

Background: Allosteric integrase (IN) inhibitors (ALLINIs) promote aberrant protein multimerization.
Results: ALLINI-2 induces protein-protein interactions, including C-terminal residues Lys-264 and Lys-266, which lead to aberrant, higher-order IN multimerization.
Conclusion: The protein-protein contacts beyond the inhibitor binding site contribute to aberrant IN multimerization.
Significance: Our findings provide structural clues and underscore the significance for exploiting IN multimerization as a new therapeutic target.
Keywords: Drug Resistance, Human Immunodeficiency Virus (HIV), Integrase, Lentivirus, Mass Spectrometry (MS), Pharmacology, HIV Integrase, Allosteric Inhibitors, Drug Resistance, Protein Multimerization
Abstract
Allosteric HIV-1 integrase (IN) inhibitors (ALLINIs) are a promising class of antiretroviral agents for clinical development. Although ALLINIs promote aberrant IN multimerization and inhibit IN interaction with its cellular cofactor LEDGF/p75 with comparable potencies in vitro, their primary mechanism of action in infected cells is through inducing aberrant multimerization of IN. Crystal structures have shown that ALLINIs bind at the IN catalytic core domain dimer interface and bridge two interacting subunits. However, how these interactions promote higher-order protein multimerization is not clear. Here, we used mass spectrometry-based protein footprinting to monitor surface topology changes in full-length WT and the drug-resistant A128T mutant INs in the presence of ALLINI-2. These experiments have identified protein-protein interactions that extend beyond the direct inhibitor binding site and which lead to aberrant multimerization of WT but not A128T IN. Specifically, we demonstrate that C-terminal residues Lys-264 and Lys-266 play an important role in the inhibitor induced aberrant multimerization of the WT protein. Our findings provide structural clues for exploiting IN multimerization as a new, attractive therapeutic target and are expected to facilitate development of improved inhibitors.
Introduction
HIV-1 integrase (IN)2 is an important therapeutic target because its function is essential for the virus life cycle. During the early stage of HIV-1 replication, a tetramer of IN assembles with reverse transcribed viral DNA ends to form the stable synaptic complex or intasome. Two catalytic reactions ensue within the nucleoprotein complex. First, a GT dinucleotide is cleaved from the 3′ terminus of each viral DNA (3′-processing). Subsequently, the recessed viral DNA ends are integrated into complementary strands of target DNA in staggered fashion (DNA strand transfer). The integration is facilitated by cellular chromatin-associated protein LEDGF/p75, which tethers the lentiviral intasomes to active genes (1–7).
Mutagenesis studies have suggested that the role of IN in the virus life cycle extends beyond integration (8–14). For example, a number of substitutions in the IN coding region have been shown to impair proper maturation of HIV-1 particles and to block the subsequent round of reverse transcription. These mutants have been classified as class II IN mutants as opposed to class I IN mutants, which selectively impair the integration step. Experiments with ectopically expressed Vpr-IN constructs have shown that catalytically defective IN bearing the D116N substitution can complement δ-IN HIV-1 to rescue reverse transcription in target cells, indicating that the correct IN structure rather than its catalytic activity is important for the late stage of HIV-1 replication (15). However, the exact role of HIV-1 IN during virion maturation remains to be elucidated.
The viral IN protein is composed of three domains (reviewed in Ref. 16). The N-terminal domain (NTD, residues 1–46) contains the zinc-binding HHCC motif, which is essential for the correct folding of this domain and function of the full-length protein (17–22). The catalytic core domain (CCD, residues 56–202) contains the DDE active site, which coordinates catalytic Mg2+ ions and mediates catalysis of both 3′-processing and strand transfer reactions (23–27). The C-terminal domain (CTD, residues 220–288) has a SH3-like fold (28–30). Each domain is essential for functional protein multimerization and directly interacts with viral DNA. LEDGF/p75 preferentially binds to a tetramer of IN by bridging the CCD-CCD interface of one dimer and the NTD of another dimer (22, 31, 32). The V-shaped cavity created by two interacting CCDs provides a key docking site for the cellular protein (7). LEDGF/p75 Asp-366 hydrogen bonds with backbone amides of Glu-170 and His-171 of one IN subunit, whereas LEDGF/p75 Ile-365, Leu-368, Phe-406, and Val-408 form hydrophobic contacts with the second IN subunit (7). Additional interactions between LEDGF/p75 and the IN NTD are mediated by electrostatic interactions (22, 32).
Allosteric HIV-1 IN inhibitors (ALLINIs) are an attractive new class of antiviral compounds for clinical development because they target the unexploited IN dimer interface and are expected to complement currently available therapies (reviewed in Ref. 33–36). These compounds bind at the IN CCD dimer interface occupying the principal LEDGF/p75 binding pocket (12, 37–42). Similar to LEDGF/p75 Asp-366, the ALLINI carboxylic acid hydrogen bonds with the main chain nitrogens of residues Glu-170 and His-171 of one IN subunit. The ALLINI methoxy group also hydrogen bonds with the Thr-174 side chain of the same subunit, whereas the ALLINI quinoline ring extends toward another subunit to establish hydrophobic interactions. Consequently, ALLINIs exhibit a dual mode of action: they stabilize interacting IN subunits and promote higher-order protein multimerization as well as inhibit IN-LEDGF/p75 binding in vitro (37, 39, 43).
In infected cells, ALLINIs impair both the early and late stage of HIV-1 replication underscoring the multifunctional nature of IN during HIV-1 replication (12, 39, 44, 45). When added to target cells, ALLINIs block 3′-processing as well as reduce LEDGF/p75-mediated integration into active transcription units (39, 46). Knockdown or knock-out of endogenous LEDGF/p75 significantly enhanced the potencies of these inhibitors in target cells, suggesting that there is competition between the cellular protein and ALLINIs for binding to HIV-1 IN during early steps of viral replication (12, 47, 48). However, the ALLINI antiviral potency is determined primarily through inhibiting the late stage of HIV-1 replication (12, 41, 42, 44, 45, 48). In producer cells, ALLINIs potently promote aberrant, higher-order multimerization of IN during virus maturation resulting in eccentric, non-infectious cores reminiscent to the phenotype seen with some class II IN mutants (8–12, 44, 45). In contrast with target cells, the absence of LEDGF/p75 had no effect on ALLINI potencies in the virus producer cells (12, 44, 47, 48). The recent discovery of pyridine-based multimerization selective IN inhibitors, which potently promoted aberrant IN multimerization in vitro and in infected cells but were not effective inhibitors of IN-LEDGF/p75 interactions, have further highlighted IN multimerization as a plausible antiviral target (46). Yet, how a small molecule binding at the IN dimer interface induces aberrant, higher-order protein multimerization is not clear.
HIV-1 genotyping in tissue culture under ALLINI selective pressure has revealed a number of mutations at the inhibitor binding site, including Y99H, L102F, A124D/T124D, A128T, H171T, and T174I substitutions (12, 38–40, 42, 44). Of these, the A128T substitution has been identified as the most prevalent change and which confers marked resistance to the majority of ALLINIs. Crystallographic studies have shown that a representative inhibitor, ALLINI-2, interacted with both WT and A128T CCDs (38). The A128T substitution subtly relocated the ALLINI-2 quinoline ring in the A128T CCD dimer co-crystal, whereas the ALLINI-2 carboxylic acid remained similarly hydrogen bonded to WT and A128T CCDs. However, size exclusion chromatography (SEC) experiments with full-length recombinant proteins have revealed that ALLINI-2 induced aberrant multimerization of WT but not A128T IN (38). Consequently, HIV-1 bearing the A128T IN substitution was remarkably resistant to ALLINI-2 (38). How ALLINI-2 binding at the CCD dimer interface promoted differential multimerization of full-length WT and A128T INs remains to be elucidated.
Here, we have used MS-based protein footprinting to monitor ALLINI-2 induced surface topology changes in full-length WT and A128T INs. Our studies show that ALLINI-2 binding promotes protein-protein interactions that extend beyond the direct inhibitor binding site and lead to aberrant multimerization of WT but not A128T IN. These findings further our understanding of HIV-1 IN multimerization as a therapeutic target and will facilitate development of improved antiretroviral agents.
EXPERIMENTAL PROCEDURES
Proteins and the Inhibitor
Full-length recombinant WT and mutant (A128T and K264A/K266A) INs containing the N-terminal His6 tag were expressed in Escherichia coli and purified as described previously (49). Similar purification protocols were used to prepare isolated IN domains (CCD, CTD, and the CCD-CTD). ALLINI-2 was synthesized as described previously (38).
Differential Hydrogen/Deuterium Exchange (HDX) MS
Solution-phase amide HDX experiments were carried out with a fully automated system (CTC HTS PAL, LEAP Technologies, Carrboro, NC; housed inside a 4 °C cabinet) as described previously (50) with slight modifications. 10 μm of the His6-HIV-1 IN (WT or A128T) protein was mixed with 1:5 molar excess of ALLINI-2 and incubated for 2 h on ice before subjecting to HDX. For the differential HDX experiments, 5 μl of either the His6-HIV-1 IN (apo) or the complex (1:5 molar mixture of IN and ALLINI-2) were mixed with 20 μl of D2O-containing HDX buffer (50 mm HEPES 7.5,1 M NaCl, 3 mm DTT) and incubated at 4 °C for 0 s, 10 s, 30 s, 60 s, 900 s, or 3,600 s. Following the on exchange reaction, unwanted forward or back exchange was minimized, and the protein was denatured by the addition of 25 μl of a quench solution (1% (v/v) TFA in 5 m urea and 50 mm TCEP). Samples were then passed through an immobilized pepsin column (prepared in-house) at 50 μl min−1 (0.1% (v/v) TFA, 15 °C), and the resulting peptides were trapped on a C18 trap column (Hypersil Gold, Thermo Fisher). The bound peptides were then gradient-eluted (5–50% CH3CN (w/v) and 0.3% (w/v) formic acid) across a 1-mm × 50-mm C18 HPLC column (Hypersil Gold, Thermo Fisher) for 5 min at 4 °C). The eluted peptides were then subjected to electrospray ionization directly coupled to a high resolution Orbitrap mass spectrometer (LTQ Orbitrap XL with electron transfer dissociation, Thermo Fisher). Each HDX experiment was carried out in triplicate.
Peptide Identification and HDX Data Processing
MS/MS experiments were performed with a LTQ linear ion trap mass spectrometer (LTQ Orbitrap XL with ETD, Thermo Fisher) over a 70-min gradient. Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis. The MS/MS raw data files were converted to .mgf files and then submitted to Mascot (Matrix Science, London, UK) for peptide identification. Peptides included in the peptide set used for HDX detection had a MASCOT score of 20 or greater. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence, and false positives were ruled out. The MS/MS spectra of all the peptide ions from the MASCOT search were further manually inspected, and only the unique charged ions with the highest MASCOT score were used in estimating the sequence coverage. The intensity weighted average m/z value (centroid) of each peptide isotopic envelope was calculated with the latest version of our in-house developed software, HDX Workbench (51).
MS-based Footprinting of Surface Accessible Lysines
The surface-accessible Lys residues were monitored as described previously (31, 52). Briefly, purified IN (4 μm WT or mutant proteins) in the absence or presence of ALLINI-2 (1:5 molar mixture of IN and ALLINI-2) was incubated on ice for 2 h and then subjected to treatments at 37 °C with 1 mm of N-hydroxysuccinimide (NHS)-biotin for 30 min. NHS-biotin treatment was carried out in buffer containing 50 mm HEPES (pH 8.0), 150 mm NaCl, and 10 mm MgCl2. The reactions were quenched with 0.1 mm UltraPureTM Tris-HCl buffer, pH 7.5 (Invitrogen). The reaction mixture was subjected to denaturing SDS-PAGE, and the IN band was visualized by Coomassie Blue staining. Protein bands were excised, destained, and subjected to in-gel proteolysis using 2.5 μg of trypsin. The tryptic peptides were mixed with α-cyano-4-hydroxy-cinnamic acid and analyzed with the Axima-CFR MALDI-ToF instrument (Shimadzu).
SEC
WT and mutant INs (20 μm) after preincubation with DMSO or 40 μm ALLINI-2 for 30 min on ice were analyzed with a Superdex 200 Increase 10/300 GL column (GE Healthcare) using 0.75 ml/min flow rate and the elution buffer containing 20 mm HEPES (pH 6.8), 1 m NaCl, 10 mm MgSO4, 0.2 mm EDTA, 10% glycerol, and 5 mm β-mercaptoethanol. The column was calibrated with the following proteins: conalbumin (75,000 Da), carbonic anhydrase (29,000 Da), ribonuclease A (13,700 Da), and aprotinin (6,500 Da). Proteins were detected by absorbance at 280 nm.
Dynamic Light Scattering (DLS)
Purified WT or mutant INs was diluted to a final concentration of 200 nm in 50 mm HEPES, pH 7.4, buffer containing 2 mm DTT, 2 mm MgCl2, and 1 m NaCl. ALLINI-2 (0.2 μm) or DMSO was added to IN at room temperature, and DLS signal was recorded at indicated time intervals using a Malvern Nano series Zetasizer instrument.
Surface Plasmon Resonance (SPR)
SPR experiments were performed using a Biacore T100 (GE Healthcare). A series S sensor chip NTA (GE Healthcare) was conditioned with 0.5 mm NiCl2 at a flow rate of 10 μl/min for 1 min followed by a 1-min wash with 3 mm EDTA at a flow rate of 10 μl/min. In parallel experiments, 4 μg/ml proteins (WT His6-IN CCD, A128T His6-IN CCD, WT HisHis6-IN CTD, or HisHis6-IN CCD-CTD) in the HBS-P buffer (GE Healthcare) were immobilized on the chip to 4400 response units. Indicated concentrations of ALLINI-2 in the HBS-P buffer with 10% DMSO were flowed over the cell for 180 s at a flow rate of 40 μl/min followed by a 5-min dissociation. The chip was regenerated with 500 mm imidazole.
IN-LEDGF/p75 Binding
LEDGF/p75 (4 μm) was incubated with 4 μm of His6-tagged WT, A128T, or K264A/K266A IN in binding buffer containing 50 mm HEPES, pH 7.1, 300 mm NaCl, 2 mm MgCl2, 20 mm imidazole, 2 mm β-mercaptoethanol, and 0.2% (v/v) Nonidet P-40 for 30 min at room temperature. Samples were pulled down with nickel-affinity resin (GE Healthcare) for 60 min on a rotator at room temperature. The resin was washed four times with the same buffer, and the bound proteins were subjected to SDS-PAGE separation and visualized by Coomassie Blue staining.
IN Activity Assays
WT, A128T, and K264A/K266A IN activities were examined using the reported homogeneous time-resolved fluorescence-based assay (38) with following modifications. 100 nm WT, A128T, or K264A/K266A IN was incubated with 50 nm Cy-5 labeled donor, 10 nm biotinylated target DNA, and 100 nm LEDGF/p75. The time-resolved fluorescence signal was recorded after the addition of europium-streptavidin using a Perkin Elmer EnSpire multimode plate reader.
RESULTS
Two complementary MS-based approaches were used to identify the inhibitor induced surface topology changes in WT and A128T INs. Amide HDX-MS allowed us to monitor alterations in hydrogen bond networks and conformational changes in the protein backbone, whereas reactivity of side chain primary amines were probed with small molecule modifier NHS-biotin. ALLINI-2 was chosen for these experiments as a representative ALLINI because the crystal structures of its binding to WT and A128T IN CCD dimers are available (38) and allowed us to compare the atomic structures with protein footprinting results for full-length HIV-1 WT and A128T IN interactions with the inhibitor.
Differential rates at which an amide hydrogen exchanges with solvent deuterium can reveal information about inhibitor induced protein conformational changes. Protection from D2O uptake results in slower exchange and indicates stronger amide hydrogen bonding suggesting that the protein region has been stabilized and is less dynamic, plastic, or flexible. Conversely, deprotection results in increased exchange, indicating that some of hydrogen bonds are disrupted, suggesting that the protein region is more flexible. Representative HDX results for ALLINI-2 interactions with WT and A128T INs are depicted in Fig. 1, and the data are summarized in Fig. 2. Near complete coverage of the HIV-1 IN primary sequence was obtained enabling detailed analysis of the full-length protein (Fig. 2). For example, significantly slower HDX rates were observed for a representative peptide fragment encompassing regions 155–178 when ALLINI-2 was added to WT or A128T INs, indicating increased protection of this protein segment against D2O uptake in the presence of the inhibitor as compared with apo-IN (Fig. 1, A and B, also indicated by the lower black side bar in Fig. 2). This region encompasses the binding pocket for ALLINIs and is in agreement with our crystal structures that show that ALLINI-2 carboxylic acid similarly hydrogen bonds with Glu-170 and His-171 backbone amides of subunit 1 in both the WT and A128T IN CCDs (38). In addition, the substituted benzene ring of the inhibitor engages in hydrophobic interactions with Tyr-99 of subunit 1 in the crystal structure (38). Accordingly, HDX experiments reveal increased protection for the peptides spanning residues 86–101 (Fig. 2, indicated by the top black side bar) suggesting direct contacts with the inhibitor. The crystal structure of WT CCD bound to ALLINI-2 also showed that the quinoline ring extends toward Ala-128 to establish additional hydrophobic interactions with subunit 2. However, the substitution of Ala with the relatively bulkier and polar Thr relocates the inhibitor away from this position. In keeping with these structural observations, ALLINI-2 binding induces increased protection in the region 121–131 of the WT IN but not the A128T IN when compared with their respective apo-INs (Fig. 2, indicated by the middle black side bar). Taken together, the inhibitor binding patterns observed with the WT and A128T IN CCDs crystal structures were closely recapitulated using HDX in the context of full-length proteins.
FIGURE 1.

Deuterium uptake plots for representative peptides of HIV-1 IN in the presence and absence of ALLINI-2. The plots depict the level of deuterium incorporation (% deuterium or % D) for indicated time points for peptide 155–178 for WT IN (A) and A128T IN (B) and for peptide 257–288 for WT IN (C) and A128T IN (D). The error bars represent the S.D. for three independent experiments.
FIGURE 2.

HDX signature comparison for WT and A128T mutant INs in the presence of ALLINI-2. Amino acid sequences, charge states, and start and end amino acid numbers of each peptide detected by MS are indicated. The schematic presentation on the left shows peptide positioning with respect to IN domains. Bars on the right indicate protein regions interpreted as directly interacting with ALLINI-2 (black) or contributing to protein-protein interactions (magenta). Deuterium uptake for each peptide is calculated as the average of % deuterium (% D) for the six time points (10, 30, 60, 300, 900, and 3600 s), and the difference in average % deuterium values between the apo-IN and ALLINI-2 bound samples is presented as a heat map with a color code given at the bottom of the figure. % Deuterium S.D. for three individual replicates is indicated in the parentheses. An asterisk indicates peptides that did not show statistically significant difference in deuterium uptake when IN + ALLINI-2 was compared with the apo-IN based on the software HDX Workbench (51), and these regions are colored gray. The regions that revealed statistically significant reduction in deuterium uptake in the presence of added inhibitor are colored green and blue according to the heat map coloring scheme used by the software HDX Workbench. The software employs a statistical (p value < 0.05 for two consecutive time points or a p value < 0.01 for a single time point) based coloring scheme to distinguish real protection differences from inherent variation in the data. Additionally, overlapping peptides covering the same region and showing a similar protection trend are used to rule out data ambiguity. NS, not significant.
Importantly, HDX experiments have identified additional IN segments affected by the inhibitor binding to full-length IN that extend beyond the direct ALLINI-2 binding sites seen in crystal structures with the CCD. The peptides spanning amino acids 224–248 were similarly affected by the inhibitor binding to WT and A128T INs (Fig. 2, indicated by the middle magenta side bar). However, two IN segments: 181–202 and 250–280, were uniquely affected by the inhibitor binding to WT IN but not the A128T variant (Fig. 2, indicated by top and bottom magenta side bars). For instance, a peptide overlapping residues 257–288 in the WT IN in complex with ALLINI-2 displays increased protection in comparison to the same peptide from its unliganded counterpart (Fig. 1C). However, HDX rates for the identical peptide remained very similar when A128T IN was compared in the presence and absence of ALLINI-2 (compare Fig. 1, C and D).
We next extended footprinting of WT and A128T INs to compare reactivity of Lys residues to a small molecule modifier, NHS-biotin, in the presence and absence of ALLINI-2 (31, 52). Of particular interest are the residues that are susceptible to modifications in free protein but are shielded from modification upon complex formation. Treatment of apo-INs with NHS-biotin revealed a very similar modification patterns for WT and A128T INs, indicating that the single amino acid substitution did not detectably alter the overall structure of IN. Ten modified Lys residues were detected in each protein, including Lys-14, Lys-159, Lys-160, Lys-186, Lys-215, Lys-219, Lys-244, Lys-258, Lys-264, and Lys-266. Of these, only a single tryptic peptide peak (264–269) containing biotinylated Lys-264 and Lys-266 was markedly diminished upon ALLINI-2 addition to WT IN (Fig. 3, compare B and C). In contrast, only a slight reduction of this peak was observed when ALLINI-2 was added to A128T IN (Fig. 3, compare E and F). These findings were highly reproducible in three independent experiments. Reactivity levels of the other nine Lys residues to NHS-biotin were not significantly affected when WT and A128T INs were compared in the absence or presence of ALLINI-2. As a representative data, the modification patterns for the peptide 216–224 containing modified Lys-219, which remained reactive to NHS-biotin in the presence and absence of the inhibitor is shown in Fig. 3. Collectively, the findings illustrated in Fig. 3 agree with and extend the HDX data (Fig. 1B), implicating the 257–280 segment and in particular Lys-264 and Lys-266 in the inhibitor induced structural changes seen in WT but not A128T IN structures.
FIGURE 3.

MS-based protein footprinting comparing surface accessible lysines in WT and A128T INs in the presence of ALLINI-2. A, untreated WT IN; B, WT IN was treated with NHS-biotin (Bio.); C, ALLINI-2 was added to WT IN, and then the complex was treated with NHS-biotin; D, untreated A128T IN; E, A128T IN was treated with NHS-biotin; F, ALLINI-2 was added to A128T IN, and then the complex was treated with NHS-Biotin. The start and end amino acid numbers of each identified peptide peaks are shown. Modified Lys residues are indicated in parentheses.
The structural changes observed in the two domains (the CCD and CTD) could be due to either direct ALLINI-2 binding to IN or inhibitor induced protein-protein interactions. To delineate these possibilities, we have examined ALLINI-2 binding to the isolated WT and A128T CCDs as well as to WT CTD and the two domain CCD-CTD using SPR (Fig. 4). The inhibitor bound to WT and A128T CCDs with comparable Kd values of 0.722 ± 0.2 μm and 1.66 ± 0.2 μm, respectively (Fig. 4, A and B), indicating that the A128T substitution did not significantly affect ALLINI-2 binding. The inhibitor bound to WT CCD-CTD with a Kd of 2.64 ± 0.4 μm (Fig. 4C), whereas it failed to bind the WT CTD (Fig. 4D), suggesting that the CTD does not contribute to ALLINI-2 binding. Although poor solubility of full-length WT IN does not permit its analysis by SPR, it should be noted that the Kd values obtained with the isolated WT CCD and CCD-CTD domains correlate well with the ALLINI-2 IC50 of ∼0.6 μm against WT HIV-1 (38). Taken together, the data in Fig. 4 along with available structural data (38) help to interpret our MS-based protein footprinting results. It is logical to conclude that the IN CCD segments indicated by black side bars in Fig. 2 are affected by direct binding of ALLINI-2 to IN, whereas the additional regions indicated by magenta side bars in Fig. 2 reflect protein-protein interactions induced by the inhibitor binding.
FIGURE 4.

SPR analysis of ALLINI-2 interactions with HIV-1 IN domains. The binding kinetics of ALLINI-2 interactions with WT IN CCD (A), A128T IN CCD (B), WT IN CCD-CTD (C), and the WT IN CTD (D). These results yielded the following Kd values: 0.722 ± 0.2 μm for WT IN CCD; 1.66 ± 0.2 μm for A128T IN CCD; 2.64 ± 0.4 μm for WT CCD-CTD, whereas no binding was seen for the isolated CTD. RU, response units.
We next examined ALLINI-2 induced HIV-1 IN multimerization using DLS (Fig. 5). This technique can detect particle aggregation and has enabled us to identify higher-order oligomerization of IN in the presence of multimerization selective pyridine-based inhibitor KF116 (46). In the absence of ALLINI-2, no aggregation of apo-IN (200 nm) was observed during the entire time course of the experiment. The lower oligomeric soluble forms (monomers, dimers, and tetramers) of IN were not detected by this technique. ALLINI-2 (200 nm) alone was also soluble in the same buffer. In contrast, light scattering recorded 1, 5, and 15 min after addition of ALLINI-2 to WT IN revealed peaks corresponding to particle sizes with diameters of 164, 342, and 615 nm, respectively (Fig. 5A), indicating an equilibrium shift toward higher-order oligomers in a time-dependent manner. Of note, these particle sizes are significantly larger than functional HIV-1 IN tetramers (with a diameter of ∼7.5 nm) seen by atomic force microscopy analysis of in vitro-assembled HIV-1 intasomes (53). We have also performed DLS experiments with IN containing either A128T (Fig. 5B) or K264A/K266A (Fig. 5C) substitutions. Strikingly, both mutant proteins exhibited remarkable resistance to ALLINI-2-induced protein multimerization.
FIGURE 5.
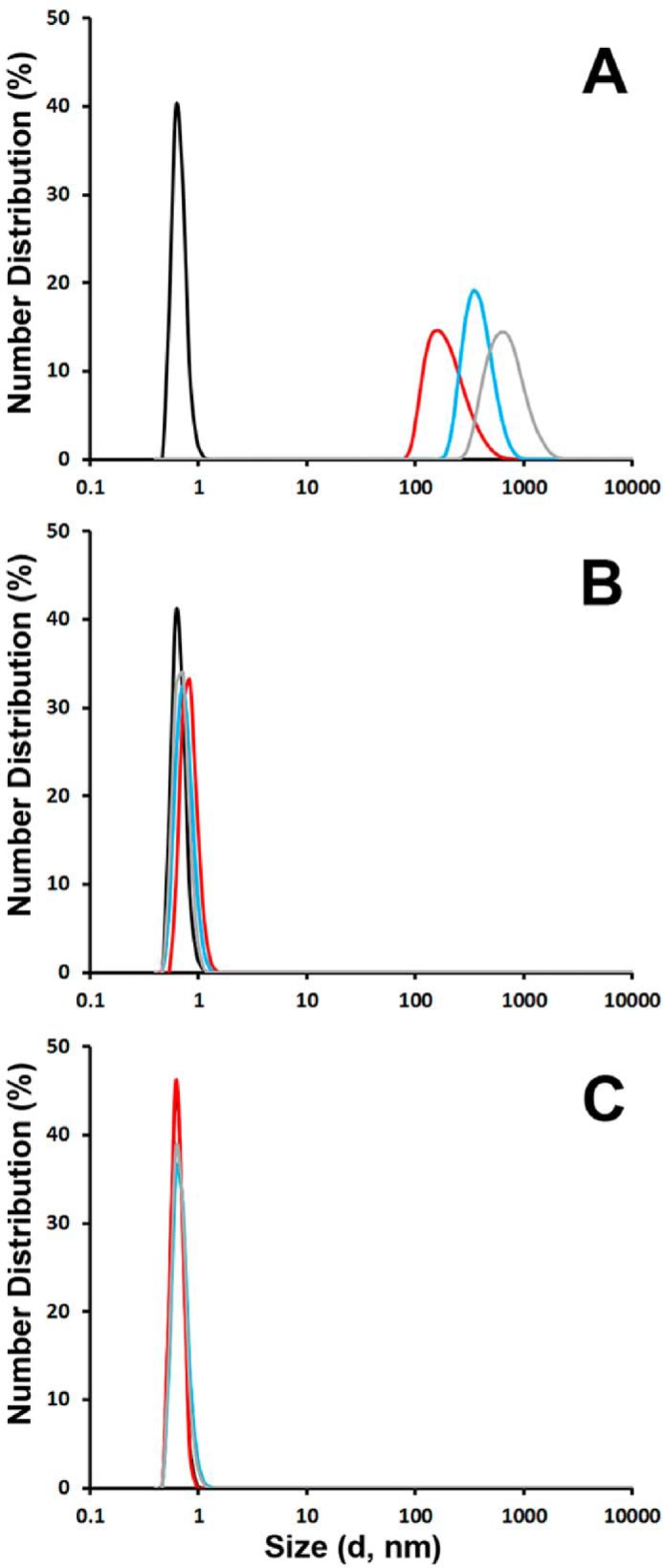
Kinetic analysis of ALLINI-2-induced IN multimerization by DLS. Size distributions of WT (A), A128T (B), and K264A/K266A (C) INs at 1 min (red), 5 min (cyan), and 15 min (gray) after addition of ALLINI-2 or after 15 min with DMSO (black). Peaks observed with WT IN + ALLINI-2 correspond to particle diameter sizes of 164 (1 min), 342 (5 min) and 615 nm (15 min). The peak with <1-nm diameter size was a background signal that was also observed with the buffer alone. Low oligomeric soluble forms of IN such as monomers, dimers, and tetramers could not be detected by this technique.
The effects of A128T and K264A/K266A substitutions were also examined by SEC. Here, tetramers and monomers were seen for WT and the mutant proteins. Addition of ALLINI-2 to WT IN markedly reduced the tetrameric form. However, higher-order oligomers seen in the DLS experiments could not be detected by SEC possibly due to the inability of large protein aggregates to migrate through the Superdex 200 matrix. In contrast, ALLINI-2 addition to A128T or K264A/K266A IN did not significantly alter the tetrameric or monomeric species.
To further examine the effects of A128T and K264A/K266A substitutions on IN structure and function, we carried out additional biochemical and biophysical characterization of the mutant proteins. MS-based protein footprinting of WT and the two mutant proteins displayed very similar surface accessibility profiles, indicating that A128T or K264A/K266A substitutions did not significantly alter the protein structure. The K264A/K266A IN mutant exhibited WT levels of LEDGF/p75 binding, whereas A128T IN displayed slightly reduced binding to the cellular cofactor (Fig. 6B). Catalytic activities of WT and A128T INs were comparable, whereas K264A/K266A IN was not active (Fig. 6C).
FIGURE 6.

Biochemical characterization of WT, A128T, and K264A/K266A INs. SEC profiles of 20 μm WT (A), A128T (B), and K264A/K264A (C) INs in the presence of 5% DMSO (upper panels) or 40 μm ALLINI-2 (lower panels). Peaks corresponding to a tetramer (T) and monomer (M) are indicated. D, IN-LEDGF/p75 binding assays. Pulldown results for WT IN+LEDGF/p75 (lane 1), A128T IN+LEDGF/p75 (lane 2), K264A/K266A IN+LEDGF/p75 (lane 3), and LEDGF/p75 alone (lane 4) are shown. Lanes 5, 6, 7, 8, and 9 depict input of LEDGF/p75, WT IN, A128T IN, K264A/K266A IN, and molecular weight markers, respectively. E, catalytic activities of WT, A128T and K264A/K266A INs.
DISCUSSION
The crystal structure of ALLINI-2 binding to the isolated CCD dimer has revealed that the inhibitor bridges between two IN subunits (38). However, how these interactions promote aberrant, higher-order IN multimerization is not clear. MS-based protein footprinting studies of full length IN, the results of which are summarized in Fig. 7A in the context of the IN structural model (54), allowed us to identify protein-protein interactions in the CCD and CTD that extend beyond the inhibitor binding sites and contribute to higher-order IN multimerization. A recent study (41), which investigated the multimerization of IN domains in the presence of ALLINI GSK1264 using sedimentation velocity and turbidity assays, has also revealed the significance of the CTD in addition to the CCD for the formation of insoluble protein aggregates. In particular, the addition of GSK1264 stimulated dimerization of INF185H(CCD) and INF185H(NTD-CCD), whereas INF185H(CCD-CTD) as well as full-length IN showed evidence of the inhibitor-induced protein aggregation (41). Our DLS analysis of ALLINI-2 interactions with the WT CCD-CTD truncated mutant did not result in detectable particle aggregates as was seen with full-length WT IN (data not shown). These discrepancies could be due to different experimental conditions employed in the two studies. For example, sedimentation assays were performed with 19 μm CCD-CTD containing the solubilizing F185H substitution and 50 μm GSK1264 (41), whereas our DLS experiments were carried out with 200 nm CCD-CTD containing the WT IN sequence and 200 nm ALLINI-2. The differences between full-length WT IN and its truncated CCD-CTD construct observed with DLS experiments argue for the importance of the unique architecture of the full-length protein for aberrant, higher-order IN multimerization.
FIGURE 7.
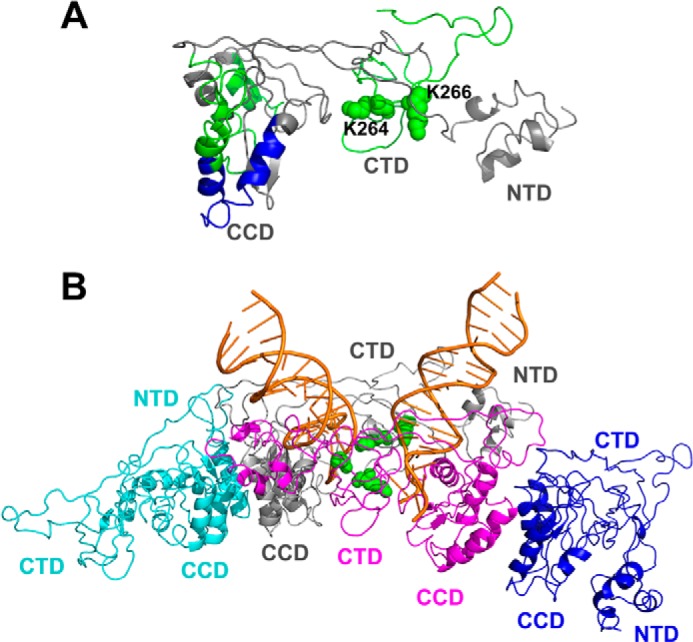
A summary of MS-based footrprinting results in the context of IN structural models. A, a single full-length IN subunit colored gray from the intasome model in Fig. 7B, which interacts with viral DNA, is shown. The peptides that exhibited reduced deuterium uptake in the presence of added ALLINI-2 are colored green and blue according to Fig. 2. Lys-264 and Lys-266 are shown as green spheres. B, the HIV-1 intasome model (54) is shown. Individual IN subunits are colored yellow, gray, magenta, and blue. Viral DNAs are colored orange. Lys-264 and Lys-266 from gray and magenta subunits that interact with viral DNA are shown as green spheres.
We show that ALLINI-2 binds WT and the drug-resistant A128T CCDs with comparable affinities (Fig. 4). Yet, WT but not the mutant IN was susceptible to inhibitor-induced aberrant multimerization (Fig. 5). Available crystal structures provide potential explanation for these observations (38). The hydrogen bonding network between ALLINI-2 and CCD subunit 1, which is the primary determinant of the inhibitor binding, is preserved for both WT and A128T CCDs (38). However, the A128T IN substitution relocates the quinoline ring away from CCD subunit 2, which could reduce the ability of ALLINI-2 to effectively bridge the two subunits and promote higher-order multimerization of the mutant IN. Yet, it is possible that ALLINI-2 can still induce limited conformational changes in A128T IN resulting in protections in the 226–241 region that were detected with HDX experiments.
We have identified two IN regions 181–202 and 250–280 uniquely protected in WT but not in the A128T protein, suggesting their role in the inhibitor-induced aberrant IN multimerization. In particular, our findings demonstrate the significance of residues Lys-264 and Lys-266 for higher-order protein multimerization. In the NMR structure of the isolated CTD, which is seen as a dimer with Lys-264 and Lys-266 being fully surface-exposed and positioned on the opposite side of the CTD-CTD interface (28–30). In the HIV-1 intasome models, which have been generated based on the analogous prototype foamy virus intasome structure, the CTD domains do not engage in protein-protein interactions and instead interact with viral DNA (10, 54, 55). In particular, Lys-264 and Lys-266 directly contact viral DNA (Fig. 7B). In keeping with these observations, K264A/K266A IN substitutions compromised catalytic activities (Fig. 6C). Furthermore, substitutions introduced at IN residues Lys-264 and Lys-266 markedly impaired HIV-1 replication (56).
In vitro ALLINIs compromise IN catalytic activities by blocking formation of the stable synaptic complex between IN and viral DNA (37). Consistently, ALLINIs impaired a step at or prior to 3′-processing in infected cells (39). These observations could be explained by our findings that ALLINI-2-induced IN multimerization compromises the surface exposure of Lys-264 and Lys-266, which are needed for IN binding to viral DNA.
At the same time, it should be noted that ALLINIs are more potent during virus particle maturation rather than during the early steps of HIV-1 replication (12, 41, 42, 44, 45). The competition between chromatin associated LEDGF/p75 and ALLINI binding to the HIV-1 IN dimer interface appears to be one significant factor reducing the inhibitor potency in target cells. Such a competition is absent or minimal during the late stage of HIV-1 replication, which allows ALLINIs to potently induce aberrant IN multimerization, leading to the formation of eccentric cores with the electron dense material being mislocalized between the translucent capsid core and particle membrane. Therefore, it would be interesting to examine contributions of the CTD fragments identified in the present study and in particular Lys-264 and Lys-266 to promote aberrant IN multimerization during the virus particle maturation during ALLINI treatment. These studies, which are currently underway in our laboratory, will help to better understand the mechanism of action of available ALLINIs and facilitate development of improved inhibitors.
Acknowledgment
We thank Dr. Dennis Bong (Ohio State University, Chemistry Department) for the use of the DLS instrument.
This work was supported, in whole or in part, by National Institutes of Health Grants AI062520 (to M. K.), GM103368 (to M. K. and P. R. G.), AI110310 (to M. K. and J. R. F.), and AI110270 (to J. J. K.).
- IN
- integrase
- NTD
- N-terminal domain
- CCD
- catalytic core domain
- CTD
- C-terminal domain
- LEDGF
- lens epithelium-derived growth factor
- ALLINI
- allosteric integrase inhibitor
- HDX
- hydrogen/deuterium exchange
- SEC
- size exclusion chromatography
- NHS
- N-hydroxysuccinimide
- DMSO
- dimethyl sulfoxide
- SPR
- surface plasmon resonance
- DLS
- dynamic light scattering.
REFERENCES
- 1. Cherepanov P., Maertens G., Proost P., Devreese B., Van Beeumen J., Engelborghs Y., De Clercq E., Debyser Z. (2003) HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 278, 372–381 [DOI] [PubMed] [Google Scholar]
- 2. Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J. R., Bushman F. (2005) A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 11, 1287–1289 [DOI] [PubMed] [Google Scholar]
- 3. Llano M., Saenz D. T., Meehan A., Wongthida P., Peretz M., Walker W. H., Teo W., Poeschla E. M. (2006) An essential role for LEDGF/p75 in HIV integration. Science 314, 461–464 [DOI] [PubMed] [Google Scholar]
- 4. Shun M. C., Raghavendra N. K., Vandegraaff N., Daigle J. E., Hughes S., Kellam P., Cherepanov P., Engelman A. (2007) LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 21, 1767–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferris A. L., Wu X., Hughes C. M., Stewart C., Smith S. J., Milne T. A., Wang G. G., Shun M. C., Allis C. D., Engelman A., Hughes S. H. (2010) Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. U.S.A. 107, 3135–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Busschots K., Vercammen J., Emiliani S., Benarous R., Engelborghs Y., Christ F., Debyser Z. (2005) The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J. Biol. Chem. 280, 17841–17847 [DOI] [PubMed] [Google Scholar]
- 7. Cherepanov P., Ambrosio A. L., Rahman S., Ellenberger T., Engelman A. (2005) Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U.S.A. 102, 17308–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engelman A., Englund G., Orenstein J. M., Martin M. A., Craigie R. (1995) Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 69, 2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Engelman A. (1999) In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 52, 411–426 [DOI] [PubMed] [Google Scholar]
- 10. Johnson B. C., Métifiot M., Ferris A., Pommier Y., Hughes S. H. (2013) A homology model of HIV-1 integrase and analysis of mutations designed to test the model. J. Mol. Biol. 425, 2133–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engelman A. (2011) Pleiotropic nature of HIV-1 integrase mutations in HIV-1 Integrase: Mechanism and Inhibitor Design (Neamati N., ed.) pp. 67–81, John Wiley & Sons, Hoboken, NJ [Google Scholar]
- 12. Jurado K. A., Wang H., Slaughter A., Feng L., Kessl J. J., Koh Y., Wang W., Ballandras-Colas A., Patel P. A., Fuchs J. R., Kvaratskhelia M., Engelman A. (2013) Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U.S.A. 110, 8690–8695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bukovsky A., Göttlinger H. (1996) Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. J. Virol. 70, 6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cannon P. M., Wilson W., Byles E., Kingsman S. M., Kingsman A. J. (1994) Human immunodeficiency virus type 1 integrase: effect on viral replication of mutations at highly conserved residues. J. Virol. 68, 4768–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu X., Liu H., Xiao H., Conway J. A., Hehl E., Kalpana G. V., Prasad V., Kappes J. C. (1999) Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 73, 2126–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jaskolski M., Alexandratos J. N., Bujacz G., Wlodawer A. (2009) Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 276, 2926–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai M., Zheng R., Caffrey M., Craigie R., Clore G. M., Gronenborn A. M. (1997) Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 4, 567–577 [DOI] [PubMed] [Google Scholar]
- 18. Zheng R., Jenkins T. M., Craigie R. (1996) Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proc. Natl. Acad. Sci. U.S.A. 93, 13659–13664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bushman F. D., Engelman A., Palmer I., Wingfield P., Craigie R. (1993) Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc. Natl. Acad. Sci. U.S.A. 90, 3428–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee S. P., Xiao J., Knutson J. R., Lewis M. S., Han M. K. (1997) Zn2+ promotes the self-association of human immunodeficiency virus type-1 integrase in vitro. Biochemistry 36, 173–180 [DOI] [PubMed] [Google Scholar]
- 21. Engelman A., Craigie R. (1992) Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66, 6361–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hare S., Di Nunzio F., Labeja A., Wang J., Engelman A., Cherepanov P. (2009) Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog. 5, e1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dyda F., Hickman A. B., Jenkins T. M., Engelman A., Craigie R., Davies D. R. (1994) Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266, 1981–1986 [DOI] [PubMed] [Google Scholar]
- 24. Jenkins T. M., Hickman A. B., Dyda F., Ghirlando R., Davies D. R., Craigie R. (1995) Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues. Proc. Natl. Acad. Sci. U.S.A. 92, 6057–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bujacz G., Alexandratos J., Qing Z. L., Clément-Mella C., Wlodawer A. (1996) The catalytic domain of human immunodeficiency virus integrase: ordered active site in the F185H mutant. FEBS Lett. 398, 175–178 [DOI] [PubMed] [Google Scholar]
- 26. Goldgur Y., Dyda F., Hickman A. B., Jenkins T. M., Craigie R., Davies D. R. (1998) Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U.S.A. 95, 9150–9154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maignan S., Guilloteau J. P., Zhou-Liu Q., Clément-Mella C., Mikol V. (1998) Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: high level of similarity of the active site with other viral integrases. J. Mol. Biol. 282, 359–368 [DOI] [PubMed] [Google Scholar]
- 28. Lodi P. J., Ernst J. A., Kuszewski J., Hickman A. B., Engelman A., Craigie R., Clore G. M., Gronenborn A. M. (1995) Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry 34, 9826–9833 [DOI] [PubMed] [Google Scholar]
- 29. Eijkelenboom A. P., Lutzke R. A., Boelens R., Plasterk R. H., Kaptein R., Hård K. (1995) The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat. Struct. Biol. 2, 807–810 [DOI] [PubMed] [Google Scholar]
- 30. Eijkelenboom A. P., Sprangers R., Hård K., Puras Lutzke R. A., Plasterk R. H., Boelens R., Kaptein R. (1999) Refined solution structure of the C-terminal DNA-binding domain of human immunovirus-1 integrase. Proteins 36, 556–564 [DOI] [PubMed] [Google Scholar]
- 31. McKee C. J., Kessl J. J., Shkriabai N., Dar M. J., Engelman A., Kvaratskhelia M. (2008) Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J. Biol. Chem. 283, 31802–31812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hare S., Shun M. C., Gupta S. S., Valkov E., Engelman A., Cherepanov P. (2009) A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 5, e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jurado K. A., Engelman A. (2013) Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev. Mol. Med. 15, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Engelman A., Kessl J. J., Kvaratskhelia M. (2013) Allosteric inhibition of HIV-1 integrase activity. Curr. Opin. Chem. Biol. 17, 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Demeulemeester J., Chaltin P., Marchand A., De Maeyer M., Debyser Z., Christ F. (2014) LEDGINs, non-catalytic site inhibitors of HIV-1 integrase: a patent review (2006–2014). Expert Opin. Ther. Pat. 24, 609–632 [DOI] [PubMed] [Google Scholar]
- 36. Fader L. D., Malenfant E., Parisien M., Carson R., Bilodeau F., Landry S., Pesant M., Brochu C., Morin S., Chabot C., Halmos T., Bousquet Y., Bailey M. D., Kawai S. H., Coulombe R., LaPlante S., Jakalian A., Bhardwaj P. K., Wernic D., Schroeder P., Amad M., Edwards P., Garneau M., Duan J., Cordingley M., Bethell R., Mason S. W., Bös M., Bonneau P., Poupart M. A., Faucher A. M., Simoneau B., Fenwick C., Yoakim C., Tsantrizos Y. (2014) Discovery of BI 224436, a noncatalytic site integrase inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 5, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kessl J. J., Jena N., Koh Y., Taskent-Sezgin H., Slaughter A., Feng L., de Silva S., Wu L., Le Grice S. F., Engelman A., Fuchs J. R., Kvaratskhelia M. (2012) A multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 287, 16801–16811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feng L., Sharma A., Slaughter A., Jena N., Koh Y., Shkriabai N., Larue R. C., Patel P. A., Mitsuya H., Kessl J. J., Engelman A., Fuchs J. R., Kvaratskhelia M. (2013) The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 288, 15813–15820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsiang M., Jones G. S., Niedziela-Majka A., Kan E., Lansdon E. B., Huang W., Hung M., Samuel D., Novikov N., Xu Y., Mitchell M., Guo H., Babaoglu K., Liu X., Geleziunas R., Sakowicz R. (2012) New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 287, 21189–21203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Christ F., Voet A., Marchand A., Nicolet S., Desimmie B. A., Marchand D., Bardiot D., Van der Veken N. J., Van Remoortel B., Strelkov S. V., De Maeyer M., Chaltin P., Debyser Z. (2010) Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6, 442–448 [DOI] [PubMed] [Google Scholar]
- 41. Gupta K., Brady T., Dyer B. M., Malani N., Hwang Y., Male F., Nolte R. T., Wang L., Velthuisen E., Jeffrey J., Van Duyne G. D., Bushman F. D. (2014) Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains. J. Biol. Chem. 289, 20477–20488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Le Rouzic E., Bonnard D., Chasset S., Bruneau J. M., Chevreuil F., Le Strat F., Nguyen J., Beauvoir R., Amadori C., Brias J., Vomscheid S., Eiler S., Lévy N., Delelis O., Deprez E., Saïb A., Zamborlini A., Emiliani S., Ruff M., Ledoussal B., Moreau F., Benarous R. (2013) Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 10, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Christ F., Shaw S., Demeulemeester J., Desimmie B. A., Marchand A., Butler S., Smets W., Chaltin P., Westby M., Debyser Z., Pickford C. (2012) Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob. Agents Chemother. 56, 4365–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Balakrishnan M., Yant S. R., Tsai L., O'Sullivan C., Bam R. A., Tsai A., Niedziela-Majka A., Stray K. M., Sakowicz R., Cihlar T. (2013) Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One 8, e74163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Desimmie B. A., Schrijvers R., Demeulemeester J., Borrenberghs D., Weydert C., Thys W., Vets S., Van Remoortel B., Hofkens J., De Rijck J., Hendrix J., Bannert N., Gijsbers R., Christ F., Debyser Z. (2013) LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sharma A., Slaughter A., Jena N., Feng L., Kessl J. J., Fadel H. J., Malani N., Male F., Wu L., Poeschla E., Bushman F. D., Fuchs J. R., Kvaratskhelia M. (2014) A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 10, e1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang H., Jurado K. A., Wu X., Shun M. C., Li X., Ferris A. L., Smith S. J., Patel P. A., Fuchs J. R., Cherepanov P., Kvaratskhelia M., Hughes S. H., Engelman A. (2012) HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 40, 11518–11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fadel H. J., Morrison J. H., Saenz D. T., Fuchs J. R., Kvaratskhelia M., Ekker S. C., Poeschla E. M. (2014) TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J. Virol. 88, 9704–9717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Larue R., Gupta K., Wuensch C., Shkriabai N., Kessl J. J., Danhart E., Feng L., Taltynov O., Christ F., Van Duyne G. D., Debyser Z., Foster M. P., Kvaratskhelia M. (2012) Interaction of the HIV-1 intasome with transportin 3 protein (TNPO3 or TRN-SR2). J. Biol. Chem. 287, 34044–34058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chalmers M. J., Busby S. A., Pascal B. D., He Y., Hendrickson C. L., Marshall A. G., Griffin P. R. (2006) Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 78, 1005–1014 [DOI] [PubMed] [Google Scholar]
- 51. Pascal B. D., Willis S., Lauer J. L., Landgraf R. R., West G. M., Marciano D., Novick S., Goswami D., Chalmers M. J., Griffin P. R. (2012) HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McKee C. J., Kessl J. J., Norris J. O., Shkriabai N., Kvaratskhelia M. (2009) Mass spectrometry-based footprinting of protein-protein interactions. Methods 47, 304–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kotova S., Li M., Dimitriadis E. K., Craigie R. (2010) Nucleoprotein intermediates in HIV-1 DNA integration visualized by atomic force microscopy. J. Mol. Biol. 399, 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kessl J. J., Li M., Ignatov M., Shkriabai N., Eidahl J. O., Feng L., Musier-Forsyth K., Craigie R., Kvaratskhelia M. (2011) FRET analysis reveals distinct conformations of IN tetramers in the presence of viral DNA or LEDGF/p75. Nucleic Acids Res. 39, 9009–9022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krishnan L., Li X., Naraharisetty H. L., Hare S., Cherepanov P., Engelman A. (2010) Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 15910–15915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu R., Limón A., Ghory H. Z., Engelman A. (2005) Genetic analyses of DNA-binding mutants in the catalytic core domain of human immunodeficiency virus type 1 integrase. J. Virol. 79, 2493–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]