggest that pioglitazone probably works to increase PPARγ activity in the rat retina through increased activity in both REC and Müller cells. We found that high glucose culturing conditions significantly reduced PPARγ activity in both human REC and rat Müller cells. Reduced PPARγ is linked to increased apoptosis because pioglitazone treatment enhanced PPARγ and lowered apoptotic rates in these cells. Others have reported a similar link using bovine retinal endothelial cells (2). Likewise, the link between PPARγ and protection against apoptosis has been shown in the study by Zhang et al., demonstrating that pioglitazone reduced glial fibrillary acidic protein levels, which is indicative of Müller cell activation and also occurs in Müller cells cultured in high glucose conditions (30). In the rat model of optic nerve crush damage, the authors also report that pioglitazone reduced Müller cell activation (29). Thus, our observations in BBZDR/Wor rats and in REC and Müller cell culture support these earlier findings and strengthen the view that REC and Müller cells mediate the protective actions of PPARγ.
Because pioglitazone improves insulin signaling in type 2 diabetic humans, we used our in vivo and in vitro models to determine which PPARγ pathways are responsible for restoring insulin signaling, thereby reducing apoptosis and improving retinal function. One of the key proteins involved in inducing insulin resistance is TNFα (31, 32). Our data clearly demonstrate that pioglitazone reduces TNFα levels, leading to a reduction in IRS-1Ser-307 phosphorylation and an increase in insulin receptor resistance through PPARγ actions. These findings were observed in vivo as well as in cultured REC and Müller cells treated with pioglitazone. Our findings are in agreement with work in cholestatic rats treated with pioglitazone for gastric ulcer (33). Similar results of decreased TNFα after pioglitazone were also noted in the fat tissues of db/db mice (34).
In addition to a direct action of TNFα on insulin resistance through phosphorylation of IRS-1 on serine 307 (24), TNFα can also induce SOCS3, which, in turn, can result in insulin resistance through phosphorylation of insulin receptor on tyrosine 960 (26). Kanatani et al. (34) report that pioglitazone reduced SOCS3 in db/db mice as well as in 3T3 adipocytes. Additional work in rats maintained on a high cholesterol fructose diet and treated with pioglitazone showed reduced levels of both TNFα and SOCS3 in serum as well as hepatic tissues (13). Taken together, our findings and the findings of others demonstrate that increases in TNFα and SOCS3 lead to insulin resistance and that pioglitazone enhancement of PPARγ can reverse these effects.
We have previously reported that increased TNFα and SOCS3 levels are associated with apoptosis in both REC and Müller cells (9, 10). Our data suggest that TNFα/SOCS3 pathways work through insulin receptor resistance to trigger apoptosis. In both REC (9) and Müller cells (10), we have previously shown that insulin receptor resistance blocks induction of the antiapoptotic factor, Akt, and thus triggers apoptosis.
In conclusion, we have used type 2 diabetic BBZDR/Wor obese rats and cell cultures of REC and Müller cells to establish the pathway by which pioglitazone enhances PPARγ to block TNFα/SOCS3-induced insulin receptor resistance and thereby protect diabetic retinal tissue from apoptosis. Additionally, pioglitazone actions to reduce apoptosis and restore insulin signaling in the retina occur in REC and Müller cells, suggesting that these two cell types are key to insulin actions in the retina. These data indicate that pioglitazone is a good insulin-sensitizing agent for patients with type 2 diabetes, specifically those with diabetic retinopathy.
This work was supported, in whole or in part, by National Institutes of Health, NEI, Grants R01-EY022330 (to J. J. S.) and PHS 3P30 EY013080 (to Dianna Johnson). This work was also supported by Juvenile Diabetes Research Foundation Grant 2-2011-597 (to J. J. S.), the Oxnard Foundation (to J. J. S.), and Research to Prevent Blindness (to James C. Fleming).
Biswas, S., Jiang, Y., and Steinle, J. J., (2014) Mol. Vis., in press.
- PPARγ
- peroxisome proliferator-activated receptor γ
- REC
- retinal endothelial cell(s)
- ERG
- electroretinogram
- PPRE
- peroxisome proliferator response element
- IR
- insulin receptor
- IRS
- insulin receptor substrate.
REFERENCES
- 1. Ng M., Fleming T., Robinson M., Thomson B., Graetz N., Margono C., Mullany E. C., Biryukov S., Abbafati C., Abera S. F., Abraham J. P., Abu-Rmeileh N. M., Achoki T., AlBuhairan F. S., Alemu Z. A., Alfonso R., Ali M. K., Ali R., Guzman N. A., Ammar W., Anwari P., Banerjee A., Barquera S., Basu S., Bennett D. A., Bhutta Z., Blore J., Cabral N., Nonato I. C., Chang J. C., Chowdhury R., Courville K. J., Criqui M. H., Cundiff D. K., Dabhadkar K. C., Dandona L., Davis A., Dayama A., Dharmaratne S. D., Ding E. L., Durrani A. M., Esteghamati A., Farzadfar F., Fay D. F., Feigin V. L., Flaxman A., Forouzanfar M. H., Goto A., Green M. A., Gupta R., Hafezi-Nejad N., Hankey G. J., Harewood H. C., Havmoeller R., Hay S., Hernandez L., Husseini A., Idrisov B. T., Ikeda N., Islami F., Jahangir E., Jassal S. K., Jee S. H., Jeffreys M., Jonas J. B., Kabagambe E. K., Khalifa S. E., Kengne A. P., Khader Y. S., Khang Y. H., Kim D., Kimokoti R. W., Kinge J. M., Kokubo Y., Kosen S., Kwan G., Lai T., Leinsalu M., Li Y., Liang X., Liu S., Logroscino G., Lotufo P. A., Lu Y., Ma J., Mainoo N. K., Mensah G. A., Merriman T. R., Mokdad A. H., Moschandreas J., Naghavi M., Naheed A., Nand D., Narayan K. M., Nelson E. L., Neuhouser M. L., Nisar M. I., Ohkubo T., Oti S. O., Pedroza A., Prabhakaran D., Roy N., Sampson U., Seo H., Sepanlou S. G., Shibuya K., Shiri R., Shiue I., Singh G. M., Singh J. A., Skirbekk V., Stapelberg N. J., Sturua L., Sykes B. L., Tobias M., Tran B. X., Trasande L., Toyoshima H., van de Vijver S., Vasankari T. J., Veerman J. L., Velasquez-Melendez G., Vlassov V. V., Vollset S. E., Vos T., Wang C., Wang S. X., Weiderpass E., Werdecker A., Wright J. L., Yang Y. C., Yatsuya H., Yoon J., Yoon S. J., Zhao Y., Zhou M., Zhu S., Lopez A. D., Murray C. J., Gakidou E. (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 10.1016/S0140-6736(14)60460-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tawfik A., Sanders T., Kahook K., Akeel S., Elmarakby A., Al-Shabrawey M. (2009) Suppression of retinal peroxisome proliferator-activated receptor γ in experimental diabetes and oxygen-induced retinopathy: role of NADPH oxidase. Invest. Ophthalmol. Vis. Sci. 50, 878–884 [DOI] [PubMed] [Google Scholar]
- 3. Malchiodi-Albedi F., Matteucci A., Bernardo A., Minghetti L. (2008) PPAR-γ, Microglial cells, and ocular inflammation: new venues for potential therapeutic approaches. PPAR Res. 2008, 295784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yau H., Rivera K., Lomonaco R., Cusi K. (2013) The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr. Diab. Rep. 13, 329–341 [DOI] [PubMed] [Google Scholar]
- 5. Iwata M., Haruta T., Usui I., Takata Y., Takano A., Uno T., Kawahara J., Ueno E., Sasaoka T., Ishibashi O., Kobayashi M. (2001) Pioglitazone ameliorates tumor necrosis factor-α-induced insulin resistance by a mechanism independent of adipogenic activity of peroxisome proliferator-activated receptor-γ. Diabetes 50, 1083–1092 [DOI] [PubMed] [Google Scholar]
- 6. Ding S. Y., Shen Z. F., Chen Y. T., Sun S. J., Liu Q., Xie M. Z. (2005) Pioglitazone can ameliorate insulin resistance in low-dose streptozotocin and high sucrose-fat diet induced obese rats. Acta Pharmacol. Sin. 26, 575–580 [DOI] [PubMed] [Google Scholar]
- 7. Joussen A. M., Doehmen S., Le M. L., Koizumi K., Radetzky S., Krohne T. U., Poulaki V., Semkova I., Kociok N. (2009) TNF-α mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol. Vis. 15, 1418–1428 [PMC free article] [PubMed] [Google Scholar]
- 8. Walker R. J., Steinle J. J. (2007) Role of β-adrenergic receptors in inflammatory marker expression in Muller cells. Invest. Ophthalmol. Vis. Sci. 48, 5276–5281 [DOI] [PubMed] [Google Scholar]
- 9. Jiang Y., Zhang Q., Soderland C., Steinle J. J. (2012) TNFα and SOCS3 regulate IRS-1 to increase retinal endothelial cell apoptosis. Cell. Signal. 24, 1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang Y., Pagadala J., Miller D., Steinle J. J. (2013) Reduced insulin receptor signaling in retinal Muller cells cultured in high glucose. Mol. Vis. 19, 804–811 [PMC free article] [PubMed] [Google Scholar]
- 11. Emanuelli B., Peraldi P., Filloux C., Chavey C., Freidinger K., Hilton D. J., Hotamisligil G. S., Van Obberghen E. (2001) SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-α in the adipose tissue of obese mice. J. Biol. Chem. 276, 47944–47949 [DOI] [PubMed] [Google Scholar]
- 12. Ueki K., Kondo T., Kahn C. R. (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell Biol. 24, 5434–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Collino M., Aragno M., Castiglia S., Miglio G., Tomasinelli C., Boccuzzi G., Thiemermann C., Fantozzi R. (2010) Pioglitazone improves lipid and insulin levels in overweight rats on a high cholesterol and fructose diet by decreasing hepatic inflammation. Br. J. Pharmacol. 160, 1892–1902 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Schmidt R. E., Dorsey D. A., Beaudet L. N., Parvin C. A., Zhang W., Sima A. A. (2004) Experimental rat models of types 1 and 2 diabetes differ in sympathetic neuroaxonal dystrophy. J. Neuropathol. Exp. Neurol. 63, 450–460 [DOI] [PubMed] [Google Scholar]
- 15. Obrosova I. G. (2009) Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics 6, 638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito I., Jarajapu Y. P., Guberski D. L., Grant M. B., Knot H. J. (2006) Myogenic tone and reactivity of rat ophthalmic artery in acute exposure to high glucose and in a type II diabetic model. Invest. Ophthalmol. Vis. Sci. 47, 683–692 [DOI] [PubMed] [Google Scholar]
- 17. Caballero S., Sengupta N., Afzal A., Chang K. H., Li Calzi S., Guberski D. L., Kern T. S., Grant M. B. (2007) Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes 56, 960–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ellis E. A., Grant M. B., Murray F. T., Wachowski M. B., Guberski D. L., Kubilis P. S., Lutty G. A. (1998) Increased NADH oxidase activity in the retina of the BBZ/Wor diabetic rat. Free Radic. Biol. Med. 24, 111–120 [DOI] [PubMed] [Google Scholar]
- 19. Ellis E. A., Guberski D. L., Somogyi-Mann M., Grant M. B. (2000) Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic. Biol. Med. 28, 91–101 [DOI] [PubMed] [Google Scholar]
- 20. Jiang Y., Zhang Q., Liu L., Tang J., Kern T. S., Steinle J. J. (2013) β2-adrenergic receptor knockout mice exhibit A diabetic retinopathy phenotype. PLoS One 8, e70555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Q., Guy K., Pagadala J., Jiang Y., Walker R. J., Liu L., Soderland C., Kern T. S., Ferry R., Jr., He H., Yates C. R., Miller D. D., Steinle J. J. (2012) Compound 49b prevents diabetes-induced apoptosis through increased IGFBP-3 levels. Invest. Ophthalmol. Vis. Sci. 53, 3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker R. J., Anderson N. M., Jiang Y., Bahouth S., Steinle J. J. (2011) Role of β-adrenergic receptor regulation of TNF-α and insulin signaling in retinal Muller cells. Invest. Ophthalmol. Vis. Sci. 52, 9527–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zerani M., Maranesi M., Brecchia G., Gobbetti A., Boiti C., Parillo F. (2013) Evidence for a luteotropic role of peroxisome proliferator-activated receptor γ: expression and in vitro effects on enzymatic and hormonal activities in corpora lutea of pseudopregnant rabbits. Biol. Reprod. 88, 62. [DOI] [PubMed] [Google Scholar]
- 24. Rui L., Aguirre V., Kim J. K., Shulman G. I., Lee A., Corbould A., Dunaif A., White M. F. (2001) Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Invest. 107, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aguirre V., Uchida T., Yenush L., Davis R., White M. F. (2000) The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 275, 9047–9054 [DOI] [PubMed] [Google Scholar]
- 26. Emanuelli B., Peraldi P., Filloux C., Sawka-Verhelle D., Hilton D., Van Obberghen E. (2000) SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 275, 15985–15991 [DOI] [PubMed] [Google Scholar]
- 27. Jiang Y., Zhang Q., Steinle J. J. (2014) Intravitreal injection of IGFBP-3 restores normal insulin signaling in diabetic rat retina. PLoS One 9, e93788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang X. Y., Xiao Y. Q., Zhang Y., Ye W. (2013) Protective effect of pioglitazone on retinal ischemia/reperfusion injury in rats. Invest. Ophthalmol. Vis. Sci. 54, 3912–3921 [DOI] [PubMed] [Google Scholar]
- 29. Zhu J., Zhang J., Ji M., Gu H., Xu Y., Chen C., Hu N. (2013) The role of peroxisome proliferator-activated receptor and effects of its agonist, pioglitazone, on a rat model of optic nerve crush: PPARγ in retinal neuroprotection. PLoS One 8, e68935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feit-Leichman R. A., Kinouchi R., Takeda M., Fan Z., Mohr S., Kern T. S., Chen D. F. (2005) Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest. Ophthalmol. Vis. Sci. 46, 4281–4287 [DOI] [PubMed] [Google Scholar]
- 31. Olson N. C., Callas P. W., Hanley A. J., Festa A., Haffner S. M., Wagenknecht L. E., Tracy R. P. (2012) Circulating levels of TNF-α are associated with impaired glucose tolerance, increased insulin resistance, and ethnicity: the Insulin Resistance Atherosclerosis Study. J. Clin. Endocrinol. Metab. 97, 1032–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldberg R. B. (2009) Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J. Clin. Endocrinol. Metab. 94, 3171–3182 [DOI] [PubMed] [Google Scholar]
- 33. Moezi L., Janahmadi Z., Amirghofran Z., Nekooeian A. A., Dehpour A. R. (2014) The increased gastroprotective effect of pioglitazone in cholestatic rats: role of nitric oxide and tumour necrosis factor alpha. Int. J. Exp. Pathol. 95, 78–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanatani Y., Usui I., Ishizuka K., Bukhari A., Fujisaka S., Urakaze M., Haruta T., Kishimoto T., Naka T., Kobayashi M. (2007) Effects of pioglitazone on suppressor of cytokine signaling 3 expression: potential mechanisms for its effects on insulin sensitivity and adiponectin expression. Diabetes 56, 795–803 [DOI] [PubMed] [Google Scholar]