


Abstract
Endogenous metabolism, environmental exposure, and treatment with some chemotherapeutic agents can all give rise to DNA alkylation, which can occur on the phosphate backbone as well as the ring nitrogen or exocyclic nitrogen and oxygen atoms of nucleobases. Previous studies showed that the minor-groove O2-alkylated thymidine (O2-alkyldT) lesions are poorly repaired and persist in mammalian tissues. In the present study, we synthesized oligodeoxyribonucleotides harboring seven O2-alkyldT lesions, with the alkyl group being a Me, Et, nPr, iPr, nBu, iBu or sBu, at a defined site and examined the impact of these lesions on DNA replication in Escherichia coli cells. Our results demonstrated that the replication bypass efficiencies of the O2-alkyldT lesions decreased with the chain length of the alkyl group, and these lesions directed promiscuous nucleotide misincorporation in E. coli cells. We also found that deficiency in Pol V, but not Pol II or Pol IV, led to a marked drop in bypass efficiencies for most O2-alkyldT lesions. We further showed that both Pol IV and Pol V were essential for the misincorporation of dCMP opposite these minor-groove DNA lesions, whereas only Pol V was indispensable for the T→A transversion introduced by these lesions. Depletion of Pol II, however, did not lead to any detectable alterations in mutation frequencies for any of the O2-alkyldT lesions. Thus, our study provided important new knowledge about the cytotoxic and mutagenic properties of the O2-alkyldT lesions and revealed the roles of the SOS-induced DNA polymerases in bypassing these lesions in E. coli cells.
INTRODUCTION
Exposure to a variety of endogenous and exogenous agents can result in damage to genomic DNA (1). Unrepaired DNA lesions may block DNA replication and transcription as well as induce mutations in these processes, which may contribute to the development of cancer and other human diseases (1).
Alkylation represents a major type of DNA damage (2). Both endogenous metabolism and environmental exposure can induce DNA alkylation, and DNA alkylation also constitutes the major mechanism of action for a number of commonly prescribed cancer chemotherapeutic agents (3,4). Hence, understanding how the alkylated DNA lesions perturb the flow of genetic information is important for revealing the implications of these lesions in the etiology for the development of human diseases. Such knowledge also forms the basis for designing better strategies for cancer treatment.
Depending on the nature of the alkylating agents involved, the size of the alkyl group adducted to nucleobases varies substantially. For instance, methylation can be induced by a number of anticancer drugs including temozolomide, dacarbazine, streptozotocin and procarbazine, and chloroethylation can be introduced by anticancer drugs including carmustine [bis-(2-chloroethyl)-nitrosourea] and lomustine [(1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea] (5). On the other hand, bulky pyridyloxobutyl (POB) and pyridylhydroxybutyl (PHB) groups can be conjugated with the O2 position of cytosine and thymine as well as the N7 and O6 positions of guanine by metabolites of tobacco carcinogens 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N’-nitrosonornicotine (NNN) (6–9). The POB and PHB adducts formed on the O6 of dG and O2 of dT are stable, and they have been detected in the esophagus, lung and liver tissues of rats treated with NNK, NNN, and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol, which is the major metabolite of NNK (10–12). In this context, it is worth noting that the size of the alkyl group also affects the distributions of alkylation products formed on different positions of nucleobases and the phosphodiester backbone (13).
Multiple lines of evidence suggest that the minor-groove O2-alkylated thymidine (O2-alkyldT) lesions are poorly repaired in mammalian systems. Despite the fact that alkylation of the O6 position of guanine is more facile than that of the O4 or O2 position of thymine (14), O2-POBdT and O2-PHBdT were found to accumulate in lung and liver tissues of NNK- and NNAL-treated F-344 rats at markedly higher levels than the corresponding adducts formed at the O6 position of guanine (11,12), suggesting the poor repair of the O2-POBdT and O2-PHBdT lesions in mammalian tissues. The inefficient repair of the minor-groove dT lesions is also manifested by the observation that O2-EtdT is present at much higher levels, and persist much longer, than O6-EtdG in tissues of rats treated with DNA ethylating agents (15–17). Additionally, O2-EtdT could be detected in human lymphocyte DNA, and cigarette smoking led to substantially elevated levels of this lesion (18).
Some replication studies have been carried out for the O2-alkyldT lesions. O2-MedT, O2-EtdT and O2-POB-dT were found to strongly block DNA replication and induce mutations in Escherichia coli cells (19,20). In this vein, our recent study showed that O2-EtdT differs markedly from its regioisomeric N3- and O4-EtdT in perturbing the efficiency and fidelity of DNA replication in E. coli cells, and the different impacts of three modified nucleosides on DNA replication are attributed to their unique chemical properties (20). In the present study, we set out to assess systematically how the size of the alkyl group incorporated to the minor-groove O2 position of thymine affects the fidelity and efficiency of DNA replication, and how replication across these lesions is modulated by SOS-induced DNA polymerases. Thus, we incorporated seven O2-alkyldT lesions with varying size of the alkyl group into single-stranded M13 genome and investigated how these lesions block DNA replication and induce mutations in Escherichia coli cells. We also assessed the roles of various SOS-induced DNA polymerases in bypassing these lesions by conducting the replication studies in E. coli cells that are deficient in one or more of these polymerases.
MATERIALS AND METHODS
Materials
All chemicals, unless otherwise specified, were from Sigma-Aldrich (St. Louis, MO) or EMD Millipore. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) was obtained from TCI America (Portland, OR). Shrimp alkaline phosphatase and [γ-32P]ATP were purchased from USB Corporation (Cleveland, OH) and Perkin Elmer (Piscataway, NJ), respectively, and all other enzymes were obtained from New England Biolabs (Ipswich, MA). All unmodified oligodeoxyribonucleotides (ODNs) were purchased from Integrated DNA Technologies (Coralville, IA). The 12-mer ODNs harboring a site-specifically incorporated O2-alkyldT were synthesized using conventional phosphoramidite chemistry, as detailed below.
M13mp7(L2) plasmid and wild-type AB1157 E. coli strains were kindly provided by Prof. John M. Essigmann, and polymerase-deficient AB1157 strains [Δpol B1::spec (Pol II-deficient), ΔdinB (Pol IV-deficient), ΔumuC::kan (Pol V-deficient), and Δpol B1::spec ΔdinB ΔumuC::kan (Pol II, Pol IV, Pol V-triple knockout)] were generously provided by Prof. Graham C. Walker.
General procedures for syntheses of O2-alkyldT
Phosphoramidite building blocks of seven O2-alkyldT lesions (Scheme 1) were synthesized by employing procedures adapted from those published previously (Scheme 2) (21). Compound 1 (500 mg, 1.26 mmol), prepared with an isolated yield of 68% according to the published procedures (22), was suspended in 30 ml of anhydrous alcohol, to which solution was subsequently added sodium bicarbonate (3.0 eq., 318 mg, 3.78 mmol). For alcohols bearing a branched alkyl group, i.e. iPrOH, sBuOH and iBuOH, a catalytic amount of sodium metal (5 mg, 0.217 mmol) was also added. The resulting mixture was refluxed for 16 h prior to being cooled to room temperature. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography with a step gradient of methanol (0–10%) in CH2Cl2 to afford the desired product.
Scheme 1.
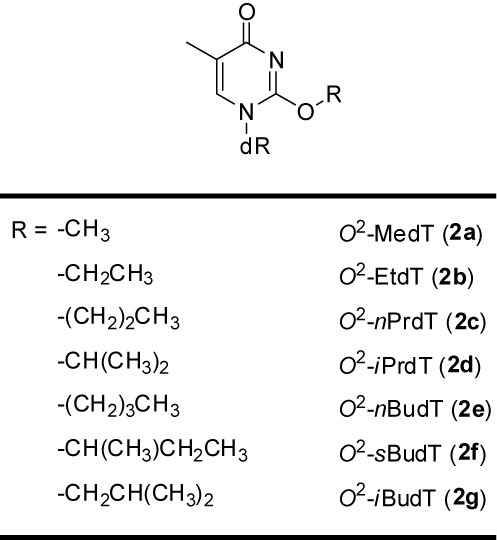
The structures of the O2-alkyldT lesions examined in the present study.
Scheme 2.
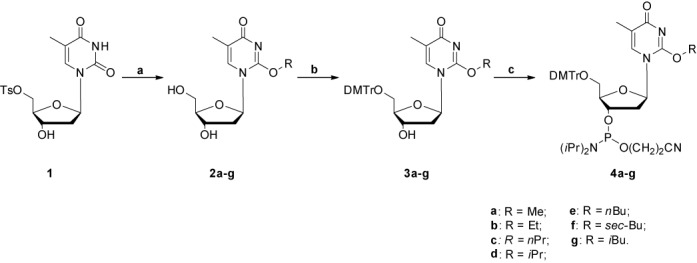
Syntheses of phosphoramidite building blocks of O2-alkylthymidine. Reagents and conditions: (a) ROH, NaHCO3 (with or without Na), reflux, 16 h; (b) DMTr-Cl, DMAP, pyridine, room temperature, 10 h; (c) 2-cyanoethyl-N,N-diisopropyl chlorophosphoramidite, DIEA, CH2Cl2, 1 h.
General procedures for dimethoxytrityl (DMTr) protection of the 5′-hydroxyl group
Compound 2a-g (100 mg) was dissolved in anhydrous pyridine (10 ml) and the solution was cooled in an ice bath. 4-Dimethylaminopyridine (DMAP, 0.5% mol) and dimethoxytrityl chloride (DMTr-Cl, 1.2 eq.) were subsequently added, and the resulting solution was stirred at room temperature for 10 h. The reaction was quenched with methanol (0.5 ml), and the solvent removed under reduced pressure. The residue was purified by silica gel column chromatography with ethyl acetate as mobile phase to yield the desired product 3a-g.
General procedures for phosphoramidite synthesis
To a round bottom flask, which contained a solution of compound 3a-g (60 mg) in dry CH2Cl2 (3.0 ml) and was stirred in an ice bath, was added N,N-diisopropylethylamine (DIEA, 2.2 eq.), followed by dropwise addition of 2-cyanoethyl-N,N-diisopropyl chlorophosphoramidite (1.2 eq.). The mixture was subsequently stirred at room temperature for 1 h under an argon atmosphere. The reaction was quenched by cooling the mixture in an ice bath followed by slow addition of methanol (0.20 ml). The solution was quickly diluted with ethyl acetate (8.0 ml). The organic layer was subsequently washed with saturated NaHCO3 (4.0 ml) and brine (4.0 ml), and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to yield 4a-g in a foam that was directly employed for ODN synthesis.
The reaction yields and spectroscopic characterizations of the above-synthesized products are provided in the online Supplementary Materials. The NMR spectra are shown in Supplementary Figures S1–S18.
ODN synthesis
The 12-mer lesion-containing ODNs 5′-ATGGCGXGCTAT-3′ (‘X’ represents O2-alkyldT) were synthesized on a Beckman Oligo 1000S DNA synthesizer (Fullerton, CA) at 1 μmol scale. The above-described phosphoramidite building block was dissolved in anhydrous acetonitrile at a concentration of 0.067 mM. Commercially available ultramild phosphoramidite building blocks were used for the incorporation of the unmodified nucleotides (Glen Research Inc., Sterling, VA, USA) following the standard ODN assembly protocol. The ODNs were cleaved from the controlled pore glass support and deprotected with concentrated ammonium hydroxide at room temperature for 1 h. The solvents were evaporated and the residues redissolved in water for high-performance liquid chromatography (HPLC) purification.
HPLC
HPLC separation was performed on an Agilent 1100 system with a Kinetex XB-C18 column (4.6 × 250 mm, 5 μm in particle size and 100 Å in pore size; Phenomenex Inc., Torrance, CA, USA). For the purification of ODNs, a triethylammonium acetate buffer (50 mM, pH 6.8, solution A) and a mixture of solution A and acetonitrile (70/30, v/v, solution B) were employed as mobile phases. The flow rate was 0.8 ml/min, and the gradient profile in terms of solution B was 5–25% in 5 min followed by 25–65% in 60 min. The HPLC traces for the purifications of the 12-mer lesion-containing ODNs are shown in Supplementary Figure S19, and electrospray ionization-mass spectrometry (ESI-MS) and tandem MS (MS/MS) characterizations of the purified lesion-containing ODNs are displayed in Supplementary Figures S20–S25.
Preparation of the lesion-carrying 22-mer ODNs
The 12-mer O2-alkyldT-containing ODNs were 5′-phosphorylated and ligated individually with a 10-mer ODN (5′-AGTGGAAGAC-3′) in the presence of a template in the ligation buffer with T4 DNA ligase and ATP at 16°C for 8 h. The resulting 22-mer ODNs were purified by denaturing PAGE.
Construction of single-stranded lesion-containing and lesion-free competitor M13 genomes
The lesion-containing and lesion-free M13mp7(L2) genomes were prepared according to the previously reported procedures (23). First, 20 pmol of single-stranded M13 genome was digested with 40 U EcoRI at 23°C for 8 h to linearize the vector. The resulting linearized vector was mixed with two scaffolds, 5′-CTTCCACTCACTGAATCATGGTCATAGCTTTC-3′ and 5′-AAAACGACGGCCAGTGAATTATAGC-3′ (25 pmol), each spanning one end of the linearized vector. To the mixture was subsequently added 30 pmol of the 5′-phosphorylated 22-mer O2-alkyldT-bearing ODNs or the competitor ODN (25-mer, 5′-AGTGGAAGACATGGCGATAAGCTAT-3′), and the DNA was annealed. The resulting mixture was treated with T4 DNA ligase at 16°C for 8 h. T4 DNA polymerase (22.5 U) was subsequently added and the mixture was incubated at 37°C for 4 h to degrade the excess scaffolds and the unligated vector. The successfully ligated M13 genomes were purified from the solution by using Cycle Pure Kit (Omega). The constructed lesion-containing genomes were normalized against the lesion-free competitor genome following published procedures (23).
Transformation of lesion-containing and competitor M13 genomes into E. coli cells
All the O2-alkyldT-containing M13 genomes, except for the O2-EtdT-harboring genome which was mixed with the competitor genome at a molar ratio of 20:1, were mixed with the competitor genome at a molar ratio of 10:1, where a constant amount (25 fmol) of competitor genome was employed. The mixtures were transformed into SOS-induced, electrocompetent wild-type AB1157 E. coli cells and the isogenic E. coli cells that are deficient in Pol II, Pol IV, Pol V or all three polymerases, following the previously published procedures (23). In this vein, the SOS system was induced by irradiating the E. coli cells with 254 nm light at a dose of 45 J/m2, as described previously (24). We chose to conduct the replication experiments in SOS-induced cells because of the relatively low bypass efficiencies for most of the O2-alkyldT lesions (vide infra), and we found that the bypass efficiency for O2-EtdT in wild-type AB1157 cells was elevated by ∼3-fold upon SOS induction. The E. coli cells were subsequently grown in lysogeny broth (LB) culture at 37°C for 6 h. The phage was recovered from the supernatant by centrifugation at 13 000 rpm for 5 min and further amplified in SCS110 E. coli cells to increase the progeny/lesion–genome ratio. The amplified phage was finally purified using the QIAprep Spin M13 kit (Qiagen) to obtain the ssM13 DNA template for polymerase chain reaction (PCR) amplification.
Quantification of bypass efficiencies and mutation frequencies
We employed the competitive replication and adduct bypass (CRAB) assay developed by Delaney et al. (23,25) to assess the bypass efficiencies of the O2-alkyldT lesions in vivo (Figure 1). The PCR amplification was carried out with the use of Phusion high-fidelity DNA polymerase for the sequence region of interest in the ssM13 DNA template. The primers were 5′-YCAGCTATGACCATGATTCAGTGAGTGGA-3′ and 5′-YTCGGTGCGGGCCTCTTCGCTATTAC-3′ (Y is an amino group). The amplification cycles were 30, each cycle consisting of 10 s at 98°C, 30 s at 65°C and 15 s at 72°C, with a final extension at 72°C for 5 min. The PCR products were purified by using Cycle Pure Kit. For the determination of bypass efficiency, a portion of the above PCR products was digested with 10 U BbsI restriction endonuclease and 1 U shrimp alkaline phosphatase in 10 μl New England Biolabs (NEB) buffer 4 at 37°C for 30 min, followed by heating at 80°C for 20 min to deactivate the shrimp alkaline phosphatase. To the above mixture was subsequently added a 15 μl solution containing 5 mM DTT, 1 μM ATP (premixed with 1.66 pmol [γ-32P]ATP) and 10 U T4 polynucleotide kinase, and the mixture was incubated at 37°C for 30 min, followed by heating at 65°C for 20 min to deactivate the T4 polynucleotide kinase. To the resulting solution was added 10 U MluCI restriction endonuclease, and the mixture was incubated at 37°C for 30 min, followed by quenching with 15 μl formamide gel loading buffer containing xylene cyanol FF and bromophenol blue dyes. The mixture was separated using 30% native polyacrylamide gel (acrylamide:bis-acrylamide = 19:1), and the DNA bands were quantified using a Typhoon 9410 variable mode imager.
Figure 1.
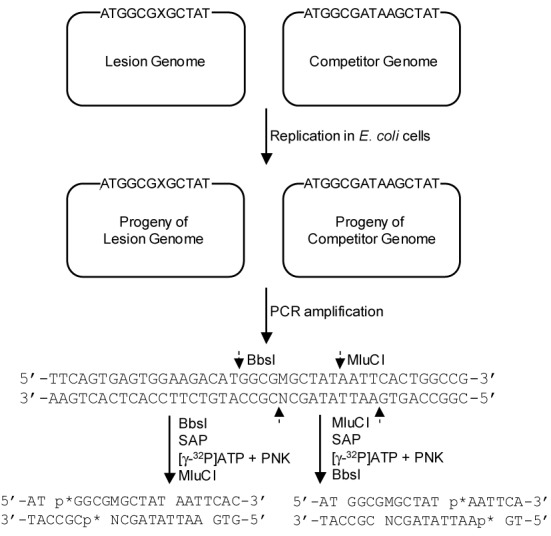
Restriction digestion and post-labeling method for determining the bypass efficiencies and mutation frequencies of O2-alkyldT lesions in E. coli cells. ‘X’ in the 22-mer DNA strand designates dT or O2-alkyldT. The cleavage sites for BbsI and MluCI restriction endonucleases are indicated with broken arrows. Only partial sequence of PCR products for the lesion-containing genome is displayed, and the PCR products of the competitor genome are not shown. For LC-MS/MS analysis, the PCR products were digested with BbsI and MluCI prior to digestion using shrimp alkaline phosphatase (SAP), and the [32P]-labeling step involving the use of T4 polynucleotide kinase (T4 PNK) was omitted.
The above restriction digestion of the PCR products gave rise to the formation of a short duplex d(p*GGCGMGCTAT)/d(AATTATAGCN), where ‘M’ represents the nucleobase incorporated at the original lesion site during DNA replication in vivo, ‘N’ is the complementary base of ‘M’ in the opposite strand, and ‘p*’ designates the [5′-32P]-labeled phosphate (Figure 1). The effect of a lesion on replication fidelity is represented by the mutation frequencies, i.e. the percentage of mutant restriction fragments in the total amounts of restriction fragments released from the PCR products of the lesion-carrying genome. The bypass efficiency was determined by using the following formula:
 |
Identification of mutant products by LC-MS/MS
The PCR products were digested with 50 U BbsI restriction endonuclease and 20 U shrimp alkaline phosphatase in 250 μl NEB buffer 4 at 37°C for 2 h, followed by deactivation of the phosphatase at 80°C for 20 min. To the mixture was added 50 U MluCI restriction endonuclease, and the solution was incubated at 37°C for 1 h. The resulting solution was extracted once with phenol/chloroform/isoamyl alcohol (25:24:1, v/v). The aqueous layer was subsequently dried in a Speed-vac, desalted with HPLC and dissolved in 20 μl water. A 10 μl aliquot was injected for liquid chromatography (LC)-MS/MS analysis on an LTQ linear ion trap mass spectrometer (Thermo Electron, San Jose, CA, USA). An Agilent Zorbax SB-C18 column (0.5 × 250 mm, 5 μm in particle size) was employed, and the gradient for LC-MS/MS analysis was 5 min of 5–20% methanol followed by 35 min of 20–50% methanol in 400 mM HFIP. The temperature for the ion-transport tube was maintained at 300°C. The mass spectrometer was set up for monitoring the fragmentation of the [M-3H]3− ions of the 10-mer [d(GGCGMGCTAT), where ‘M’ represents A, T, C or G], and d(AATTATAGCN), with ‘N’ being A, T, C or G. The fragment ions detected in the MS/MS were assigned manually.
RESULTS
The major objectives of the present study were to explore how the O2-alkyldT lesions with varying sizes of the alkyl group compromise DNA replication and to define the roles of SOS-induced DNA polymerases in bypassing these lesions in E. coli cells. To this end, we first synthesized ODNs harboring site-specifically incorporated O2-alkyldT lesions (Scheme 1) and characterized these ODNs by ESI-MS and MS/MS analyses (Supplementary Figures S20–S25). Here, we use d(ATGGCGXGCTAT) (‘X’ represents O2-nPrdT) as an example to illustrate how we use ESI-MS and MS/MS to confirm the site of O2-nPrdT incorporation. Negative-ion ESI-MS showed that the m/z value for the [M – 3H]3− ion of the ODN (Supplementary Figure S21a) is 14 Thomson unit higher than the calculated m/z value of the unmodified d(ATGGCGTGCTAT), which is consistent with the presence of an O2-nPrdT in this ODN. The MS/MS of the [M – 3H]3− ion (m/z 1238.5) of the ODN showed the formation of a series of wn and [an-Base] ions (Supplementary Figure S21b). The measured molecular masses for the w2, w3, w4 and w5 ions were the same as the calculated ones for the corresponding ions of the unmodified ODN, whereas the w6, w7, w8, w9 and w11 ions exhibited 42 Da higher in molecular mass than those of the respective fragment ions formed from the unmodified ODN. These results are in agreement with the presence of an O2-nPrdT at the 7th position in this ODN. The above conclusion is further supported by the observed masses for the [an-Base] ions; the measured molecular masses for the [a8-G], [a9-C] and [a11-A] ions are 42 Da higher, whereas the measured molecular masses for the [a3-G], [a4-G], [a5-C], [a6-G] and [a7-X] ions are the same as the calculated ones for the corresponding fragment ions for the unmodified d(ATGGCGTGCTAT).
We subsequently ligated these ODNs individually into single-stranded M13 genome, and assessed the bypass efficiencies and mutation frequencies of the O2-alkyldT lesions by using a modified version of the CRAB and restriction endonuclease and post-labeling (REAP) assays (Figure 1) (26). In this assay, the progeny genomes emanating from in vivo replication were amplified by PCR, and the resulting PCR products were digested with two restriction enzymes, i.e. MluCI and BbsI, and the ODNs released from the restriction digestion were analyzed by LC-MS/MS and denaturing PAGE to identify the replication products, as described recently (20). In this regard, with the use of 30% native PAGE, we were able to resolve [5′-32P]-labeled d(p*GGCGTGCTAT) (non-mutagenic product, 10mer-T) from the corresponding products carrying a T→A or T→G mutation, i.e. d(p*GGCGAGCTAT) (10mer-A) and d(p*GGCGGGCTAT) (10mer-G) (Figures 1 and 2a and Supplementary Figure S27a). Nevertheless, the non-mutagenic product co-migrated with the corresponding product bearing a T→C mutation at the lesion site, i.e. d(p*GGCGCGCTAT) (10mer-C, Figure 2a and Supplementary Figure S27a). To determine the frequency of T→C mutation, we reversed the order of the two restriction enzyme digestion so that the bottom strand, namely d(AATTATAGCN), could be selectively radiolabeled. In doing so, we could completely resolve the product emanating from T→C mutation at the lesion site, i.e. d(AATTATAGCG) (10mer-G), from the non-mutagenic product (10mer-A) and the products with a T→A (10mer-T) or T→G (10mer-C) mutation (Figure 2b and Supplementary Figure S27b). Thus, by monitoring separately the products from the two strands, we were able to quantify the frequencies of nucleobase substitutions.
Figure 2.

Native PAGE (30%) for monitoring the bypass efficiencies and mutation frequencies of O2-alkyldT lesions in SOS-induced AB1157 E. coli cells that are proficient in translesion synthesis or deficient in Pol II, Pol IV or Pol V. (a) Gel image showing the 13-mer and 10-mer products released from the top strand (lesion-containing strand) of the PCR products of the progeny of the competitor genome and the control or lesion-carrying genome, where 10mer-A, 10mer-C, 10mer-G and 10mer-T represent the [5′-32P]-labeled standard ODNs 5′-GGCGMGCTAT-3′, with ‘M’ being A, C, G and T, respectively, and 10mer-CA designates TG→CA tandem double mutation. (b) Gel image showing the 13-mer and 10-mer products released from the bottom strand (opposite to lesion-containing strand) of the PCR products of the progeny of the competitor genome and the control or lesion-carrying genome, where 10mer-A, 10mer-C, 10mer-G and 10mer-T represent the [5′-32P]-labeled standard ODNs 5′-AATTATAGCN-3′, with ‘N’ being A, C, G and T, respectively, and 10mer-TG designates 5′-AATTATAGTG-3′. The lesion-containing genomes were mixed individually with the competitor genome at molar ratios of 10:1 or 20:1 in the transfection experiments (see the Materials and Methods section).
We further confirmed the identities of the above restriction digestion products by using LC-MS/MS, where we monitored the fragmentation of the [M – 3H]3− ions of d(GGCGMGCTAT) and d(AATTATAGCN), where ‘M’ and ‘N’ designate the nucleotides inserted at the initial damage site and the opposing nucleotide in the complementary strand, respectively. Our LC-MS/MS data revealed that, similar as what we found recently for the O2-EtdT (20), all other six O2-alkyldT lesions display promiscuous miscoding properties, where T→A, T→C and T→G mutations were observed (representative LC-MS and MS/MS data are shown in Supplementary Figures S28–S30). Additionally, O2-iPrdT induced a unique type of mutation, the TG→CA tandem mutation (Supplementary Figures S28–S30).
After identifying the replication products, we determined the bypass efficiencies of the O2-alkyldT lesions by measuring the ratio of the total amounts of products from the lesion-containing genome over the damage-free competitor genome and normalized this ratio against the corresponding ratio obtained for the damage-free control genome, as described in the Materials and Methods section. Our results showed that, in SOS-induced wild-type E. coli cells, the bypass efficiency decreases with the size of the alkyl group (Figure 3).
Figure 3.
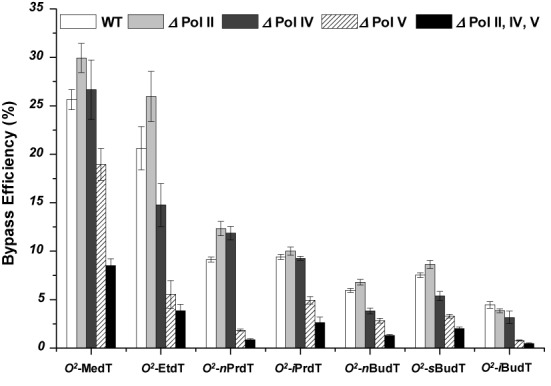
The bypass efficiencies of the O2-alkyldT lesions in E. coli strains that are proficient in translesion synthesis or deficient in Pol II, Pol IV, Pol V or all three SOS-induced DNA polymerases. The data represent the means and standard deviations of results from at least three independent replication experiments.
To define the roles of the SOS-induced DNA polymerases in bypassing the O2-alkyldT lesions, we conducted the replication experiments in E. coli strains deficient in various SOS-induced DNA polymerases. Our results showed that the removal of Pol II did not affect appreciably the bypass efficiencies for any of the O2-alkyldT lesions; however, pronounced drops in bypass efficiencies were observed in Pol V-null cells, supporting that Pol V plays a major role in bypassing the minor-groove O2-alkyldT lesions (Figure 3).
We also determined the mutation frequencies of the O2-alkyldT lesions in wild-type and polymerase-deficient AB1157 E. coli strains (Figure 4). Interestingly, the T→G mutation was totally abolished when the replication experiments were conducted in E. coli cells that are deficient in Pol IV or Pol V (Figure 4c), underscoring the mutual involvement of Pol IV and Pol V in the mutagenic incorporation of dCMP opposite the minor-groove O2-alkyldT lesions and the subsequent extension of the mismatched primer terminus. Additionally, depletion of Pol V resulted in the complete loss of T→A mutation for all O2-alkyldT lesions (Figure 4b). Thus, Pol V is essential for the misincorporation of dTMP opposite these lesions. Individual depletion of any of the SOS-induced DNA polymerases did not lead to the elimination of the T→C mutation for any of the O2-alkyldT lesions, whereas simultaneous deletion of all three SOS-induced DNA polymerases rendered the T→C mutation undetectable for O2-MedT, O2-nBudT and O2-sBudT (Figure 4a). These results suggest the redundant roles of Pol II, Pol IV and Pol V in the misincorporation of dGMP opposite these three O2-alkyldT lesions. On the other hand, no marked difference in T→C mutation was observed for O2-EtdT, O2-nPrdT, O2-iPrdT or O2-iBudT in wild-type or triple knockout AB1157 cells (Figure 4a), suggesting the involvement of other polymerase(s) (e.g. Pol III) in the generation of the T→C mutation for these lesions. Aside from the absence of difference in bypass efficiencies, the mutation frequencies for all the seven O2-alkyldT lesions were indistinguishable in the wild-type and Pol II-null AB1157 cells (Figure 4), supporting that Pol II is not responsible for bypassing the minor-groove O2-alkyldT lesions.
Figure 4.
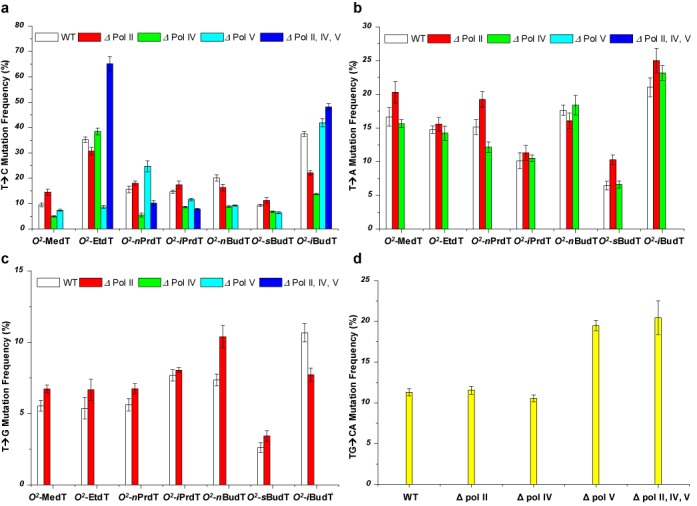
Mutation frequencies of the O2-alkyldT lesions in E. coli cells. Shown are the frequencies for the T→C (a), T→A (b) and T→G (c) mutations observed for the O2-alkyldT lesions, and the TG→CA mutation found for O2-iPrdT (d). The data represent the means and standard deviations of results from three independent replication experiments.
DISCUSSION
We assessed systematically the cytotoxic and mutagenic properties of the O2-alkyldT lesions and our results led to several important conclusions. First, we found that the O2-alkyldT lesions are strong impediments to DNA replication in E. coli cells, with the blocking effect increasing with the size of the alkyl group. This may reflect the greater difficulty experienced by the DNA polymerases in accommodating the more bulky O2-alkyldT lesions into their active sites, which results in less productive nucleotide incorporation at or near the lesion site.
Second, our results demonstrated that the nucleotide insertions opposite the O2-alkyldT lesions are promiscuous, where all three types of single-base substitutions, namely T→A, T→C and T→G mutations, were observed for each of the O2-alkyldT lesion. The promiscuous nucleotide misinsertion opposite the O2-alkyldT lesions is in line with the notion that the incorporation of an alkyl group to the O2 position of thymine may render the nucleobase unfavorable in pairing with any of the four normal nucleobases (27).
Third, we observed that the misincorporation of dTMP opposite these lesions required Pol V, whereas dCMP misinsertion necessitates both Pol IV and Pol V. In this respect, it is worth noting that replication across the (+)-trans-anti diastereomer of the benzo[a]pyrene-7,8-diol-9,10-epoxide (BPDE) adduct on minor-groove N2 position of dG necessitates the mutual action of Pol IV and Pol V (28), whereas only Pol IV is essential for the replicative bypass of the corresponding (−)trans-anti diastereomer (28–30). Future structural studies on complexes between Pol IV/Pol V and lesion-containing primer templates will provide important mechanistic insights into the unique mutagenic nucleotide incorporations mediated by Pol V alone, or by the combined action of Pol IV and Pol V.
It is also worth noting the difference in replicative bypass of the O2-alkyldT lesions with the more extensively studied minor-groove N2-alkyldG lesions. In this vein, a site-specifically incorporated N2-(1-carboxyethyl)-2′-deoxyguanosine was found not to block strongly the DNA replication in wild-type AB1157 cells, and depletion of Pol IV, but not Pol II or Pol V, led to a marked drop in bypass efficiency for this lesion (26). In addition, biochemical experiments revealed that Pol IV and its human ortholog, i.e. DNA polymerase κ, are capable of inserting the correct nucleotide (dCMP) opposite a number of N2-dG lesions at high efficiency (26,31–34). This is in keeping with the findings made from the X-ray crystal structure of human DNA polymerase κ in complex with the primer–template, which reveals the capacity of the active site of the polymerase in accommodating the minor-groove modifications at the primer–template junction (35). Pol IV may be capable of fitting the O2-alkyldT lesions into its active site. Different from alkylation on the N2 position of dG, which does not disrupt significantly the hydrogen bonding properties of the guanine base, the inability of the O2-alkyldT lesions to form favorable base pair with any of the four canonical nucleotides may impede productive nucleotide incorporation opposite the lesion site. This may account for the lack of significant involvement of Pol IV in bypassing the minor-groove dT lesions.
Taken together, our systematic shuttle vector-based study on a group of structurally defined O2-alkyldT lesions provided important new knowledge about the impact of this under-investigated group of DNA lesions on the efficiency and accuracy of DNA replication in vivo. Our results also demonstrated that the cytotoxic properties of these DNA lesions depend on the size of the alkyl group and the mutagenic properties of these lesions are modulated by the nature of SOS-induced DNA polymerases involved.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank Prof. Graham C. Walker for providing the E. coli strains and Prof. John M. Essigmann for providing the initial M13 vector.
FUNDING
National Institutes of Health (NIH) [ES025121 to Y.W.]. Funding for open access charge: NIH [R01 ES025121].
Conflict of interest statement. None declared.
REFERENCES
- 1.Friedberg E.C., Walker G.C., Siede W., Wood R.D., Schultz R.A., Ellenberger T. DNA Repair and Mutagenesis. Washington, D.C: ASM Press; 2006. [Google Scholar]
- 2.Singer B., Grunberger D. Molecular Biology of Mutagens and Carcinogens. New York: Plenum Press; 1983. [Google Scholar]
- 3.Sedgwick B., Bates P.A., Paik J., Jacobs S.C., Lindahl T. Repair of alkylated DNA: recent advances. DNA Repair. 2007;6:429–442. doi: 10.1016/j.dnarep.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Helleday T., Petermann E., Lundin C., Hodgson B., Sharma R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 5.Gerson S.L. MGMT: its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 6.Wang L., Spratt T.E., Liu X.K., Hecht S.S., Pegg A.E., Peterson L.A. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1997;10:562–567. doi: 10.1021/tx9602067. [DOI] [PubMed] [Google Scholar]
- 7.Wang M., Cheng G., Sturla S.J., Shi Y., McIntee E.J., Villalta P.W., Upadhyaya P., Hecht S.S. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem. Res. Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]
- 8.Upadhyaya P., Sturla S.J., Tretyakova N., Ziegel R., Villalta P.W., Wang M., Hecht S.S. Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem. Res. Toxicol. 2003;16:180–190. doi: 10.1021/tx0256376. [DOI] [PubMed] [Google Scholar]
- 9.Hecht S.S., Villalta P.W., Sturla S.J., Cheng G., Yu N., Upadhyaya P., Wang M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with DNA. Chem. Res. Toxicol. 2004;17:588–597. doi: 10.1021/tx034263t. [DOI] [PubMed] [Google Scholar]
- 10.Lao Y., Yu N., Kassie F., Villalta P.W., Hecht S.S. Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)- and (S)-N’-nitrosonornicotine. Chem. Res. Toxicol. 2007;20:246–256. doi: 10.1021/tx060208j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lao Y., Yu N., Kassie F., Villalta P.W., Hecht S.S. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2007;20:235–245. doi: 10.1021/tx060207r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Upadhyaya P., Kalscheuer S., Hochalter J.B., Villalta P.W., Hecht S.S. Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2008;21:1468–1476. doi: 10.1021/tx8001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singer B. O-alkyl pyrimidines in mutagenesis and carcinogenesis: occurrence and significance. Cancer Res. 1986;46:4879–4885. [PubMed] [Google Scholar]
- 14.Shrivastav N., Li D., Essigmann J.M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010;31:59–70. doi: 10.1093/carcin/bgp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Den Engelse L., De Graaf A., De Brij R.J., Menkveld G.J. O2- and O4-ethylthymine and the ethylphosphotriester dTp(Et)dT are highly persistent DNA modifications in slowly dividing tissues of the ethylnitrosourea-treated rat. Carcinogenesis. 1987;8:751–757. doi: 10.1093/carcin/8.6.751. [DOI] [PubMed] [Google Scholar]
- 16.Brent T.P., Dolan M.E., Fraenkel-Conrat H., Hall J., Karran P., Laval L., Margison G.P., Montesano R., Pegg A.E., Potter P.M., et al. Repair of O-alkylpyrimidines in mammalian cells: a present consensus. Proc. Natl. Acad. Sci. USA. 1988;85:1759–1762. doi: 10.1073/pnas.85.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronstein S.M., Skopek T.R., Swenberg J.A. Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 1992;52:2008–2011. [PubMed] [Google Scholar]
- 18.Chen H.J., Wang Y.C., Lin W.P. Analysis of ethylated thymidine adducts in human leukocyte DNA by stable isotope dilution nanoflow liquid chromatography-nanospray ionization tandem mass spectrometry. Anal. Chem. 2012;84:2521–2527. doi: 10.1021/ac203405y. [DOI] [PubMed] [Google Scholar]
- 19.Jasti V.P., Spratt T.E., Basu A.K. Tobacco-specific nitrosamine-derived O2-alkylthymidines are potent mutagenic lesions in SOS-induced Escherichia coli. Chem. Res. Toxicol. 2011;24:1833–1835. doi: 10.1021/tx200435d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai Q., Wang P., Wang Y. Cytotoxic and mutagenic properties of regioisomeric O2-, N3- and O4-ethylthymidines in bacterial cells. Carcinogenesis. 2014 doi: 10.1093/carcin/bgu085. doi:10.1093/carcin/bgu1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y.Z., Swann P.F. Oligodeoxynucleotides containing O2-alkylthymine—synthesis and characterization. Tetrahedron Lett. 1994;35:303–306. [Google Scholar]
- 22.Reist E.J., Goodman L., Benitez A. Synthesis of some 5′-thiopentofuranosylpyrimidines. J. Org. Chem. 1964;29:554–558. [Google Scholar]
- 23.Delaney J.C., Essigmann J.M. Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Methods Enzymol. 2006;408:1–15. doi: 10.1016/S0076-6879(06)08001-3. [DOI] [PubMed] [Google Scholar]
- 24.Neeley W.L., Delaney S., Alekseyev Y.O., Jarosz D.F., Delaney J.C., Walker G.C., Essigmann J.M. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J. Biol. Chem. 2007;282:12741–12748. doi: 10.1074/jbc.M700575200. [DOI] [PubMed] [Google Scholar]
- 25.Delaney J.C., Essigmann J.M. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14051–14056. doi: 10.1073/pnas.0403489101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan B., Cao H., Jiang Y., Hong H., Wang Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen N., Wang J., Wang P., Jiang Y., Wang Y. In vitro replication studies on O2-methylthymidine and O4-methylthymidine. Chem. Res. Toxicol. 2012;25:2523–2531. doi: 10.1021/tx300325q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Napolitano R., Janel-Bintz R., Wagner J., Fuchs R.P. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen X., Sayer J.M., Kroth H., Ponten I., O'Donnell M., Woodgate R., Jerina D.M., Goodman M.F. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7,8-diol 9,10-epoxide A and G adducts. J. Biol. Chem. 2002;277:5265–5274. doi: 10.1074/jbc.M109575200. [DOI] [PubMed] [Google Scholar]
- 30.Seo K.Y., Nagalingam A., Miri S., Yin J., Chandani S., Kolbanovskiy A., Shastry A., Loechler E.L. Mirror image stereoisomers of the major benzo[a]pyrene N2-dG adduct are bypassed by different lesion-bypass DNA polymerases in E. coli. DNA Repair. 2006;5:515–522. doi: 10.1016/j.dnarep.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 31.Jarosz D.F., Godoy V.G., Delaney J.C., Essigmann J.M., Walker G.C. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki N., Ohashi E., Kolbanovskiy A., Geacintov N.E., Grollman A.P., Ohmori H., Shibutani S. Translesion synthesis by human DNA polymerase κ on a DNA template containing a single stereoisomer of dG-(+)- or dG-(-)-anti-N2-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene) Biochemistry. 2002;41:6100–6106. doi: 10.1021/bi020049c. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y., Yuan F., Wu X., Wang M., Rechkoblit O., Taylor J.S., Geacintov N.E., Wang Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000;28:4138–4146. doi: 10.1093/nar/28.21.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minko I.G., Harbut M.B., Kozekov I.D., Kozekova A., Jakobs P.M., Olson S.B., Moses R.E., Harris T.M., Rizzo C.J., Lloyd R.S. Role for DNA polymerase κ in the processing of N2-N2-guanine interstrand cross-links. J. Biol. Chem. 2008;283:17075–17082. doi: 10.1074/jbc.M801238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lone S., Townson S.A., Uljon S.N., Johnson R.E., Brahma A., Nair D.T., Prakash S., Prakash L., Aggarwal A.K. Human DNA polymerase κ encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell. 2007;25:601–614. doi: 10.1016/j.molcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.