

Synopsis
Malignant neoplasms are consistently among the top four leading causes of death in all age groups in the United States, despite a concerted effort toward developing novel therapeutic approaches[1]. Our understanding of and therapeutic strategy for treating each of these neoplastic diseases has been elevated through decades of research on the genetics, signaling pathways, and cellular biology that govern tumor cell initiation, progression and maintenance. Much of this work has concentrated on post-translational modifications and abnormalities at the DNA level, including point mutations, amplifications/deletions, and chromosomal translocations, and how these aberrant events affect the expression and function of protein-coding genes. Only recently has a novel class of conserved gene regulatory molecules been identified as major contributors to malignant neoplastic disease. This review focuses on how these small non-coding RNA molecules, termed microRNAs (miRNAs), can function as oncogenes or tumor suppressors, and how the misexpression of miRNAs and dysregulation of factors that regulate miRNAs contributes to the tumorigenic process. Specific focus is given to more recently discovered regulatory mechanisms that go awry in cancer, and how these changes alter miRNA expression, processing, and function.
Introduction
MicroRNAs (miRNAs) are members of a larger class of non-coding RNA that regulate a wide array of biological processes[2], and concordantly have been found to be heavily dysregulated in diseases, including cancer[3, 4]. Importantly, aberrant miRNA expression levels are linked to both facilitating and abrogating the tumorigenic process. They do so through their ability to control the expression of thousands of protein-coding and non-coding genes, and through their ability to regulate transcription of genes via promoter-associated RNAs (paRNAs)[5]. Currently there are 1872 annotated human miRNA precursor genes that are processed into ~2,578 mature miRNA sequences (http://www.mirbase.org), many of them with unknown functions. The abundance of these small yet powerful RNAs and the diversity of their function make miRNAs an exciting class of under-studied regulatory molecules. Further, as outlined below, these molecules have the potential to be used as biomarkers and therapeutic agents.
MiRNAs are transcribed from individual genes containing their own promoters or intragenically from spliced portions of protein-coding genes (some, called miRtrons, actually use the splicing machinery in their processing)[6]. Like protein-coding genes, miRNAs with their own promoters are almost exclusively transcribed byRNA polymerase II and their primary transcripts (pri-miRNAs) are5′capped and 3′polyadenylated[7]. However, unlike their protein-coding counterparts, miRNA function is driven by sequence complementary (Figure 1). The functional ~20–22nt sequence, referred to as the mature miRNA, is sequentially processed from the pri-miRNA by the RNaseIII family enzymes, DROSHA and DICER, the first step converting the pri-miRNA into the shorter pre-miRNA ~85nt stem-loop. The pre-miRNA product of DROSHA cleavage is exported to the cytoplasm, where DICER processes it to the ~20–22nt miRNA/miRNA* duplex (* indicates the passenger strand, while the other complementary stand is referred to as the mature or guide strand). The miRNA guide strand associates with a protein complex termed the RNA induced silencing complex (RISC) and guides RISC to target messenger RNAs (mRNAs) via base-pairing between the miRNA and the cognate target mRNA sequence[8]. In many cases base pairing occurs between nucleotides 2–8, the seed region of the miRNA; however, a recent unbiased technique to identify miRNA/mRNA duplexes identified four additional non-canonical binding clusters[9]. Collectively, greater than 35% of the binding events were independent of nucleotides 2–8, highlighting complexity in miRNA target identification. Regardless, once the miRNA is bound to its target mRNA translation is inhibited through one of two methods: mRNA degradation/destabilization (thought to require full complementation between the miRNA/mRNA duplex) or translational repression (mediated by imperfect complementarity to the target mRNA)[10]. Since miRNA binding to mRNAsis often achieved with imperfect complementarity, each miRNA canbind to and regulate multiple protein-coding mRNAs and non-coding RNAs. Therefore, aberrant expression of a single miRNA can deleteriously affect the translation of multiple genes within a cell, leading to profound phenotypic responses.
Figure 1. Biogenesis of miRNAs.
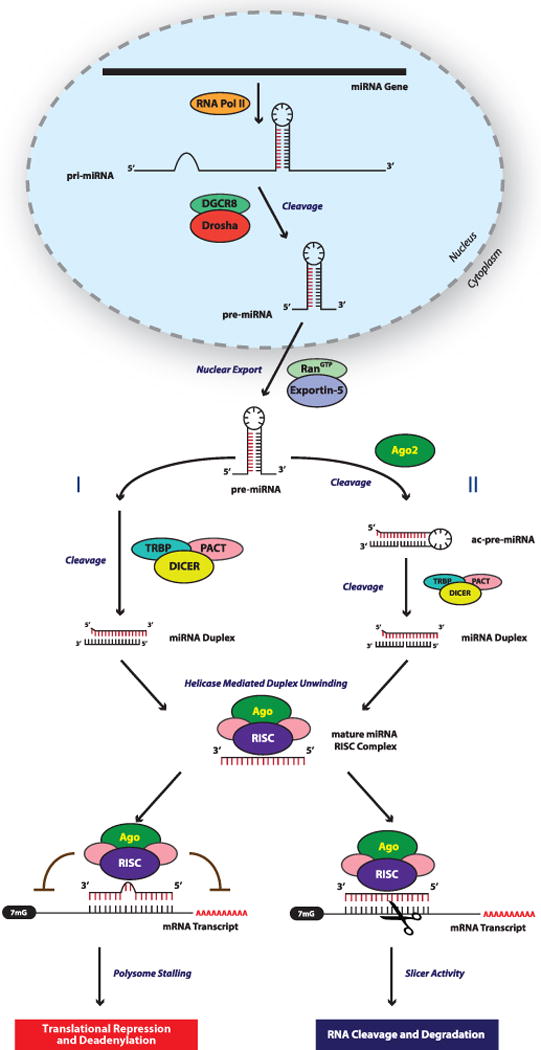
In the canonical miRNA biogenesis pathway miRNAs are transcribed by RNA Polymerase II into long miRNA primary transcripts termed pri-miRNAs. These pri-miRNAs serve as substrates for Drosha and DGCR8, the former cleaves the flanking single stranded RNA to generate an ~85ntprecursor miRNA (pre-miRNA). After nuclear export to the cytoplasm via RanGTP/Exportin-5, the majority of pre-miRNAs are directly processed by Dicer and are subsequently cleaved to generate an ~22nt miRNA duplex (I). However, pre-miRNAs can be cleaved by Ago2 to generate an ac-pre-miRNA, which is then recognized by Dicer (II). Once the miRNA-duplex is unwound and the passenger strand degraded, the guide strand (highlighted in red), is incorporated into RISC, which contains Ago and other proteins. A miRNA loaded RISC mediates gene silencing via mRNA target cleavage and degradation, or translational repression and deadenylation depending on the complementarity between the miRNA and the targeted mRNA transcript. It should be appreciated that there are additional non-canonical ways in which miRNAs can be generated (i.e. miRtrons), and that miRNAs can function is non-classical ways.
While unbeknownst to scientists for centuries, we now appreciate that miRNA expression can classify disease states, predict sensitivity to therapeutics, and function themselves as therapeutic entities. This is best exemplified in the cancer field, where misexpression of miRNAs alters numerous aspects of cellular function such as differentiation, apoptosis, and survival signaling, linking miRNAs to disease pathology. In this review, we focus on the role of miRNAs as oncogenes or tumor suppressors, how these regulatory entities can themselves be regulated, and how the dysregulation of miRNAs can promote pathological and morphological phenotypes such as therapeutic relapse. While impossible to cover all of the advancements in the miRNA field, we will attempt to highlight some recent findings that broaden our conceptual notions with regard to miRNA function and their dysregulation in cancer.
miRNA Biogenesis in Cancer
Over the past decade, numerous studies have shown that miRNAs are misexpressed in tumors when compared to normal tissue[11, 12]. Additionally, some studies showed a general down-regulation of miRNAs in tumors. In lieu of highlighting individual miRNAs and their particular roles in driving or antagonizing tumorigenic processes, we focus here on the miRNA biogenesis machinery itself and how various components of the machinery, posttranscriptional miRNA changes, and alterations in RNA sequences are dysregulated in cancer.
Drosha and Dicer
Roles for both DROSHA and DICER in the tumorigenic process have been clearly defined through multiple reports identifying their misexpression in human tumor tissue and through detailed analysis of their function in genetically engineered mouse models. A seminal study performed by Merritt et al., determined that high DICER and DROSHA mRNA levels are associated with increased median survival (>11 years, vs. 2.66 years) in ovarian cancer[13]. Furthermore, Karube et al. found that reduced DICER mRNA levels correlated with shortened post-operative survival in lung cancer patients[14]. These findings are complemented by animal studies where single copy loss of Dicer1 in a KrasLSL-G12Dmutantlung cancer mouse model was sufficient to reduce overall survival, while homozygous deletion of Dicer1 did not result in an exaggeration of this effect[15]. In fact, tumors that did develop in animals that carried the homozygous floxed Dicer allele retained a functional copy of Dicer; homozygous null tumors were not identified. The study also found that Dicer functions as a haploinsufficient tumor suppressor given that human tumors harbored lower levels of DICER but were never found to be completely devoid of the protein. In both human samples with low DICER expression and in the Dicer1fl/+ mice, a global reduction of miRNA levels was observed as expected. This pro-tumorigenic effect mediated by single copy loss of DICER is perplexing, given that homozygous mutations in tumor suppressor genes are usually advantageous to tumor cells (Figure 2A). Although others have observed this phenomenon, for instance, TAp63 directly transactivates the Dicer promoter, and in TAp63−/− mice metastatic tumors develop that harbor aberrantly low but not absent Dicer levels[16]. This is an important finding given loss of p63 has been reported to be associated with tumor metastasis[17]. Therefore, it is likely that some amount of miRNA activity is required for normal cellular growth and development and that complete loss is lethal. Indeed, Dicer1-null mice die at E7.5[18], while Dicer1-hypomorphic mice are viable albeit with morphologic abnormalities. Furthermore, injection of Dicer1-deficient ES cells into nude mice failed to generate tumors[19]. These studies suggestan essential developmental role for DICER, which may or may not be completely miRNA-dependent[20]. Therefore, while loss of DROSHA and DICER may not be an initiating event in tumorigenesis, loss of miRNA expression by reduced biogenesis activity may allow for a more permissive tumorigenic environment (Figure 2B).
Figure 2. Consequencesof Aberrant miRNA Biogenesis in Cancer.
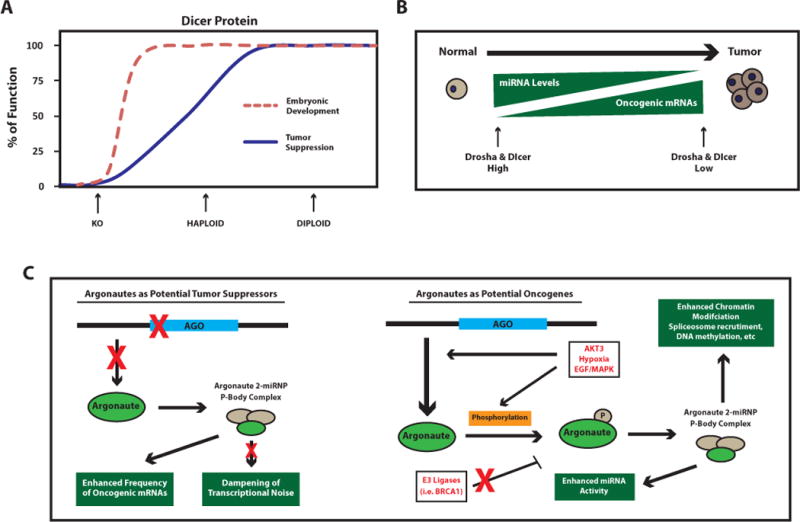
(A) Depicted is the Knudson’s model of haploinsufficiency for a tumor suppressor gene (adapted from Figure 1 [147]). The controlled expression of Dicer has important functions in both developmental and tumorigenic processes. Red dashed line indicates a haploid dose of Dicer expression is sufficientfor embryonic development, while haploidy is insufficient for tumor suppression. Given that tumors retain some Dicer expression and Dicer loss is embryonic lethal, complete loss (KO gene dosage) of Dicer is presumably detrimental to both normal and tumor cell viability. As noted in the text, tumor suppressor genes such as Rb are haplosufficient for tumor suppression (see red-dashed line), and usually require KO gene dosage to impair tumor suppressive activity. (B)In general, reduced miRNA levels are associated with tumor development, in part through decreased miRNA biogenesis proteins such as Drosha and Dicer. This suggests a majority of miRNAs harbor tumor suppressive functions by controlling the expression of genes with oncogenic activity when aberrantly expressed. Whether loss of Drosha/Dicer is an initiating event that allows for a permissive tumorigenic environment, or if established tumors actively suppress these proteins to maintain a tumorigenic state is still unclear. (C) Ago2 can function either as a tumor suppressor (left panel), or as an oncogene (right panel) depending on cell context. As a tumor suppressor, loss of Ago2 would result in reduced formation of Ago2-miRNP complexes and P-body formation, ultimately reducing miRNA activity. As a result, normal gene regulation and dampening of transcriptional noise would decrease and potentially lead to enhanced abundance of aberrantly expressed transcripts that harbor oncogenic activity. In some cancers, AGO2 expression is elevated suggesting an oncogenic role. While the mechanism is unclear, it is known that EGF/MAPK signaling pathways promote the phosphorylation of Ago2 and its localization to P-bodies. Presumably the resulting enhancement in miRNA activity reduces certain tumor suppressive transcripts highly abundant in those tumor cells, or through other Ago2-mediated mechanisms such as chromatin modification. Since proteasome inhibitors reduce AGO2 levels[26], other mechanisms for gain of Ago2 expression in cancer could be through loss of certain E3 ligases such as BRCA1, which is know to be mutated or lost in certain breast cancers.
Argonaute
Argonaute proteins, key components of RISC, induce endonuclease cleavage of the mRNA and miRNA passenger strand through intrinsic catalytic activity[8]. Similar to DICER and DROSHA, dysregulation of argonaute proteinsalso occurs in cancer. For example, three of the four mammalian argonaute genes, AGO3 (EIF2C3), AGO1 (EIF2C1), and AGO4 (EIF2C4) are frequently deleted in Wilms tumor of the kidney and are associated with neuro-ectodermal tumors[3, 21, 22]. In contrast to the generalized downregulation of Drosha and Dicer in tumors, the expression of AGO proteins are regulated in a cell-context dependent manner (Figure 2C). For instance, a recent study highlighted that in melanoma cell lines AGO2 (EIF2C2) expression was lower in both primary and metastatic melanoma samples as compared to normal epidermal melanocytes[23]. However, observations from human breast and colon tumor samples indicated that gain of AGO2 expression associates with a more aggressive phenotype and distant metastasis. Lan Li et al. reported the gain of AGO2 protein expression in colon cancer samples and in advanced tumors with distant metastasis compared to adjacent normal tissue[24]. In addition, Blenkinron et al. found that both ERα-negative breast tumors and the basal-like subset of breast tumors, which are more aggressive and metastatic compared to other breast cancer subtypes harbored significantly elevatedAGO2 mRNA levels[25]. Indeed, the ectopic expression of AGO2 in ERα-positive MCF-7 breast cancer cells resulted in loss of epithelial markers such as E-cadherin, increased growth rates, and an enhanced ability to migrate in a wound healing assay, suggesting a role for AGO2 in disease progression[26]. Furthermore, AGO2 function is directly modulated by EGFR signaling in ERα-negative breast cancer under states of hypoxic stress. Here, EGFR can directly bind and phosphorylate residue Y393 of AGO2[27].p-Y393-AGO2 enhances cell survival and invasiveness of breast cancer cells, and correlates with poorer overall survival in breast cancer patients. However, it is unclear if this a universal phosphorylation event in all cancer types, given that other groups have found AGO2 to be phosphorylated on other residues such asY529[28], and S387 via AKT3[29]. The question also still remains as to whether the alteration in AGO2 levels in cancer is directly linked to gain or loss of miRNA activity. Unlike DICER, AGO2 has been linked with other biochemical process such as chromatin modification and facilitation of spliceosome recruitment, DNA methylation, modulation of RNA polymerase II elongation rate, and promoter-directed transcriptional gene silencing[30, 31].
Lin-28
LIN28/LIN28B proteins are RNA binding proteins that harbor a cold-shock domain (CSD) and two CysCysHisCys (CCHC) zinc finger domains, providing the RNA-binding function[32]. While normally only expressed at high levels during development, these small proteins are unique in their ability to convert human somatic cells to pluripotency when co-expressed with OCT4, NANOG and SOX2[33]. Indeed, LIN28 is highly expressed in embryonic stem (ES) cells as well as cancer stem cells in numerous cancer types, including ovarian, breast, and NSCLC[34–36]. For this reason, LIN28 is viewed as an oncogene, which can be targeted by the tumor suppressive miRNA let-7. Interestingly, it was discovered that a double negative feedback loop existed whereby let-7 repressed LIN28 through canonical targeting of the LIN28 3′UTR, and LIN28 itself could bind to a conserved terminal loop of pri- and pre-let-7, preventing the processing of mature let-7 miRNAs via DROSHA and DICER, respectively (Figure 3A). A separate LIN28 mechanism involves the terminal uridylation (addition of uridine nucleotides) of the pre-let-7 through recruitment of the TUTase/TUT4 complex, which blocks DICER processing, and leads to pre-let-7 degradation[36–38].
Figure 3. Mechanisms Disrupting or Promoting miRNA Biogenesis in Cancer.

(A) LIN28 mediated regulation of let-7. Left panel depicts LIN28 binding to the stem-loop region of either pri-let-7 or pre-let-7, blocking the miRNA biogenesis machinery from recognizing their respective substrates, and ultimately reducing mature let-7 levels. Right panel depicts a separate but not mutually exclusive mechanism, whereby LIN28 recruits TUT4 to the pre-let-7 substrate resulting in polyuridylation and degradation. It is unclear if other miRNAs are controlled by LIN28. (B) Depiction of how dead-box RNA helicases can control miRNA biogenesis. Specifically, p72 and p68 aid in Drosha-mediated processing of pri-miRNA transcripts, and when these biogenesis factors recruit other proteins such as ERα or SMADs, processing activity can be inhibited or promoted, respectively. Other proteins such as KSRP and NF45/NF90 represent other classes of biogenesis factors, but function in a similar manner to alter biogenesis activity.
Adding to this complexity, there are signaling-mediated regulatory circuits that control let-7 levels in a LIN28-dependent fashion. Inflammatory signaling molecules such as NF-κB and IL6 are pro-tumorigenic, and can promote cell growth, motility, and malignant transformation via STAT3 activation. This signaling axis can also reduce the tumor suppressive let-7 by activating LIN28 transcription[39]. Furthermore, let-7 directly targets IL6 and LIN28, thereby antagonizing let-7 transcriptional repression mediated by inflammatory signaling, as well as subsequent STAT3 activation. A second LIN28-dependent mechanism involves the oncogenic c-Myc transcription factor. Initially c-Myc was reported to transcriptionally negatively regulate let-7 expression directly[40] and by modulating IMP1 expression[41]. However c-Myc can also transactivate LIN28B, a LIN28 homolog, resulting in repression of let-7[42]. Given that LIN28 may bind to RNA in a sequence not structure-dependent manner, it remains unclear as to whether other miRNAs are controlled by LIN28.
More direct evidence supporting the oncogenic role of LIN28 comes from studies where forced expression of LIN28 in NIH/3T3 cells resulted in colony formation in soft agar and growth of small tumors in nude mice[43]. Conversely, the loss of LIN28B in K562 cells reduced tumor growth and promoted differentiation. However, if LIN28 functions as a bona fide oncogene, then Lin28a transgenic mice ought to harbor a greater propensity for developing tumors as compared to littermate controls. The initial phenotype that emerged from these mice was instead a bit more complex. Lin28a transgenic mice have larger body sizes, greater organ weights and bone mineral density, and also show a delay in the onset of puberty[44]. Some of these phenotypes support the hypothesis that LIN28 promotes expansion of transit-amplifying or tissue stem cells. Additionally, Lin28a transgenic mice have increased glucose utilization, which may provide an explanation for the heightened energy demands these mice would require for this overgrowth phenotype. Yet these mice did not appear to develop outright tumors, even though stem cell expansion and aerobic glycolysis are processes crucial for tumorigenesis.
Despite these initial findings, it appears that LIN28 may function as an oncogene ina cell-context dependent manner. Mice harboring an inducible form of Lin28a (iLin28aTg) possess enhanced tissue regenerative capacity; specifically, epidermal would healing and hair regrowth, as well as faster digit repair rates[45]. Importantly, in these iLin28 mice, the bioenergetic state is altered during tissue repair such that these cells undergo greater mitochondrial-mediated oxidative phosphorylation. This is contrary to the suggestion that LIN28 promotes aerobic glycolysis, a tumorigenic process, and supports the notion that LIN28 is important for normal physiologic bioenergetics processes. However, when Lox-TetOn- LIN28B mice were crossed with Wt1-Cre mice to express LIN28B specifically in the intermediate mesoderm of the kidney, robust development of renal tumor formation similar in morphology to human Wilms tumors was observed[46]. Interestingly, the use of other kidney lineage specific Cre models did not recapitulate this phenotype, suggesting aberrant expression of LIN28Bin an early renal progenitor cell may result in Wilms tumor formation. Given these recent findings, it would be interesting to determine if progenitor cells in other tissues harboring aberrant LIN28 expression can also promote tumor formation, and whether this process requires the expected inactivation of let-7.
Regulation of Other miRNA Biogenesis Pathway Factors in Cancer
Like most proteins, DROSHA and DICER work collectively with other factors to achieve a cellular response; therefore, dysregulation of these additional factors can also alter DROSHA and DICER activity and contribute to neoplastic disease. For example, studies have highlighted the essential role of p68 and p72 (DEAD-box RNA helicases) in regulating miRNA processing during the pri-miRNA to pre-miRNA processing stage via DROSHA (Figure 3B). In mice, RNA-chromatin immunoprecipitation assays identified that p68 and p72 are within the Drosha complex, and that loss of p68 and p72 results in embryonic lethality, similar to the phenotype observed in the Dicer knockout mice[47]. More specifically, estrogen-receptor α (ERα) was shown to associate with p72 when 17β-estradiol was present. This association prevented DROSHA from interacting with p72 and processing the pri-miRNA, essentially through competitive inhibition[48, 49]. This is a crucial finding given that in breast cancer, estrogen status is one of the most informative clinical diagnostic tools, and dictates treatment regimens for breast cancer patients. Furthermore, this data suggests that in ERα+ breast tumors, there may be a global reduction of a subset of miRNAs that may be present in other breast tumor subtypes.
A similar, but activating function, is observed when R-SMAD associates with p68 and Drosha to enhance miRNA processing (Figure 3B)[50, 51]. Since the activity of DROSHA is contained in the nuclear compartment, shuttling of R-SMAD into the nucleus can enhance DROSHA processing activity of some pri-miRNAs in a sequence-specific manner. Indeed following TGFβ signaling, a fundamental pathway heavily dysregulated in cancer, R-SMAD enters the nucleus and cooperates with DROSHA to enhance the processing of the oncogenic miRNA, miR-21[52]. The result, at least in vascular smooth muscle cells, is that R-SMAD signaling stimulates the biogenesis of miR-21 subsequently repressing programmed cell-death-4 (PDCD4). This process occurs in other systems as well and involves binding of SMAD to a conserved RNA sequence in other miRNAs to induce their processing such as miR-105, miR-199a, and miR-421[53].
KSRP is a KH-type splicing regulator protein that has been implicated in regulating both DROSHA and DICER processing of miRNAs[54]. Specifically, KSRP can associate with both DROSHA and DICER complexes, and knockdown of KSRP in HeLa cells led to a reduction of mature miRNAs including let-7. While the mechanism of action is still unclear, data suggests KSRP aides in the enzymatic cropping activity of DROSHA and DICER on the respective pri/pre-miRNA substrates. Conversely, nuclear factors NF45 and NF90 can reduce pre-let-7 levels by directly binding the pre-miRNA stem loop, in turn preventing DROSHA binding[55].
RNA binding proteins (RBPs) can complex with biogenesis proteins directly to promote or inhibit miRNA activity within a cell. Specifically AUF1 is an RBP that can recognize DICER mRNA, in turn decreasing the production of the DICER protein[56, 57]. As expected, this results in a global loss of mature miRNAs, and importantly, in tumor samples DICER levels were found to be low, and AUF1 levels high. Another RBP, HuR can alter AGO2 activity, by facilitating targeting of the miRNA-loaded RISC complex to its respective mRNA targets[58, 59]. It is still unclear as to whether this is through direct interaction between HuR and AGO2, or whether HuR stabilizes transcripts and due to higher transcript abundance indirectly results in a greater targeting frequency by RISC. Regardless, it is interesting that at almost every level of miRNA biogenesis, there are regulatory processes present to keep miRNA abundance and activity in check, and when these proteins are dysregulated in cancer it results in aberrant miRNA expression patterns.
Finally, and perhaps not surprising, miRNAs themselves are negative regulators of DICER. The miRNA family, miR-103/107 can attenuate miRNA biogenesis through reducing endogenous levels of DICER[60]. High levels of miR-103/107 have been observed in breast cancer specimen and are associated with metastatic disease and poor clinical outcome contributing to an epithelial-to-mesenchymal phenotype.
Regulating the Regulators – How Many Ways?
Over the past decade it has become clear that miRNAs can serve as master regulators that target numerous genes within a single pathway, or multiple genes within a variety of pathways feeding into a single cellular phenotype. Initially, what was less well understood was how these master regulators are themselves controlled beyond the standard biogenesis pathways discussed above. Recent studies have begun to shed light on this topic, and have opened a new realm of possibilities for how small RNAs can act in concert with proteins to regulate complex cellular processes.
Transcriptional Activation and Repression
While some miRNAs are transcribed by RNA polymerase III[61], a majority of miRNAs are transcribed by RNA polymerase II[62], and are subjected to the same regulatory processes as mRNA transcripts. Large-scale mapping studies identified highfrequencies of CpG islands, TATA boxes, TFIIB recognition sites, and the presence of histone modifications within miRNA promoters (Figure 4A)[62, 63]. Furthermore, many key transcription factors regulate the expression of primary miRNA transcripts, including known oncogenes and tumor suppressors. For example, c-Myc binding to E-box elements in the promoter of the mir-17~92gene induces transcriptional activation of mir-17~92[64]. In addition, c-Myc can also promote the transcription of E2F1[65], which in turn is repressed by miR-17~92, generating a feedback loop whereby E2F1 levels can be easily regulated. Given that E2F1 is a crucial cell cycle regulator, the ability to tightly regulate its expression is critical for maintaining cellular homeostasis. In other cases, and in line with its oncogenic role, c-Myc imposes negative transcriptional activity on certain tumor suppressive miRNAs such as mir-15a, mir-34, and let-7 families[40].
Figure 4. Factors Regulating miRNA Abundance.
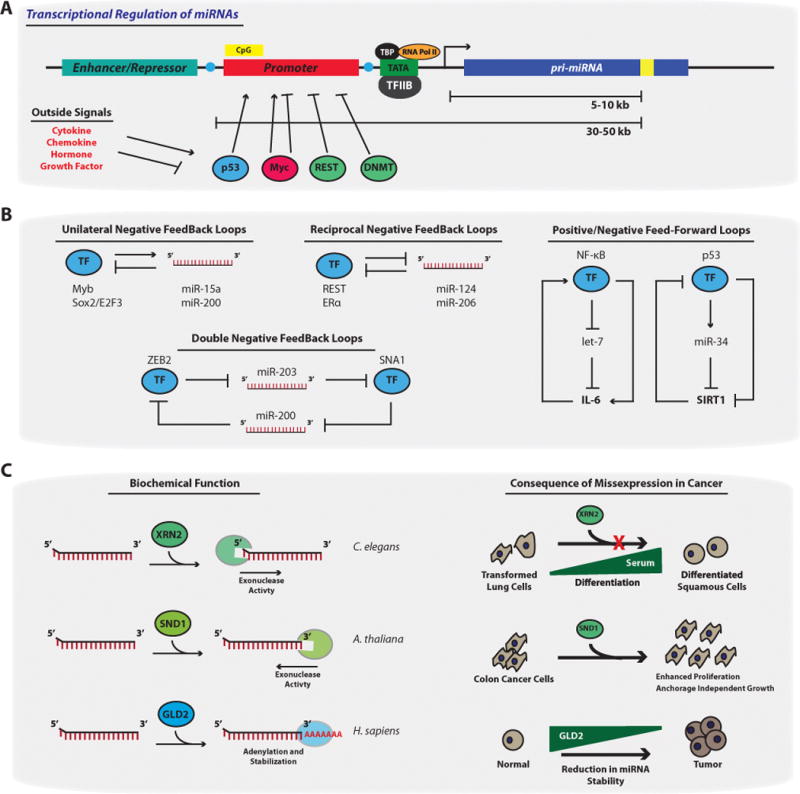
(A) Transcriptional regulation of miRNAs. Similar to other RNA polII transcripts, miRNA genes harbor TATA boxes and TFIIB recognition sites upstream of the transcription start site (TSS) (arrow). MiRNA genes contain upstream enhancer/repressor elements, and promoter regions, indicating miRNAs are subjected to CpG promoter methylation (yellow CpG Box), histone modification (blue circles), as well as other regulatory events. Transcription factors (TFs) such as p53, Myc, and REST can bind to canonical sites within miRNA promoters to either promote or repress transcriptional activation. These TFs, and therefore the miRNA genes, can be misexpressed if the chemokine, hormone, or growth factor signaling pathways that control these TFs becomes dysregulated. Importantly, while the transcriptional start site (TSS) of miRNA genes are sometimes ~5–10kb away from the pre-miRNA sequence (yellow box)[148], the promoter regions can be up to 50kb away, making it difficult to study transcriptional regulation of particular miRNAs. (B)Examples of transcriptional networks that control miRNA expression patterns[149]. Unilateral negative feedback loops occur when certain TFs promote miRNAs thatare responsible for dampeningthe activity of the same TFs. Reciprocal and/or double negative feedback loops occur when certain TFs repress miRNAs that promote the negative regulation of the same TFs[150]. These networks result in either the oscillatory or stable expression of both the TF and the miRNA. This is in contrast to TFs involved in feed forward loops, which regulate miRNAs in a manner that reinforces the initial signaling activity (i.e. NF-κB enhancement of IL-6 through repression of let-7 levels). (C) Factors that control miRNA stability and decay. XRN2 represents a class of 5′ → 3′ exonucleases that can reduce miRNA levels and was initially identified in C. elegans. However, in an immortalized human lung epithelial cellmodel, serum-mediated differentiation of these cells is blocked by forced XRN2 expression. SND1 a member of the 3′ → 5′ exonucleases has similar activity on miRNAs in A. thaliana. The over-expression of SDN1 enhances colon cancer proliferation and anchorage independent growth by controlling APC expression. While gain of exonucleases occurs during tumorigenesis, in part to reduce miRNA levels, the loss of miRNA-stabilizing proteins such as GLD2 have also been noted to occur. Here, GLD2 promotes polyadenylation of miRNAs and therefore protects miRNAs from exonuclease activity.
The miR-34-p53 regulatory axis is another example of how transcriptional regulation of a miRNA can result in fine-tuning of gene expression levels (Figure 4B), in this case through a feed-forward system to mediate tumor suppressive phenotypes[66, 67]. The miR-34 family of miRNAs, including miR-34a/b/c, promotes cell cycle arrest, cell senescence, and apoptosis in cancer, similar to the phenotypes mediated by p53[68]. In p53 KO mice, whole body irradiation failed to induce miR-34a expression, an observation noted to occur in WT mice, which suggests that miR-34a levels depend upon p53. Indeed, p53 can directly bind the promoter of mir-34aand mir-34b/c and induce their expression, and miR-34a can indirectly induce p53 by targeting SIRT1, a negative regulator of p53 via deacetylation. Other regulatory pathways that affect the expression of miR-34a in cancer include NF-κB, which can transcriptionally enhances miR-34a levels[69], and CEBPα, which when mutated results in lower miR-34a levels, enhanced expression of miR-34a-targets such as E2F3, in turn resulting in acute myeloid leukemia[70].
miR-21 is highly expressed in a variety of cancers including breast, ovarian, colorectal, lung, and leukemia[71], and targets tumor suppressors such as PTEN[72]and TPM1[73]. Much is known about the transcriptional control of miR-21. Despite being located in an intron of the TMEM49 gene, approximately 7 different transcription factor binding sites are present within ~500bp upstream of mir-21, some of which are recognized by transcription factors that are mutated or dysregulated in cancer, such as p53, CEBPα, and STAT3[71, 74]. These sites are functional, since cells treated with agents that stimulate or inhibit the activity of these transcription factors promote stimulation or repression of pri-miR-21 levels respectively. Additionally, some of these transcription factors are members of a regulatory feedback network regulated by miR-21. For instance, arsenite can transform human embryonic lung fibroblast (HELF) cells via activation of ERK and JNK, promoting NF-κB and c-Jun mediated transcriptional activation of miR-21[75]. Furthermore, miR-21 subsequently inhibits PDCD4 and SPRY1, which negatively regulate ERK/JNK signaling, thereby maintaining a transformed state.
Other examples of negative feedback loops with transcriptional regulation of miRNAs at the hub of these interactions includemir-200 and ZEB1-SIP1[76], mir-124 and REST[77], mir-206 through SRF[78] and ERα[79], and the mir-106b cluster by E2F[80]. Finally, these transcriptional regulatory networks can be modulated by outside signals, such as PDGF and TGFβ[81], suggesting miRNA activity is kept in check inside the cell and that miRNAs can promote switches in cellular gene expression patterns upon growth factor and cytokine stimulation.
Promoter Methylation of miRNA Genes
The field of epigenetics has expanded greatly in the past decade, and with it the discovery that miRNA genes can be epigenetically silenced through DNA methylation. The first evidence for miRNA silencing via methylation came from microarray studies on T24 bladder cancer cells treated with or without 5-aza-2′-deoxycytidine (5-aza-dC), a DNA methyltransferase inhibitor[82]. One of the miRNAs shown to be strongly upregulated after treatment-induced DNA demethylation was miR-127. This miRNA was also shown to be downregulated in primary prostate, bladder, and colon cancer samples as compared to matched normal tissues. Further experimental evidence indicates miR-127 functions as a tumor suppressor by targeting the proto-oncogene BCL6. Other studies supporting epigenetic silencing of miRNAs found mir-124 and mir-34b/c to be hypermethylated in colorectal cancer cells through disruption of the DNA methyltransferase genes (DMNT1 and DNMT3B)[83]. Furthermore, 5-aza-dC can restore endogenous miR-34 expression in colorectal cancer cells harboring a hypermethylatedmir-34 promoter[84].
Promoter methylation represents a significant means of silencing various tumor suppressor miRNAs in the cell (Figure 4A). For example in non-small cell lung cancer (NSCLC), mir-34b/c methylation and accompanying low levels of mature miR-34b/c are associated with higher rates of recurrence and decreased overall survival[85]. In breast cancer, mir-34 b/c methylation results in enhanced self-renewal capacity of breast cancer tumor-initiating cells[86], presumably due to loss of miR-34b/c levels given that ectopic miR-34c expression abrogated these processes. Other miRNA genes known to be methylated in solid cancers are mir-34a[87], mir-145, mir-200, mir-9, and mir-193a[83]. As epigenetic cancer therapeutics are emerging as a new class of anti-tumorigenic agents, it will be important to consider how these agents effect miRNA gene expression, and whether these effects might help to generate therapeutic efficacy through restoration of miRNA expression.
While certain tumor suppressive miRNAs harbor promoter-hypermethylated CpG islands, there are also reports identifying promoter hypomethylation of microRNAs. Although the let-7 family is largely considered to be a tumor suppressive series of miRNAs, let-7a-3 displays oncogenic activity in lung cancer by supporting anchorage-independent growth and the upregulation of oncogenic genes such as CDK6, CXCL5, and PCNA[88]. In support of its oncogenic role, the promoter of let-7a-3 was hypomethylated in lung cancer samples compared to normal tissue.miR-200a/b is another tumor suppressive miRNA that in pancreatic cancer possesses oncogenic activity. Here, the levels of miR-200b are elevated in the serum of patient samples[89], and in pancreatic cancer cells the mir-200a/b promoter is hypomethylated [90]. Therefore both DNA hyper-and hypo-methylation will serve as useful biomarkers for detecting cancer, and predicting patient outcomes.
miRNA Stability and Decay
While miRNAs can be regulated at any stage during the biogenesis pathway what is less understood is the regulatory processes governing the decay rates and the resulting half-lives of miRNAs (Figure 4C). In Caenorhabditis elegans, degradation of miRNAs is mediated by the 5′-3′ exoribonuclease XRN2[91], and in plants SDN1 is a 3′–5′ exonuclease that degrades ssRNA in a sequence dependent manner[92]. Other proteins such as HEN1 can methylate the last ribose molecule on miRNAs, resulting in the protection of miRNA from degradation by exonucleases such as SDN1[93]. Importantly, these proteins are highly conserved in mammalian cells, and some appear to be aberrantly expressed in several cancer subtypes. A GWAS study on spontaneous lung tumor incidence in inbred mice identified a SNP, rs27328255, to be strongly associated in cis with Xrn2 transcript levels[94]. Additionally, forced expression of XRN2 inhibits differentiation and promotes proliferation of immortalized human lung epithelial cells. In another study, SDN1 was found to be elevated over 5-fold in colon cancer samples compared to normal tissue, and over-expression of SDN1 enhanced colon cancer proliferation and anchorage independent growth by controlling APC expression[95]. While, it has been shown that RRP41 and XRN1 exonucleases can destabilize miR-382 in HEK293 cells[96], there has been no direct evidence as of yet linking the degradation of miRNAs via exonucleases to tumorigenic processes.
Similar to mRNA, miRNAs can be mono or polyadenylated on their 3′ end, resulting in stabilization of the miRNA. Monoadenylation of miRNA is mediated by GLD2[97, 98], a cytoplasmic poly(A) polymerase, which confers protection from exonuclease activity in cancer cell lines. Furthermore, the process of monoadenylation is in direct competition with oligo-uridylation. An example of this involves the TUT4 let-7 axis[99], where the uridylatedlet-7miRNA via TUT4 fails to be recognized by DICER and undergoes degradation. In contrast to the ribonucleases mentioned above, meta-analysis indicates that these RNA modifying enzymes, such as GLD2, are downregulated in several cancers including a variety of lung and digestive carcinomas, as well as invasive breast cancer[100, 101]. While not all cancers harbor loss of GLD2, in those that do it would interesting to determine whether the reduced stability of tumor suppressive miRNAs is playing a biological role in promoting tumorigenesis. Finally, there are some sequence motif dependent processes that mediate miRNA decay, such as AU-rich elements within the mature miRNA sequence. Hwang et al. identified a hexanucleotide element that directs miRNA nuclear import[102], but also affected miRNA stability, though the exact mechanism is still unclear.
RNA Binding Factors Controlling miRNA Levels and Activity
The concept of an RNA binding protein (RBP) regulating miRNA levels should not be surprising given that miRNA biogenesis requires RNaseIII enzymes to sequentially process a pri-miRNA to a mature miRNA. The discovery of the let-7/LIN28 axis however has opened up the possibility that other classes of RBPs could interact with the secondary structure conferred by a miRNA transcript or via sequence specificity to regulate the stability and function of these miRNAs (Figure 5A).
Figure 5. Factors Effecting miRNA Targeting.
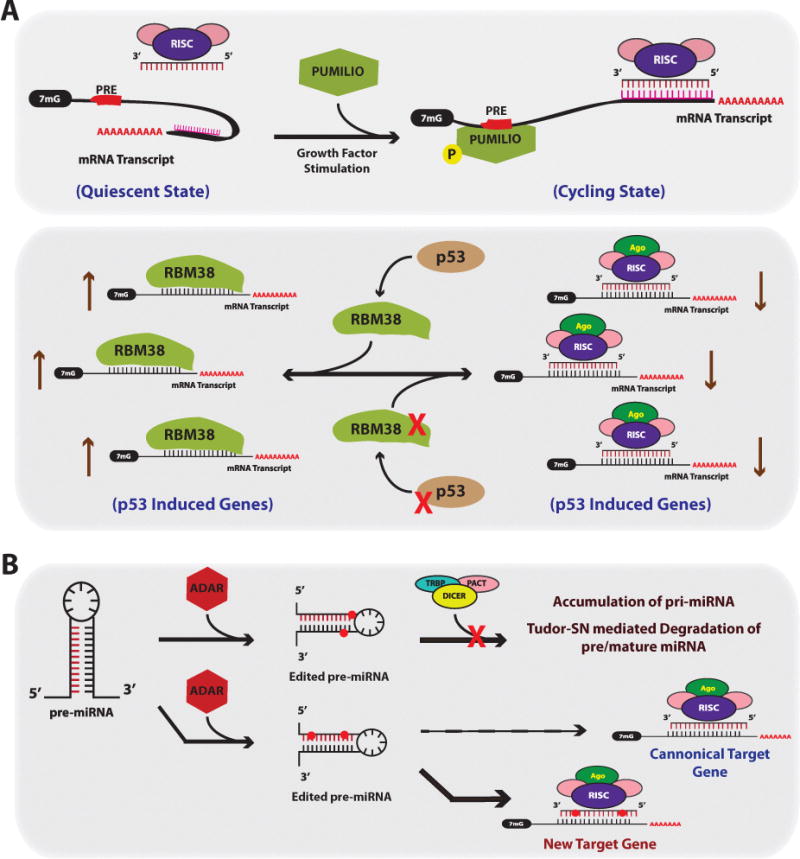
(A) RNA binding proteins can alter miRNA activity and targeting. Top Panel; certain predicted miRNA:mRNA pairing interactions may not occur under certain cell states such as quiescence, presumably due to complex or secondary structures in the mRNA preventing miRNA-RISC binding. After growth factor stimulation, transcripts that harbor PUMILIO Response Elements (PREs), will bind to PUMILIO resulting in a change in the secondary structure of the mRNA such that the predicted miRNA target site becomes accessible for RISC binding. Bottom Panel; RBM38 is an RBP that inhibits miRNA function by directly competing for RISC binding in U-rich regions in the 3′UTRs of mRNA transcripts. RBM38 is induced in a p53 dependent manner after induction of DNA damage and protects p53-induceed genes from miRNAs. In cancer, inactivating mutations in p53 and/or loss of RBM38 would result in aberrant targeting and reduction in p53 induced genes. (B) RNA editing controls both miRNA levels and proper targeting. ADAR can mediate adenosine to inosine editing of miRNA residues. Edited residues near the stem loop of the miRNA results in an unrecognized substrate for Dicer, and subsequently results in accumulation of pri-miRNA and Tudor-SN mediated miRNA degradation. Edited residues residing within the mature miRNA sequence results in new target gene recognition given the edited adenosine to inosine now functions as a guanine.
PUMILIO, a RBP not involved in miRNA biogenesis but rather in promoting miRNA activity[103], can induce conformational changes in the target mRNA allowing for greater accessibility of the RISC complex. Specifically, Kedde et al. demonstrated in MCF-7 breast cancer cell lines that PUMILIO-induced conformational change in the 3′UTR of p27Kip1 gave miR-221/222 better access to the cognate target site within p27Kip1[104]. The involvement of PUMILIO has been observed in other systems such as bladder cancer, where miR-502 and miR-125b target E2F3 in the presence of PUMILIO[105]. Interestingly, Murata et al. used a screening method to find PUMILIO regulatory elements [PREs] (UGUAXAUA) within 3′UTRs[106], and found that 3′UTRs harboring putative PREs were lost due to proximal polyadenylation usage. Presumably if PREs exist in other 3′UTRs, PUMILIO could be a global regulator of miRNA activity. This is also important given that our current methodology for finding miRNA target sites is through target prediction software, and does not take into account target site accessibility with competing RBPs such as LIN28 or PUMILIO.
RBM38 is an RBP that serves to inhibit miRNA function during states of stress in breast cancer. RBM38 is induced in a p53 dependent manner after induction of DNA damage, and binds to U-rich regions in the 3′UTRs of numerous p53 target genes, in turn protecting them from miRNA-mediated AGO2 degradation[107]. This indicates that RBM38 is a crucial mediator of p53 function. Interestingly, the promoter of RBM38 is methylated in many p53 wild type tumors[108], suggesting that some tumors abrogate p53 signaling not by mutation of p53, but through miRNA-targeting of crucial p53 mediators. This RBM38/RBP form of competition for miRNA binding sites can be found in other tumor systems as well. In the leukemic setting, miR-328 serves as a decoy by interfering with hnRNP E2 binding to CEBPα. MiR-328 serves as a tumor suppressor by targeting PIM1; however, miR-328 can also be recognized by the translational regulator protein hnRNP E2[109]. This recognition prevents hnRNP E2 from translationally repressing the master myeloid differentiation regulator CEBPα. The consequence is that cells with high levels of miR-328 will bind hnRNP E2, in turn protecting CEPBα and promoting the differentiation of leukemic blast cells.
HuR is another RNA binding protein that cooperates and competes with miRNA activity[110]. HuR is a ubiquitously expressed protein, a member of the ELAV family of proteins, and can predominately stabilize mRNA transcripts by binding AU-rich elements (ARE). HuR is mainly restricted to the nucleus; however, under states of cell stress HuR translocates to the cytoplasm to promote stabilization of mRNA transcripts[111, 112]. MiRNAs were first shown to be affected by HuR when CAT-1 translation was repressed by miR-122, specifically in the context of HuR binding to an ARE in the CAT-1 3′UTR[113]. A presumed mechanism for this observation, at least under states of cell stress, is that HuR could promote CAT-1 mRNA release from P-bodies, where miR-122 would normally be mediating repressive activity on the transcript. In subsequent studies, miR-331 and miR-16 were found to controlCOX-2 and ERBB-2 transcript levels via competition with HuR binding[114, 115]. Similarly, HuR promote smiRNA activityas let-7-mediated targeting and repression of the c-Myc oncogene required the presence of HuR[116]. This positive regulatory loop mediated by HuR binding was also observed with miR-19 and the RhoB mRNA[117]. Finally, HuR can also be modulated by miRNAs in cancer. For example, miR-519, a tumor suppressive miRNA, promotes anti-proliferative properties in cervical, colon, and ovarian cancer lines through targeting and reducing HuR transcripts[118]. Furthermore, miR-519 and HuR levels are inversely correlated in many cancer samples, specifically miR-519 levels are low and HuR levels are elevated[119]. As more RBPs are found to control miRNA activity there will evolve a need to better categorize their respective activity in an effort to identify universal mechanisms of action that can be exploited for therapeutic potential.
miRNA Editing
Adenosine deaminases (ADARs) are a family of enzymes that mediate adenosine to inosine editing of RNA molecules. This process results in RNA and protein diversity in higher eukaryotes by altering codons in the mRNA[120, 121]. These enzymes classically recognize dsRNA structures within an mRNA transcript while still in the nucleus; however, certain miRNAs can also be edited in the brain and other tissues. Specifically, cDNAs obtained from multiple tissues indicate that pri-miR-22 is heavily edited[122]. Furthermore, cells ectopically expressing ADAR1 promoted the editing of these same residues on the pri-miR-22.
Editing of pri-miRNAs would have major consequences for miRNA biogenesis and function (Figure 5B). First, DICER activity on the pre-miRNA substrate heavily depends on proper base pairing within the stem region. If adenosines were edited to inosines near the region where DICER cleaves, then further processing of the miRNA would be compromised. Consistent with this notion, an artificially edited pri-miR-142 expressed in HEK-293 cells could not be processed, resulting in accumulation of the primary miRNA transcript and degradation of mature miR-142 via Tudor-SN, an inosine-specific ribonuclease[123].
More importantly, editing within the 20–22mer portion of the mature miRNA would alter the target mRNA repertoire for that particular miRNA. For example, adenosines, which normally base pair with uridine, if edited to inosine, would function as guanine and base pair with cytidine. A study by Wang et al. exemplifies this point, finding that miR-376a* editing was significantly reduced in high grade human glioma samples[124]. In this model, U87 glioma cells containing stable expression of an unedited miR-376a*A were highly metastatic, while cells expressing the edited (miR-376*G) form were not as invasive. Furthermore miR-376a*A targeted RAP2A, a gene that produces a protein involved in both the regulation of dendrite and axonal branching as well as cancer cell migration, while the edited miR-376a*G targeted AMFR (a novel player in glioma). The restoration of these two genes in each respective model can rescue the phenotypes observed, suggesting that differential targeting of the unedited/edited forms of miR-376, rather than just the gain or loss of the targets themselves, drive these phenotypes.
There are other examples of RNA editing in cancer (see review by Skarda et al.[125]), as well as databases compiling particular miRNA sequences subject to RNA editing[126]. Overall, it is clear that RNA editing is modified in tumors on a global scale; however, more work is required to understand the role of miRNA editing in the processes of tumorigenesis. Eventually, this knowledge could be exploited for diagnostic, prognostic, and therapeutic value.
The ceRNA Network
Competing endogenous RNAs (ceRNAs) regulate miRNA activity on a particular target mRNA by competing with other RNA transcripts for miRNA binding, and furthermore co-regulate each other in a complex ceRNA network (Figure 6)[127]. Recent examples of this network highlight the role of transcribed pseudogenes, which are evolutionary remnants of ancestral genes that do not code for protein, yet contain target sites for miRNAs and therefore serve asmiRNA sponges, allowing for known miRNA-regulated protein coding transcripts to escape miRNA regulation. The PTEN-PTENP1 regulatory axis was the first ceRNA network shown to exist in human cancer[128]. PTENP1 is a pseudogene highly homologous to PTEN, a tumor suppressor in cancer. Since the 3′UTR of PTENP1 is highly conserved, especially within the miRNA binding sites to PTEN, miRNAs that bind to PTEN could be sequestered away by PTENP1. In this way PTENP1functions as an endogenous competitor allowing for enhanced translation of PTEN. Indeed miR-17, miR-21, and miR-26 miRNA families can target both genes, and importantly PTENP1 can also function as a tumor suppressor. Lastly, the expression of PTENP1 results in elevated PTEN levels and inhibition of growth in prostate cancer lines.
Figure 6. The miRNA ceRNA Network.
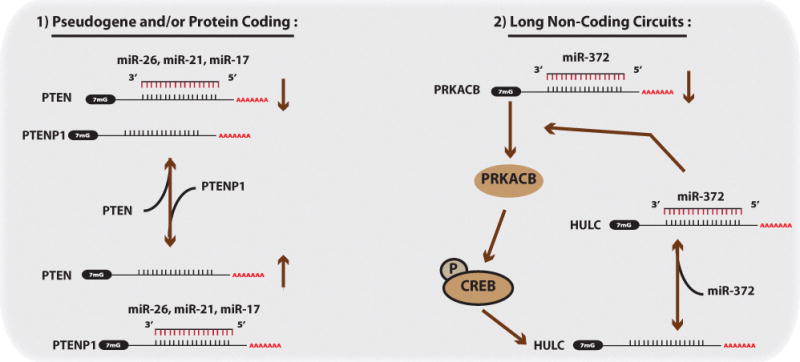
In the ceRNA network, ceRNAs can affect miRNA targeting primarily by acting as sponges. 1) Given that many genes also have a number of pseudogenes with conserved miRNA binding sites, as is the case with PTEN, the abundance of the pseudogene (PTENP1) will influence how strong the regulatory action of a particular miRNA is on the intended mRNA target (in this case PTEN). However, the system is oscillatory in that when PTENP1 are lower the PTEN levels, miRNAs will preferentially target PTEN, and vice versa. 2) Long noncoding RNAs can also be apart of this network. PRKACB can normally be targeted by miR-372; however, in response PRKACB can induce HULC long-noncoding RNA levels by phosphorylating and activating CREB. HULC serves as a sponge for miR-372, thereby preventing the miR-372 regulation of PRKACB.
Since this discovery, other ceRNA networks have been found in prostate cancer, glioblastoma, and melanoma. There have been PTEN ceRNA networks that include genes such as VAPA and ZEB[129, 130], and others such as KRAS-KRAS1P, CD44-CDC42, and HULC-CREB[131]. The long non-coding RNA HULC serves as a ceRNA by inhibiting miR-372 activity and functions as a sponge allowing for the de-repression of PRKACB. PRKACB can then phosphorylate CREB and in turn promote the expression of HULC in auto-regulatory fashion. Recent work also indicates that the mRNA of HMGA2, which is highly expressed during development and in tumor tissue, can sequester let-7 away from its cellular targets[132]. This sequestration promotes the progression of NSCLC.
It will be important to examine the expression profile of these ceRNAs in cancer samples, given that dysregulation of these “miRNA sponges” can produce striking phenotypes. Furthermore, this complex communication between pseudogenes, miRNAs, and mRNA transcripts affects the testing of putative miRNA targets given that certain cells may have high levels of a particular pseudogene acting as a sponge, or target decoy. New databases (i.e. ceRDB) are emerging to identify ceRNA interactions on a genome-wide scale[133, 134]. Additionally, elucidating ceRNA biology in the normal cells from which the cancer arises will be crucial since Denzler et al. recently found that in normal hepatocytes modulating the abundance of one canonical miR-122 target (AldoA) did not significantly affect other miR-122 target genes[135]. Furthermore, global miR-122 target site abundance was not sufficiently different enough in a metabolic liver disease model to alter miR-122-mediated gene repression, arguing ceRNAs are not involved in this biology. However, if ceRNA levels can approach miRNA target site abundance, as may be the case in the PTEN story described above, then ceRNAs may indeed regulate miRNA activity. Therefore, more effort will be required to determine the extent of the ceRNA network in cancer and the implications for the pathophysiology of this disease.
Single Nucleotide Polymorphisms / Mutations
Single nucleotide polymorphisms (SNPs) are naturally occurring variations in the DNA sequence that have been studied in great detail, and found to predict disease associations for particular populations. A number of studies have identified how miRNA regulation of mRNA transcripts can be disrupted by the presence of certain SNPs within those respective targets.
Specifically, Saunders et al. identified polymorphisms which occur predominately in the DNA sequence flanking pre-miRNAs but less so within the miRNA sequences themselves[136]. It appears that SNPs distribute near putative miRNA target sites, though this cannot be fully appreciated given that not all miRNA targets have been experimentally validated. Specifically, a bioinformatics approach was used to scan putative miRNA target sites within the 3′UTR of respective mRNA genes, and this data set was overlaid with SNP array data[136–138]. A theme that emerged was that SNPs within mRNAs disrupting miRNA:mRNA binding are under positive selection pressureso as to avoid miRNA-mediated downregulation of gene expression. However, this can lead to the perpetuation of SNPs that may contribute to disease phenotypes. In NSCLC, KRAS is an oncogene with identified SNPs that associate with risk of developing the disease. Specifically, a SNP in a putative let-7 binding site (LCS) of KRAS not only was associated with a 2.3-fold increase risk for developing NSCLC, but also led to disruption of let-7 binding to KRAS[139]. Other examples of SNPs affecting miRNA-target interactions include, miR-221 and KIT in papillary thyroid carcinoma and melanoma[140, 141], and the miR-189 SLITRK1 interaction in Tourette’s syndrome[142].
SNPs found in pre-miRNAs themselves can disrupt the processing of these miRNAs since proper nucleotide base-pairing is crucial for proper miRNA biogenesis[143]. An example of this is a germ line mutation 7nt downstream of mir-16 in CLL that abolishes the expression of both miR-15a and miR-16-1[144]. Furthermore, a SNP in mir-125a can block pri-miR-125a processing, reducing miRNA-mediated repression of LIN28 target RNA[145]. Sequence variations can also be found in the transcripts coding for biogenesis proteins such as DICER[146], and these SNPs can associate with diseases including cancer. While it is exciting that a single nucleotide variation in the DNA can affect miRNA biogenesis and activity, further effort is required to understand the functionality of miRNA-SNP interactions, and whether these interactions can promote tumorigenic events.
Conclusions
As highlighted in this review, the pervasive dysregulation of miRNA-mediated gene regulatory networks are evident in many cancer models. MiRNAs function as master regulators and signal modulators by fine tuning gene expression, a process that when disrupted, allows for a permissive tumorigenic state. While it has been known for sometime that miRNAs can regulate mRNA transcripts, and that disruption of this process can result in tumorigenesis, what was less understood was how miRNAs were themselves regulated. Transcription of miRNA genes primarily depends upon RNA polymerase II, and so it should be no surprise that miRNAs are under similar regulatory constraints as protein coding transcripts (i.e. transcriptional activation/inhibition, and DNA methylation). However, the surprising finding in the past few years has been the extent to which miRNAs are regulated at the posttranscriptional level, either by RNA binding proteins, RNA editing, or modulating miRNA decay, and how tumor cells can subvert these regulatory processes.
Furthermore, it appears that there is a complex ceRNA communication network and it will be interesting to see how this RNA network in all its complexity, can contribute to specific cellular phenotypes such as transformation, epithelial to mesenchymal transition, and generation of cancer stems cells. Given this complexity, the tumorigenic environment may rely more on the disruption of RNA-mediated gene regulatory networks than was previously appreciated. Indeed, it is known that miRNA expression profiles can classify tumor subtypes more effectively than mRNA profiles[11], suggesting a substantial role for miRNAs in the regulation of malignant biological processes by regulating mRNA transcripts that result in tumor progression.
Finally, expression of miRNAs can convert a noisy transcriptional network into a concerted or dedicated bimodal switch system that can direct cell fates. Furthermore, the identification of miRNAs that can promote cancer stem cell formation, or protect these cells from insults such as chemotherapeutic agents will be an important discovery. Those miRNAs could then be targeted by antisense-based therapies in the clinic to sensitize the cancer stem cell population to current clinical regimens for the particular tumor type. Overall, there has been a recent explosion of knowledge in miRNA biology, and as more discoveries are made in the non-coding RNA field, hopefully some of these findings can soon be translated into promising clinical advances.
Acknowledgments
This work was supported by grants to FS from the NIH (R01 CA157749 and R01 CA131301). BDA was supported by a training grant (5T32 HG003198-10). ALK was supported by a young investigator award from the NIH (K99/R00 CA178091-01). We thank Carlos Stahlhut and Minlee Kimfor critical reading of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest to disclose.
References
- 1.Hamilton BE, Hoyert DL, Martin JA, Strobino DM, Guyer B. Annual summary of vital statistics: 2010–2011. Pediatrics. 2013;131:548–58. doi: 10.1542/peds.2012-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 4.Kasinski AL, Slack FJ. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa FF. Non-coding RNAs: Meet thy masters. Bioessays. 2010;32:599–608. doi: 10.1002/bies.200900112. [DOI] [PubMed] [Google Scholar]
- 6.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 2011;12:846–60. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 7.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–60. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macfarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics. 2010;11:537–61. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 11.Lu J, Getz G, Miska Ea, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando Aa, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, Urbauer D, Pennacchio LA, Cheng JF, Nick AM, et al. Dicer, Drosha, and Outcomes in Patients with Ovarian Cancer N. Engl J Med. 2008;359:2641–50. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, Yatabe Y, Takamizawa J, Miyoshi S, Mitsudomi T, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–5. doi: 10.1111/j.1349-7006.2005.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, Jacks T. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–4. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–90. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F, Jacks T. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–73. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–7. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 19.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukasawa M, Morita S, Kimura M, Horii T, Ochiya T, Hatada I. Genomic imprinting in Dicer1-hypomorphic mice. Cytogenet Genome Res. 2006;113:138–43. doi: 10.1159/000090825. [DOI] [PubMed] [Google Scholar]
- 21.Carmell MA, Xuan Z, Zhang MQ, Hannon GJ. The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev. 2002;16:2733–42. doi: 10.1101/gad.1026102. [DOI] [PubMed] [Google Scholar]
- 22.Nelson P, Kiriakidou M, Sharma A, Maniataki E, Mourelatos Z. The microRNA world: small is mighty. Trends Biochem Sci. 2003;28:534–540. doi: 10.1016/j.tibs.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Völler D, Reinders J, Meister G, Bosserhoff AK. Strong reduction of AGO2 expression in melanoma and cellular consequences. Br J Cancer. 2013;109:3116–24. doi: 10.1038/bjc.2013.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Yu C, Gao H, Li Y. Argonaute proteins: potential biomarkers for human colon cancer. BMC Cancer. 2010;10:38. doi: 10.1186/1471-2407-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, Barbosa-Morais NL, Teschendorff AE, Green AR, Ellis IO, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8:R214. doi: 10.1186/gb-2007-8-10-r214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams BD, Claffey KP, White BA. Argonaute-2 expression is regulated by epidermal growth factor receptor and mitogen-activated protein kinase signaling and correlates with a transformed phenotype in breast cancer cells. Endocrinology. 2009;150:14–23. doi: 10.1210/en.2008-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497:383–7. doi: 10.1038/nature12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rüdel S, Wang Y, Lenobel R, Körner R, Hsiao HH, Urlaub H, Patel D, Meister G. Phosphorylation of human Argonaute proteins affects small RNA binding. Nucleic Acids Res. 2011;39:2330–43. doi: 10.1093/nar/gkq1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng Y, Sankala H, Zhang X, Graves PR. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem J. 2008;413:429–36. doi: 10.1042/BJ20080599. [DOI] [PubMed] [Google Scholar]
- 30.Ameyar-Zazoua M, Rachez C, Souidi M, Robin P, Fritsch L, Young R, Morozova N, Fenouil R, Descostes N, Andrau JC, et al. Argonaute proteins couple chromatin silencing to alternative splicing. Nat Struct Mol Biol. 2012;19:998–1004. doi: 10.1038/nsmb.2373. [DOI] [PubMed] [Google Scholar]
- 31.Ender C, Meister G. Argonaute proteins at a glance. J Cell Sci. 2010;123:1819–23. doi: 10.1242/jcs.055210. [DOI] [PubMed] [Google Scholar]
- 32.Kallen AN, Ma J, Huang Y. Does Lin28 Antagonize miRNA-Mediated Repression by Displacing miRISC from Target mRNAs? Front Genet. 2012;3:240. doi: 10.3389/fgene.2012.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 34.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–8. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell. 2010;140:445–9. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, Ng SB, Chng WJ. LIN28/LIN28B: An emerging oncogenic driver in cancer stem cells. Int J Biochem Cell Biol. 2013;45:973–978. doi: 10.1016/j.biocel.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 37.Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008;32:276–84. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 38.Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang TC, Yu D, Lee YS, Wentzel Ea, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS, Feig C, Xu J, Burge CB, Peter ME. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008;68:2587–91. doi: 10.1158/0008-5472.CAN-08-0264. [DOI] [PubMed] [Google Scholar]
- 42.Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM, Minn AJ, Rosner MR. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009;28:347–58. doi: 10.1038/emboj.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–8. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, Takeuchi A, Grasemann C, Rinn JL, Lopez MF, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010;42:626–30. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shyh-Chang N, Zhu H, Yvanka de Soysa T, Shinoda G, Seligson MT, Tsanov KM, Nguyen L, Asara JM, Cantley LC, Daley GQ. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell. 2013;155:778–92. doi: 10.1016/j.cell.2013.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, Shukrun R, Charlton J, Sebire N, Mifsud W, et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev. 2014;28:971–82. doi: 10.1101/gad.237149.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, Mihara M, Naitou M, Endoh H, Nakamura T, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–11. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 48.Yamagata K, Fujiyama S, Ito S, Ueda T, Murata T, Naitou M, Takeyama KI, Minami Y, O’Malley BW, Kato S. Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol Cell. 2009;36:340–7. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe M, Yanagisawa J, Kitagawa H, Takeyama K, Ogawa S, Arao Y, Suzawa M, Kobayashi Y, Yano T, Yoshikawa H, et al. A subfamily of RNA-binding DEAD-box proteins acts as an estrogen receptor alpha coactivator through the N-terminal activation domain (AF-1) with an RNA coactivator, SRA. EMBO J. 2001;20:1341–52. doi: 10.1093/emboj/20.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Warner DR, Bhattacherjee V, Yin X, Singh S, Mukhopadhyay P, Pisano MM, Greene RM. Functional interaction between Smad, CREB binding protein, and p68 RNA helicase. Biochem Biophys Res Commun. 2004;324:70–6. doi: 10.1016/j.bbrc.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 51.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis-Dusenbery BN, Hata A. MicroRNA in Cancer: The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer. 2010;1:1100–14. doi: 10.1177/1947601910396213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–84. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010–4. doi: 10.1038/nature08025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sakamoto S, Aoki K, Higuchi T, Todaka H, Morisawa K, Tamaki N, Hatano E, Fukushima A, Taniguchi T, Agata Y. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol. 2009;29:3754–69. doi: 10.1128/MCB.01836-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zucconi BE, Wilson GM. Modulation of neoplastic gene regulatory pathways by the RNA-binding factor AUF1. Front Biosci (Landmark Ed. 2011;16:2307–25. doi: 10.2741/3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abdelmohsen K, Tominaga-Yamanaka K, Srikantan S, Yoon JH, Kang MJ, Gorospe M. RNA-binding protein AUF1 represses Dicer expression. Nucleic Acids Res. 2012;40:11531–44. doi: 10.1093/nar/gks930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–34. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 59.Höck J, Weinmann L, Ender C, Rüdel S, Kremmer E, Raabe M, Urlaub H, Meister G. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007;8:1052–60. doi: 10.1038/sj.embor.7401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141:1195–207. doi: 10.1016/j.cell.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 61.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 62.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22:3172–83. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corcoran DL, Pandit KV, Gordon B, Bhattacharjee A, Kaminski N, Benos PV. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One. 2009;4:e5279. doi: 10.1371/journal.pone.0005279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 65.Coller HA, Forman JJ, Legesse-Miller A. “Myc’ed messages”: myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genet. 2007;3:e146. doi: 10.1371/journal.pgen.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1, and p53: The feedback loop. Cell Cycle. 2009;8:712–715. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- 67.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–9. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 69.Li J, Wang K, Chen X, Meng H, Song M, Wang Y, Xu X, Bai Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol Biol. 2012;13:4. doi: 10.1186/1471-2199-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pulikkan JA, Peramangalam PS, Dengler V, Ho PA, Preudhomme C, Meshinchi S, Christopeit M, Nibourel O, Müller-Tidow C, Bohlander SK, et al. C/EBPα regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood. 2010;116:5638–49. doi: 10.1182/blood-2010-04-281600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13:39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–58. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–36. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 74.Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 75.Shen L, Ling M, Li Y, Xu Y, Zhou Y, Ye J, Pang Y, Zhao Y, Jiang R, Zhang J, et al. Feedback regulations of miR-21 and MAPKs via Pdcd4 and Spry1 are involved in arsenite-induced cell malignant transformation. PLoS One. 2013;8:e57652. doi: 10.1371/journal.pone.0057652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 77.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103:2422–7. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–96. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 79.Adams BD, Furneaux H, White BA. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol. 2007;21:1132–47. doi: 10.1210/me.2007-0022. [DOI] [PubMed] [Google Scholar]
- 80.Thangavel C, Boopathi E, Ertel A, Lim M, Addya S, Fortina P, Witkiewicz AK, Knudsen ES. Regulation of miR106b cluster through the RB pathway: mechanism and functional targets. Cell Cycle. 2013;12:98–111. doi: 10.4161/cc.23029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal. 2009;7:18. doi: 10.1186/1478-811X-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 83.Esteller Badosa M, Suzuki H, Maruyama R, Yamamoto E, Kai M. DNA methylation and microRNA dysregulation in cancer. Mol Oncol. 2012;6:567–578. doi: 10.1016/j.molonc.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008;68:4123–32. doi: 10.1158/0008-5472.CAN-08-0325. [DOI] [PubMed] [Google Scholar]
- 85.Wang Z, Chen Z, Gao Y, Li N, Li B, Tan F, Tan X, Lu N, Sun Y, Sun J, et al. DNA hypermethylation of microRNA-34b/c has prognostic value for stage I non-small cell lung cancer. Cancer Biol Ther. 2011;11:490–6. doi: 10.4161/cbt.11.5.14550. [DOI] [PubMed] [Google Scholar]
- 86.Yu F, Jiao Y, Zhu Y, Wang Y, Zhu J, Cui X, Liu Y, He Y, Park EY, Zhang H, et al. MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J Biol Chem. 2012;287:465–73. doi: 10.1074/jbc.M111.280768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Körner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 88.Brueckner B, Stresemann C, Kuner R, Mund C, Musch T, Meister M, Sültmann H, Lyko F. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007;67:1419–23. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- 89.Li A, Omura N, Hong SM, Vincent A, Walter K, Griffith M, Borges M, Goggins M. Pancreatic cancers epigenetically silence SIP1 and hypomethylate and overexpress miR-200a/200b in association with elevated circulating miR-200a and miR-200b levels. Cancer Res. 2010;70:5226–37. doi: 10.1158/0008-5472.CAN-09-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li A, Omura N, Hong SM, Vincent A, Walter K, Griffith M, Borges M, Goggins M. Pancreatic cancers epigenetically silence SIP1 and hypomethylate and overexpress miR-200a/200b in association with elevated circulating miR-200a and miR-200b levels. Cancer Res. 2010;70:5226–37. doi: 10.1158/0008-5472.CAN-09-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009;461:546–9. doi: 10.1038/nature08349. [DOI] [PubMed] [Google Scholar]
- 92.Ramachandran V, Chen X. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science. 2008;321:1490–2. doi: 10.1126/science.1163728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X. Methylation as a crucial step in plant microRNA biogenesis. Science. 2005;307:932–5. doi: 10.1126/science.1107130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lu Y, Liu P, James M, Vikis HG, Liu H, Wen W, Franklin A, You M. Genetic variants cis-regulating Xrn2 expression contribute to the risk of spontaneous lung tumor. Oncogene. 2010;29:1041–9. doi: 10.1038/onc.2009.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsuchiya N, Nakagama H. MicroRNA, SND1, and alterations in translational regulation in colon carcinogenesis. Mutat Res. 2010;693:94–100. doi: 10.1016/j.mrfmmm.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 96.Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, Kiledjian M. Differential regulation of microRNA stability. RNA. 2010;16:1032–9. doi: 10.1261/rna.1851510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara SI, Baba T, Suzuki T. Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009;23:433–8. doi: 10.1101/gad.1761509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.D’Ambrogio A, Gu W, Udagawa T, Mello CC, Richter JD. Specific miRNA stabilization by Gld2-catalyzed monoadenylation. Cell Rep. 2012;2:1537–45. doi: 10.1016/j.celrep.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thornton JE, Chang HM, Piskounova E, Gregory RI. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7) RNA. 2012;18:1875–85. doi: 10.1261/rna.034538.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.D’Ambrogio A, Nagaoka K, Richter JD. Translational control of cell growth and malignancy by the CPEBs. Nat Rev Cancer. 2013;13:283–90. doi: 10.1038/nrc3485. [DOI] [PubMed] [Google Scholar]
- 101.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hwang HW, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 103.Nolde MJ, Saka N, Reinert KL, Slack FJ. The Caenorhabditis elegans pumilio homolog, puf-9, is required for the 3′UTR-mediated repression of the let-7 microRNA target gene, hbl-1. Dev Biol. 2007;305:551–63. doi: 10.1016/j.ydbio.2007.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JAF, Elkon R, Agami R. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–20. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- 105.Miles WO, Tschöp K, Herr A, Ji JY, Dyson NJ. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012;26:356–68. doi: 10.1101/gad.182568.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Murata Y, Wharton RP. Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos. Cell. 1995;80:747–56. doi: 10.1016/0092-8674(95)90353-4. [DOI] [PubMed] [Google Scholar]
- 107.Léveillé N, Elkon R, Davalos V, Manoharan V, Hollingworth D, Oude Vrielink J, le Sage C, Melo CA, Horlings HM, Wesseling J, et al. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun. 2011;2:513. doi: 10.1038/ncomms1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ciafrè SA, Galardi S. microRNAs and RNA-binding proteins: a complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013;10:935–42. doi: 10.4161/rna.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R, Liu S, Schwind S, Santhanam R, Hickey CJ, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140:652–65. doi: 10.1016/j.cell.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ciafre SA, Galardi S. microRNAs and RNA-binding proteins: A complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013;10:935–42. doi: 10.4161/rna.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jain RG, Andrews LG, McGowan KM, Gao F, Keene JD, Pekala PP. Hel-N1, an RNA-binding protein, is a ligand for an A + U rich region of the GLUT1 3′ UTR. Nucleic Acids Symp Ser. 1995:209–11. [PubMed] [Google Scholar]
- 112.Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci U S A. 1998;95:15293–8. doi: 10.1073/pnas.95.26.15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–24. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 114.Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem. 2011;286:41442–54. doi: 10.1074/jbc.M111.301481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10:167–80.A. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009;42:125–30. doi: 10.5483/bmbrep.2009.42.3.125. [DOI] [PubMed] [Google Scholar]
- 117.Glorian V, Maillot G, Polès S, Iacovoni JS, Favre G, Vagner S. HuR-dependent loading of miRNA RISC to the mRNA encoding the Ras-related small GTPase RhoB controls its translation during UV-induced apoptosis. Cell Death Differ. 2011;18:1692–701. doi: 10.1038/cdd.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci U S A. 2008;105:20297–302. doi: 10.1073/pnas.0809376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Abdelmohsen K, Kim MM, Srikantan S, Mercken EM, Brennan SE, Wilson GM, Cabo R de, Gorospe M. miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle. 2010;9:1354–9. doi: 10.4161/cc.9.7.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maas S, Rich A, Nishikura K. A-to-I RNA editing: recent news and residual mysteries. J Biol Chem. 2003;278:1391–4. doi: 10.1074/jbc.R200025200. [DOI] [PubMed] [Google Scholar]
- 121.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–46. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–7. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scadden ADJ. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol. 2005;12:489–96. doi: 10.1038/nsmb936. [DOI] [PubMed] [Google Scholar]
- 124.Choudhury Y, Tay FC, Lam DH, Sandanaraj E, Tang C, Ang BT, Wang S. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J Clin Invest. 2012;122:4059–76. doi: 10.1172/JCI62925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Skarda J, Amariglio N, Rechavi G. RNA editing in human cancer: review. APMIS. 2009;117:551–7. doi: 10.1111/j.1600-0463.2009.02505.x. [DOI] [PubMed] [Google Scholar]
- 126.Laganà A, Paone A, Veneziano D, Cascione L, Gasparini P, Carasi S, Russo F, Nigita G, Macca V, Giugno R, et al. miR-EdiTar: a database of predicted A-to-I edited miRNA target sites. Bioinformatics. 2012;28:3166–8. doi: 10.1093/bioinformatics/bts589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–95. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–57. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38:5366–83. doi: 10.1093/nar/gkq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N, Winslow MM, Downward J. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014;505:212–7. doi: 10.1038/nature12785. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 133.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–52. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sarver AL, Subramanian S. Competing endogenous RNA database. Bioinformation. 2012;8:731–3. doi: 10.6026/97320630008731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA Hypothesis with Quantitative Measurements of miRNA and Target Abundance. Mol Cell. 2014 doi: 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saunders MA, Liang H, Li WH. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci U S A. 2007;104:3300–5. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–6. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 138.Jin Y, Lee C. Single Nucleotide Polymorphisms Associated with MicroRNA Regulation. Biomolecules. 2013;3:287–302. doi: 10.3390/biom3020287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, Muller RU, Straka E, Su L, Burki EA, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–40. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2005;102:19075–80. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Godshalk SE, Paranjape T, Nallur S, Speed W, Chan E, Molinaro AM, Bacchiocchi A, Hoyt K, Tworkoski K, Stern DF, et al. A Variant in a MicroRNA complementary site in the 3′ UTR of the KIT oncogene increases risk of acral melanoma. Oncogene. 2011;30:1542–50. doi: 10.1038/onc.2010.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–20. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- 143.Yu Z, Li Z, Jolicoeur N, Zhang L, Fortin Y, Wang E, Wu M, Shen SH. Aberrant allele frequencies of the SNPs located in microRNA target sites are potentially associated with human cancers. Nucleic Acids Res. 2007;35:4535–41. doi: 10.1093/nar/gkm480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Calin George Adrian, MD PhD, Ferracin Manuela, PhD, Cimmino Amelia, MD PhD, Di Leva Gianpiero, PhD, Shimizu Masayoshi, BS, Wojcik Sylwia E, MSc, Iorio Marilena V, PhD, Visone Rosa, PhD, Sever Nurettin Ilfer, PhD, Fabbri M Muller., MD A MicroRNA Signature Associated with Prognosis and Progression in Chronic Lymphocytic Leukemia. NEJM. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 145.Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16:1124–31. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- 146.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cook WD, McCaw BJ. Accommodating haploinsufficient tumor suppressor genes in Knudson’s model. Oncogene. 2000;19:3434–8. doi: 10.1038/sj.onc.1203653. [DOI] [PubMed] [Google Scholar]
- 148.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22:3172–83. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Herranz H, Cohen SM. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 2010;24:1339–44. doi: 10.1101/gad.1937010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Moes M, Le Béchec A, Crespo I, Laurini C, Halavatyi A, Vetter G, Del Sol A, Friederich E. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PLoS One. 2012;7:e35440. doi: 10.1371/journal.pone.0035440. [DOI] [PMC free article] [PubMed] [Google Scholar]