

Abstract
We report the design, synthesis, and biological evaluation of imidazopyridine-based peptidomimetics based on the substrate consensus sequence of Akt, an AGC family serine/threonine kinase hyperactivated in over 50% of human tumors. Our ligand-based approach led to the identification of novel substrate mimetic inhibitors of Akt1 featuring an unnatural extended dipeptide surrogate. Compound 11 inhibits Akt isoforms in the submicromolar range and exhibits improved proteolytic stability relative to a parent pentapeptide.
Keywords: Kinase inhibitor, Peptide mimic, Dipeptide surrogate, β-strand mimic
The use of peptidomimetics for the inhibition of kinase-substrate interactions is not widely pursued due to the perceived challenges associated with precise topological mimicry, chemical synthesis, and bioavailability. However, the disruption of these interactions holds several potential advantages over ATP-site targeting approaches, including distinct target selectivity and improved therapeutic profiles through inhibition of specific regulatory mechanisms.1–3
Akt (PKB) is one example of a kinase that has been targeted by several different small molecule approaches.4–8 Akt is an important regulator of cell growth, cell-cycle progression, transcription and metabolism, and is known to phosphorylate over 20 endogenous substrates.5,9–12 Many of these substrates are intimately involved the induction of apoptosis and the arrest of cell proliferation, and are inactivated upon phosphorylation by Akt. Overall, enhanced Akt activity through increased expression, upstream amplification of PI3K, or loss of PTEN, its most important negative regulator, is observed in over 50% of all human solid tumors.13–17 Akt has thus emerged as an attractive target for the development of novel anticancer therapeutics.4,6,7,18–22
Most small molecules block Akt activity by direct inhibition of the ATP-binding site, interfering with cellular localization (via inhibition of the Pleckstrin Homology domain), or through allosteric binding. Recently, mimics of the consensus substrate peptide of Akt have also emerged as lead compounds for further development. While achieving ligand complementarity in the relevant protein-protein interaction (PPI) region is expected to be more topochemically demanding, such inhibitors may also exhibit better selectivity relative to PH and ATP-binding domain antagonists. Early work in this area focused polypeptides exhibiting IC50 values in the low to sub-micromolar range (~10–0.1).23–25 A co-crystal structure of Akt1 bound to a substrate peptide in the presence of an ATP-competitive inhibitor revealed that the peptide adopts a highly extended conformation in the binding cleft.26 Efforts to reduce peptide character while maintaining the bioactive conformation have led to the identification of additional pseudosubstrate Akt1 inhibitors.27–31
Our group recently reported inhibitors of Akt1 based on a consensus sequence incorporating an azabicycloalkane dipeptide surrogate.30 Here, we describe the design and synthesis of a series of imidazopyridine-based peptidomimetics with enhanced potency and proteolytic stability. The undecapeptide Akt substrate GRPRTSSFAEG (Crosstide) was used as a lead structure and the central Thr7-Ser8 dipeptide was identified as a candidate site for conformational constraint (Figure 1).
Figure 1.

Design of peptidomimetic Akt inhibitors
The general synthesis of Akt substrate mimics is depicted in Scheme 1. The imidazo[1,2-a]pyridine (IP)-based dipeptide surrogate32 was prepared by bromination of β-ketoester 1 and subsequent condensation with 2,3-diaminopyridine. Amidation of the IP N-terminus with protected amino acids required stirring in the presence of EDC in DCM for 24–48 hr for optimal yields. The addition of auxiliary base or the use of other common coupling conditions (HBTU/HOBt, HATU, PyBOP, COMU, DEPBT) resulted in significantly lower conversion. The slow rate of amidation also precluded direct coupling to various N-protected arginine derivatives, all of which underwent intramolecular cyclization prior to reacting with the IP amine. In contrast, 2 was efficiently coupled to Cbz-Orn(Boc)-OH, Cbz-Lys(Boc)-OH, and Cbz-Har(Boc)2-OH without any observable lactam formation. Arginine derivatives were prepared via Boc acidolysis and subsequent guanidinylation using Goodman’s reagent to give protected tripeptide mimics 3b and 3d.
Scheme 1.

Synthesis of imidazo[1,2-a]pyridine-based inhibitors.
Incorporation of various C-terminal fragments was achieved by removal of the allyl ester protecting group and condensation with amino acid and dipeptide derivatives. Notably, the dipeptide amides used in the condensation reaction were efficiently prepared by simple aminolysis of the corresponding Bocprotected dipeptide methyl esters (see Supplementary Data). We found this procedure to be a convenient and racemization-free method to produce a variety of protected peptide amides. After coupling to the IP-containing fragment, Boc group removal with TFA/DCM was followed by column chromatography to afford inhibitors 4–31.
All compounds were assayed in vitro for their ability to inhibit the phosphorylation of Crosstide by Akt1 in the presence of 10 µM 33P-labeled ATP (dose-response experiments were repeated 3 times, and IC50 values and 95% confidence intervals were calculated based on a variable slope four parameter model). As shown in Table 1, truncation of the lead substrate down to tetrapeptide mimics 4–7 afforded compounds with no appreciable Akt1 inhibitory activity at 20 µM. Pentapeptide mimic 8, which incorporates the native Ser9-Phe10 motif was also inactive in vitro. Replacement of Ser9 (native phosphorylation site) with the more hydrophobic Leu (9) or Phe (10) residues led to a dramatic increase in potency against Akt1. Optimal potency against Akt1 (IC50 = 0.64 µM) was achieved with derivative 11, which incorporates a Val-Phe-NHBn C-terminal dipeptide subunit.
Table 1.
Structure-activity relationships for compounds 4–31.
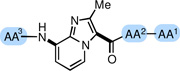 | |||||
|---|---|---|---|---|---|
| Compound | AA3 | AA2 | AA1 | Akt1 IC50 (µM) |
95% CI (µM) |
| 4 | Cbz-Arg | Val-NHBn | - | >20 | - |
| 5 | Cbz-Arg | Val-OBn | - | >20 | - |
| 6 | Cbz-Arg | Hyp(Bn)-OBn | - | >20 | - |
| 7 | Cbz-Arg | pip(N-Bz) | - | >20 | - |
| 8 | Cbz-Arg | Ser | Phe-NHBn | >20 | - |
| 9 | Cbz-Arg | Leu | Phe-NHBn | 1.59 | 1.43 –1.78 |
| 10 | Cbz-Arg | Phe | Phe-NHBn | 1.13 | 1.03 –1.24 |
| 11 | Cbz-Arg | Val | Phe-NHBn | 0.64 | 0.55 –0.74 |
| 12 | Moc-Arg | Val | Phe-NHBn | >20 | |
| 13 | Cbz-Ala | Val | Phe-NHBn | >20 | - |
| 14 | Cbz-Arg | Ala | Phe-NHBn | >20 | - |
| 15 | Cbz-Arg | Val | Ala-NHBn | >20 | - |
| 16 | Cbz-Arg | Val | Phe-NHMe | >20 | - |
| 17 | Cbz-Arg | (D)Val | Phe-NHBn | >20 | - |
| 18 | Cbz-Arg | Val | (D)Phe-NHBn | >20 | - |
| 19 | Cbz-Orn | Val | Phe-NHBn | 3.06 | 2.17 –4.33 |
| 20 | Cbz-Lys | Val | Phe-NHBn | 1.92 | 1.72 –2.15 |
| 21 | Cbz-Har | Val | Phe-NHBn | 1.06 | 0.95 –1.20 |
| 22 | Cbz-Arg | Val | Phg-NHBn | 1.53 | 1.29 –1.82 |
| 23 | Cbz-Arg | Val | (4-F)Phe-NHBn | 2.00 | 1.65 –2.43 |
| 24 | Cbz-Arg | Val | homoPhe-NHBn | 1.59 | 1.38 –1.83 |
| 25 | Cbz-Arg | Val | Phe-OBn | 5.27 | 4.78 –5.80 |
| 26 | Cbz-Arg | Val | Phe-OMe | >20 | - |
| 27 | Cbz-Arg | Val | Phe-NH(4-F)Bn | 0.83 | 0.53 –1.30 |
| 28 | Cbz-Arg | Val | Phe-NH(4-Me)Bn | 1.02 | 0.83 –1.25 |
| 29 | Cbz-Arg | Val | Phe-NHi-Bu | 0.67 | 0.57 –0.79 |
| 30 | Cbz-Arg | Val | Tic-NHBn | >20 | - |
| 31 | Cbz-Arg | Val | (N-Me)Phe-NHBn | >20 | - |
In order to obtain preliminary SAR for peptidomimetic 11, we prepared analogs 12–16, in which pharmacophores were iteratively deleted. In general, these deletions were not well tolerated and resulted in a significant (>30-fold) loss of potency. The activity of compound 11 was also found to be remarkably sensitive to the incorporation of d-Val or d-Phe residues as in 17 and 18.
We next prepared a series of side chain analogs to determine optimal pharmacophores within 11. Derivatives 19–21 harbor an N-terminal Orn, Lys, or Har residue, preserving the important basic pharmacophore at position 6 of the substrate consensus sequence. Orn and Lys side chain analogs 19 and 20 showed slightly weaker potency against Akt1, while the guanidine (Har)-containing analog 21 exhibited an IC50 of 1.06 µM. Phe10- mutated variants 22–24, also inhibited Akt1 with IC50 values in the 1–2 µM range. Benzyl ester 25 displayed significantly weaker inhibition of Akt1 relative to 11. As in the case of methyl amide 16, methyl ester 26 showed no significant activity at 20 µM concentration. However, other more hydrophobic substitutions at C-terminus of 11 were well tolerated, with i-Bu derivative 29 displaying almost equivalent potency in vitro. Attempts to constrain the C-terminal backbone as in 30 and 31 had a deleterious effect on activity.
In order to confirm the ability of 11 to block the phosphorylation of the Akt substrate peptide, we employed a non-radioactivity based secondary assay using Akt from cell lysates. HEK293 cells were thus transfected with Myc-tagged Akt1 and the protein was immunoprecipitated using anti-Myc antibody. Incubation of the immobilized enzyme with a substrate GSK3β fusion protein in the presence or absence of inhibitors was followed by detection of p-GSK3β by immunoblots. Asshown in Figure 2, the control Akt inhibitor MK2206 completely blocks phosphorylation of GSK3β at 5 µM. Our substrate mimetic inhibitor, 11, significantly inhibits p-GSK3β at concentrations above 1 µM relative to DMSO control. The approximate IC50 value for 11 observed by immunoblots (between 0.3 and 3 µM) was in good agreement with that obtained in the 33P-ATP kinase assay above.
Figure 2.

Phosphorylation of GSK3β fusion protein by Myc-tagged Akt1 immunoprecipitated from cell lysates.
Given that 11 was designed to mimic the consensus substrate sequence of Akt, we anticipated that it would show significant activity against each of the Akt isoforms. When evaluated against Akt2 and Akt3 in vitro, 11 exhibited IC50 values of 0.76 and 0.13 µM, respectively (Figure 5). We also tested compound 11 against seven AGC kinase family members in the hotspot assay to provide preliminary selectivity information. At 10 µM concentration 11 exhibited less than 50% inhibition of PDK1, PKCα, LATS1, ROCK1, and RSK1. The activity of PKA and MSK1 were inhibited 54 and 65%, respectively by 11 (see Supporting Information). These data points were obtained using a concentration of 11 15-fold greater that its Akt1 IC50 value, suggesting that the lead inhibitor is moderately selective against closely related kinase target
Figure 5.

Proteolytic degradation of 11 and a parent pentapeptide.
To gain insight into the potential interaction between Akt1 and 11 we performed computational docking experiments with the crystal structure of Akt1 bound to a GSK3β substrate peptide in the presence AMP-PNP (pdb 1O6K). An RMSD of 0.51 Å was observed upon selfdocking of the substrate peptide using the GLIDE XP protocol. Docking of compound 11 revealed good overlap with the backbone and side chain geometries of Arg6, Ser9, and Phe10 in the native ligand (Figure 3). Important hydrogen bonding interactions with Gly312, Glu279, and Glu236 in Akt1 are also maintained in the docked structure of 11.
Figure 3.

In vitro inhibition of Akt isoforms by 11.
Although peptides represent useful starting points for the development of kinase inhibitors, their susceptibility to proteolytic degradation remains a major drawback. To determine the impact of the IP dipeptide surrogate on compound stability, we synthesized the fully peptidic analog of 11 (Cbz-Arg-Thr-Ser- Val-Phe-NHBn) and subjected both compounds to time-course digestion experiments with chymotrypsin and pronase. The imidazopyridine-based peptidomimetic 11 was considerably more stable toward digestion than the control peptide over 48 h at 37 °C (Figure 4). Crosstide was likewise degraded within 0.5 h upon incubation with either protease (data not shown).
Figure 4.

(A) Binding of compound 11 to Akt1 (pdb 1O6K) as determined by GLIDE XP docking. (B) Overlay of docked conformation of 11 (green) with the bound X-ray conformation of a substrate peptide (orange). Key interactions of 11 with Akt1 are shown.
In summary, we have described a ligand-based approach toward the discovery of novel substrate mimetic inhibitors of Akt. These pentapeptide mimics are based on a bicyclic aromatic core structure that was recently developed as a versatile dipeptide surrogate.32 We have previously shown that imidazo[1,2-a]pyridine scaffolds can promote an extended conformation when incorporated into peptidic host structures. Substitution of the Thr7-Ser8 dipeptide in Crosstide with this motif led to the identification of lead inhibitor 11, which inhibits Akt-mediated GSK3β phosphorylation with an IC50 of 0.64 µM. Compound 11 also displays enhanced stability toward proteolytic degradation relative to its parent peptides. Efforts to further optimize 11 in the pursuit of substrate mimetic Akt inhibitors suitable for in vivo applications are currently underway.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R21CA167215) and by the Anna Valentine Cancer Fund (FIG award). We gratefully acknowledge the Moffitt Chemical Biology Core, Molecular Modeling Unit (NIH P30CA76292) for carrying computational docking of compound 11.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Supporting information associated with this article, including experimental procedures and copies of 1H NMR spectra for compounds 4–31, can be found in the online version at http://dx.doi.org/xxxxx.
References and notes
- 1.Arkin MR, Whitty A. Curr. Opin. Chem. Biol. 2009;13:284. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- 2.Bogoyevitch MA, Fairlie DP. Drug Discov. Today. 2007;12:622. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Kirkland LO, McInnes C. Biochem. Pharmacol. 2009;77:1561. doi: 10.1016/j.bcp.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 4.Barnett SF, Bilodeau MT, Lindsley CW. Curr. Top. Med. Chem. 2005;5:109. doi: 10.2174/1568026053507714. [DOI] [PubMed] [Google Scholar]
- 5.Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. Oncogene. 2005;24:7482. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- 6.Collins I. Anti-Cancer Agent Med. Chem. 2009;9:32. doi: 10.2174/187152009787047734. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Echeverria C, Sellers WR. Oncogene. 2008;27:5511. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 8.Kumar CC, Madison V. Oncogene. 2005;24:7493. doi: 10.1038/sj.onc.1209087. [DOI] [PubMed] [Google Scholar]
- 9.Staal SP. Proc. Natl. Acad. Sci. USA. 1987;84:5034. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Staal SP, Hartley JW, Rowe WP. Proc. Natl. Acad. Sci. USA. 1977;74:3065. doi: 10.1073/pnas.74.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. Science. 1991;254:274. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 12.Du K, Tsichlis PN. Oncogene. 2005;24:7401. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
- 13.Altomare DA, Testa JR. Oncogene. 2005;24:7455. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 14.Hsu JH, Shi Y, Hu L, Fisher M, Franke TF, Lichtenstein A. Oncogene. 2002;21:1391. doi: 10.1038/sj.onc.1205194. [DOI] [PubMed] [Google Scholar]
- 15.Zinda MJ, Johnson MA, Paul JD, Horn C, Konicek BW, Lu ZH, Sandusky G, Thomas JE, Neubauer BL, Lai MT, Graff JR. Clin. Cancer Res. 2001;7:2475. [PubMed] [Google Scholar]
- 16.Page C, Lin HJ, Jin Y, Castle VP, Nunez G, Huang M, Lin J. Anticancer Res. 2000;20:407. [PubMed] [Google Scholar]
- 17.Crowell JA, Steele VE, Fay JR. Mol. Cancer. Ther. 2007;6:2139. doi: 10.1158/1535-7163.MCT-07-0120. [DOI] [PubMed] [Google Scholar]
- 18.Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Benedetti Panici P, Mancuso S, Neri G, Testa JR. Int. J. Cancer. 1995;64:280. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 19.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. Proc. Natl. Acad. Sci. USA. 1992;89:9267. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heerding DA, Safonov IG, Verma SK. Annu. Rep. Med. Chem. 2007;42(42):365. [Google Scholar]
- 21.Li Q. Expert Opin. Ther. Pat. 2007;17:1077. [Google Scholar]
- 22.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Bioorg. Med. Chem. Lett. 2005;15:761. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Litman P, Ohne O, Ben-Yaakov S, Shemesh-Darvish L, Yechezkel T, Salitra Y, Rubnov S, Cohen I, Senderowitz H, Kidron D, Livnah O, Levitzki A, Livnah N. Biochemistry. 2007;46:4716. doi: 10.1021/bi061928s. [DOI] [PubMed] [Google Scholar]
- 24.Luo Y, Smith RA, Guan R, Liu X, Klinghofer V, Shen J, Hutchins C, Richardson P, Holzman T, Rosenberg SH, Giranda VL. Biochemistry. 2004;43:1254. doi: 10.1021/bi034515p. [DOI] [PubMed] [Google Scholar]
- 25.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. J. Biol. Chem. 2000;275:36108. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, Barford D. Mol. Cell. 2002;9:1227. doi: 10.1016/s1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- 27.Freeman NS, Tal-Gan Y, Klein S, Levitzki A, Gilon C. J. Org. Chem. 2011;76:3078. doi: 10.1021/jo102422x. [DOI] [PubMed] [Google Scholar]
- 28.Kayser KJ, Glenn MP, Sebti SM, Cheng JQ, Hamilton AD. Bioorg. Med. Chem. Lett. 2007;17:2068. doi: 10.1016/j.bmcl.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Kayser-Bricker KJ, Glenn MP, Lee SH, Sebti SM, Cheng JQ, Hamilton AD. Bioorg. Med. Chem. 2009;17:1764. doi: 10.1016/j.bmc.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ranatunga S, Del Valle JR. Bioorg. Med. Chem. Lett. 2011;21:7166. doi: 10.1016/j.bmcl.2011.09.079. [DOI] [PubMed] [Google Scholar]
- 31.Tal-Gan Y, Hurevich M, Klein S, Ben-Shimon A, Rosenthal D, Hazan C, Shalev DE, Niv MY, Levitzki A, Gilon C. J. Med. Chem. 2011;54:5154. doi: 10.1021/jm2003969. [DOI] [PubMed] [Google Scholar]
- 32.Kang CW, Sun Y, Del Valle JR. Org. Lett. 2012;14:6162. doi: 10.1021/ol302850n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.