

Abstract
Background
The premature infant gut has low individual but high inter-individual microbial diversity compared with adults. Based on prior 16S rRNA gene surveys, many species from this environment are expected to be similar to those previously detected in the human microbiota. However, the level of genomic novelty and metabolic variation of strains found in the infant gut remains relatively unexplored.
Results
To study the stability and function of early microbial colonizers of the premature infant gut, nine stool samples were taken during the third week of life of a premature male infant delivered via Caesarean section. Metagenomic sequences were assembled and binned into near-complete and partial genomes, enabling strain-level genomic analysis of the microbial community.
We reconstructed eleven near-complete and six partial bacterial genomes representative of the key members of the microbial community. Twelve of these genomes share >90% putative ortholog amino acid identity with reference genomes. Manual curation of the assembly of one particularly novel genome resulted in the first essentially complete genome sequence (in three pieces, the order of which could not be determined due to a repeat) for Varibaculum cambriense (strain Dora), a medically relevant species that has been implicated in abscess formation.
During the period studied, the microbial community undergoes a compositional shift, in which obligate anaerobes (fermenters) overtake Escherichia coli as the most abundant species. Other species remain stable, probably due to their ability to either respire anaerobically or grow by fermentation, and their capacity to tolerate fluctuating levels of oxygen. Metabolic predictions for V. cambriense suggest that, like other members of the microbial community, this organism is able to process various sugar substrates and make use of multiple different electron acceptors during anaerobic respiration. Genome comparisons within the family Actinomycetaceae reveal important differences related to respiratory metabolism and motility.
Conclusions
Genome-based analysis provided direct insight into strain-specific potential for anaerobic respiration and yielded the first genome for the genus Varibaculum. Importantly, comparison of these de novo assembled genomes with closely related isolate genomes supported the accuracy of the metagenomic methodology. Over a one-week period, the early gut microbial community transitioned to a community with a higher representation of obligate anaerobes, emphasizing both taxonomic and metabolic instability during colonization.
Keywords: Binning, Community genomics, Genome reconstruction, Human gut, Metagenomics, Microbial dynamics, Premature infant, Varibaculum cambriense, Time series binning
Background
The human adult microbiota consists of 10-fold more cells than the human body (the majority reside in the gut) and 100-fold more genes than the human genome [1-3]. The gut microbiota are involved in host nutrient acquisition [4], regulation and development of the host immune system [5,6], and the modulation of host gene expression [7]. All of these influences have the potential to seriously affect human health. Aberrations in gut microbiota membership and community structure, termed microbial dysbiosis, have been associated with obesity [8] and diseases such as inflammatory bowel disease [9], both type 1 and type 2 diabetes [10,11], and necrotizing enterocolitis in premature infants [12-14]. Although previous studies have focused on gut colonization [15,16], few have shown the process in a high-resolution manner [17,18]. Thus, much is still not known about the diversity, metabolic potential, or roles of early gut colonizers.
Although the gut microbiota of infants is characterized by high levels of inter-individual diversity (beta diversity), community composition begins to look like that of adults within the first year of life [15]. In comparison with both adults and infants, premature infants have especially low individual diversity (alpha diversity), making them ideal subjects for high-resolution (species or strain-level) community genomics approaches [17,18]. Continued study of microbial colonization in the gut of premature infants may yield further insights into the details of this process and the implications of disease-associated microbial dysbiosis.
Community genomics, the use of genomes sequenced from natural microbial communities to understand the structure and metabolism of the community, has been successful in environments with varying levels of diversity [17-24]. Recently, this approach has been applied to the human microbiome, where the genomes of abundant bacterial species were assembled from a premature infant [17] and, most recently, where increased sequencing depth allowed for genomes to be assembled for both high-abundance and low-abundance members of the microbial community found in the gut of another premature infant (including genomes for members that make up less than 0.05% of the microbial community) [18]. Both of these studies involved analysis of strain-level variation within the human gut microbiome. In human adults, a draft genome of Shiga-Toxigenic Escherichia coli O104:H4 was assembled from metagenome data taken from individuals involved in an outbreak, providing strain-level resolution of this pathogen [25]. Strain-level analysis of microbial communities contrasts strongly with 16S rRNA-based fingerprinting methods that characterize communities with phylum to genus-level resolution. This is primarily due to the added benefit of being able to directly determine the metabolic potential of strains in a particular community (which need not have been previously studied), and to identify metabolic variation between strains that may have highly similar or even identical 16S rRNA gene sequences [26]. In general, the study of infants enables development of an understanding of microbial colonization in humans, and can provide genomes for biologically and medically relevant, and oftentimes novel, species directly from their source environments (without the bias of isolation or cell sorting, or single cell manipulation and genome amplification steps).
Here, we investigate gut colonization in a relatively healthy premature infant during the third week of life with the objectives of comparing genomic novelty between the natural consortia and isolate strains, recovery and analysis of genomes from previously uncharacterized community members, and metabolic analysis of the microbial community. This period of early gut colonization was targeted for intensive sample collection because it is believed that aberrant colonization near this time can contribute to the pathogenesis of necrotizing enterocolitis (which was not observed in this infant). Our approach involves reconstructing complete and near-complete genomes from DNA extracted from fecal samples to enable prediction of the roles of specific species and strains in the community. Time series abundance analysis is a key component of the approach because shifts in community composition can be detected, and also because organism abundance patterns greatly increase the accuracy with which assembled fragments can be assigned to specific organisms (binning; [18]). We show that, even in the human gut, where many species can be represented by reference genomes, there are organisms with genomic potential not represented by reference sequences. Specifically, we report the first genome for the genus Varibaculum, a genus that has been implicated in human abscess formation, but that has not been associated with the human gut [27].
Methods
Patient, samples, and sequencing
We studied the colonization of the gut of a male (birth weight 1,205 g) born via Caesarean section during the 31st week of pregnancy to a mother with chronic hypertension and superimposed pre-eclampsia. The patient was born at Comer Children’s Hospital at the University of Chicago. He was administered total parenteral nutrition (TPN) soon after admission, but started bolus nasogastric feeding on his second day of life (DOL). The patient was weaned from TPN as he began increasing feeds with fortified breast milk. TPN was discontinued on DOL 6. The patient reached full feeds on DOL 8, continuing to be fed on fortified breast milk. The patient was never intubated but did briefly receive supplemental oxygen. He received antibiotics (ampicillin and gentamicin) only during the first 48 hours of life. Stool samples were collected on the following days of life: 14, 15, 18, 19, and 20. Samples were collected twice daily, except for DOL 14, and stored at −80°C. The patient was discharged in good health on DOL 53.
Microbial DNA was extracted from frozen fecal samples using the QIAamp DNA Stool mini-Kit (Qiagen) with modifications [28]. DNA was sequenced on an Illumina HiSeq2000 sequencer for 101 cycles from each DNA fragment end using the TruSeq SBS sequencing kit (version 2). Sequencing data were handled with pipeline 1.7 according to the manufacturer’s instructions (Illumina, San Diego, CA). The protocol for sample collection and processing was approved by the Institutional Review Board of The University of Chicago (IRB #15895A). All samples were collected with the consent of the infant’s mother.
Metagenome assembly, binning, and annotation
Environmental shotgun DNA sequences for all samples were processed and assembled as previously described [18]. Sequences were quality trimmed and human DNA was filtered out prior to assembly. Sequences from all samples were co-assembled in a multistep, iterative approach, in which optimal parameters (coverage and k-mer length) for assembly of genomes from specific populations or groups of populations were selected. Velvet [29] was used to assemble the data and the resulting assembly was subjected to quality controls that detect mis-assemblies based on regions of zero insert coverage.
All scaffolds longer than 400 bp were annotated by first predicting open reading frames (ORFs) using the metagenome implementation of Prodigal [30] and then searching translated ORFs against the UniProt UniRef90 database [31] using USEARCH [32] with an E-value threshold of 0.001. The coverage of each scaffold was determined by mapping reads using BowTie [33] with the parameters -best and -e 200. Coverage was calculated as the total number of sequence bases mapped to a scaffold divided by the length of the scaffold.
Clustering of scaffolds into genome bins was conducted as previously described [18]. Specifically, the Databionic implementation of an emergent self-organizing map (ESOM; [34]) was used to cluster scaffolds longer than 400 base pairs (bp) based on their time series abundance patterns (after first breaking scaffolds into 1.5 kbp fragments) (Additional file 1). This allowed us to bin scaffolds into near-complete and partial genomes. Genome bins were assessed in part by coloring fragments clustered on the ESOM based on the best BLAST [35] hit of each scaffold against the NCBI NT database [36] (Additional file 2: Figure S1). Bins were manually extracted by contouring fields on the ESOM. Genome completeness was determined by comparing the length of each putative genome with the most closely related reference genome and by searching for 26 universal single copy marker genes (Additional file 3 and [37]).
Manual curation of microbial genome bins
Each genome bin was evaluated based on its size, coverage, and the presence of single copy genes. Single copy genes were used to estimate genome completeness and to determine whether multiple genomes were being clustered into a single bin. Incomplete bins may be the result of several factors: (i) low coverage can prevent an entire genome from being assembled; (ii) sequence variation can cause genomic regions unique to strains to assemble separately, sometimes generating very small contigs that cannot be binned; (iii) strain-specific genomic regions will be binned separately from shared regions if the time series abundance patterns of the strains differ; and (iv) inherent noise in coverage calculations can result in scaffolds representing a genome being placed in separate bins. Bins with similar taxonomic affiliations were evaluated to determine whether their scaffolds were split into different bins owing to strain variation or noise in coverage calculations. Such bins were subsequently combined into a single genome bin if they did not contain redundant single copy genes. Other bins with similar taxonomic affiliations but with overall different time series abundance patterns were considered to be the result of strain variation.
The genome assembled and binned for V. cambriense was reassembled and manually curated. This involved analyzing the read mapping for the entire metagenome assembly and capturing the reads (along with their pairs) that mapped to scaffolds in the V. cambriense bin. These reads were then used in several Velvet assemblies, in which the parameters for k-mer length and expected coverage were altered. Genome size was estimated based on the size of the bin and used to evaluate the Velvet assemblies. The best assembly based on expected length and N50 was checked for mis-assemblies (scaffolds were split at regions with zero insert coverage) and then manually curated.
The genome for V. cambriense was manually curated using a suite of in-house scripts designed to extend scaffolds by recruiting paired-reads that extend from existing scaffolds. These paired-reads were assembled independently using Velvet. Their resulting contigs were compared with existing scaffolds, based on their sequence similarity determined by BLAST. Regions of high similarity between the newly assembled contigs and existing scaffolds oftentimes reveal scaffolds that could be combined with one another [18].
Open reading frames for the manually curated V. cambriense genome were annotated using an in-house pipeline that includes BLAST-based homology searches against the NCBI NR [36], KEGG [38], UniRef90, and COG [39] databases, in addition to HMM-based functional domain recognition searches using InterProScan [40]. Metabolic analysis of the functional predictions for the V. cambriense genome was completed using Pathway Tools [41] and KEGG [42] (Additional file 4 and Additional file 5).
Plasmid and phage genomes
Plasmid genomes were identified by searching for potentially circular scaffolds by computing the Needleman-Wunsch [43] global alignment for the first and last 100 bp of the scaffold. Scaffolds with high overlap identity were further analyzed by searching those scaffolds for genes indicative of plasmids (such as plasmid replication and maintenance proteins). Putative phage fragments were identified by searching for phage-related genes (such as the capsid or tail fibers) on unbinned scaffolds and on scaffolds binned along with a genome. Phage scaffolds binned with bacterial genomes have significantly higher coverage than the bacterial-associated scaffolds.
Plasmids were associated with individual species in the community by comparing the abundance patterns of each plasmid with each species and by leveraging plasmid phylogenetic annotations. To narrow down the list of possible host organisms for each plasmid, Pearson correlation coefficients were calculated on the abundance pattern of each plasmid compared with each bacterial species. Putative phage fragments were associated with species based on initial ESOM binning, searching for integration sites in reconstructed genomes, and by comparing the relative abundance patterns of phage with bacterial species (assisted by the Pearson correlation coefficient).
Plasmid novelty and diversity were determined by building a phylogenetic tree of plasmid replication proteins. Sequences representative of closely related plasmid replication proteins were acquired by searching the NCBI NR database using BLAST. The amino acid sequences were aligned using MUSCLE [44] and a phylogenetic tree was reconstructed using FastTree2 [45] with the Jones-Taylor-Thornton model of amino acid evolution and by assuming a single rate of evolution for each site (known as the “CAT” approximation) [46]. Local support values were calculated with the Shimodaira-Hasegawa test [47] and the tree was formatted using FigTree [48].
Coverage and abundance calculations
Coverage was calculated for each scaffold based on mapped reads. Absolute abundance, average coverage, and relative abundance were calculated for reconstructed genomes at each time point, in order to represent changes in microbial community structure (Additional file 2: Tables S2-5). Genome coverage was calculated as the number of bases mapped to the genome divided by the total length of the genome. Relative abundance was calculated for a genome by taking the average coverage of the genome and normalizing it by the sum of the average coverage values for all genomes. Thus, relative abundance is the abundance of a genome taken as a percent of the total abundance of all genomes. Absolute abundance was calculated for each genome by dividing the total number of sequence bases that mapped to the genome by the total number of bases associated with reads used in the assembly. Rank abundance was calculated from the relative abundance of each genome from the combined read mapping of the time points in each phase of community colonization (phases were defined after observing community abundance patterns across the time series). Plots of relative abundance were created using the R plot function [49].
Microbial community composition based on EMIRGE 16S rRNA genes
EMIRGE [50] was used to reconstruct 16S rRNA gene sequences from the metagenomic data (Additional file 6). The closest relatives of each sequence were found by searching a TaxCollector [51] version of the Ribosomal Database Project (RDP) database [52] and the GreenGenes [53] database using BLAST. Reconstructed 16S rRNA gene sequences were connected with genomes based on several criteria. First, paired-end sequences were used to link scaffolds carrying fragments of 16S rRNA gene sequences to genome scaffolds. Then, coverage and taxonomic information, from both marker genes and for the genome overall, were used to refine associations when paired-read connections were inconclusive. More 16S rRNA gene sequences were reconstructed by EMIRGE than could be represented by genome bins; thus, a subset of low-abundance 16S rRNA gene sequences were assumed to be either incorrectly reconstructed or from very rare community members, and thus were disregarded during further analyses.
Reconstructed 16S rRNA gene sequences were aligned along with sequences from their closest relatives and additional species previously reported to be in the infant gut. Sequences were aligned with PyNAST [54] using the GreenGenes [53] alignment of operational taxonomic units (OTUs) classified at 97% sequence similarity as a template. FastTree2 was used to construct the phylogenetic tree using the generalized time-reversible model for nucleotide evolution [55] and the CAT approximation. Local support values were calculated with the Shimodaira-Hasegawa test. The tree was rooted with the 16S rRNA gene sequence for Halobacterium salinarum and formatted using FigTree.
Comparison of genomes with reference genomes
Each complete, near-complete, and partial bacterial and plasmid genome was compared to the genome of its closest sequenced relative. Both complete and draft genomes from NCBI were used in the comparison. The most closely related bacterial genomes were determined using reconstructed 16S rRNA gene sequences, ribosomal protein L5, ribosomal protein S15, and hits to other protein sequences in UniRef90. Aligning reconstructed plasmid genomes to all available sequenced plasmid genomes identified their most closely related relatives. Once selected, each genome was compared to its reference genome. The shared amino acid identity between each reconstructed and reference genome was calculated as the average amino acid identity of reciprocal best USEARCH hits between the two genomes (putative orthologs).
Evaluating community oxygen tolerance and respiration capability
Community oxygen tolerance and respiration capacity were evaluated based on the presence of specific genes in the metagenome. To assess the oxygen utilization capacity of the community, all predicted ORFs were searched for cytochrome c oxidase, cytochrome bd oxidase, and heme-copper cytochrome oxidase genes based on assignments from UniRef90. To evaluate the potential for the community to use various terminal electron acceptors in anaerobic respiration, UniRef90 annotations were searched for the presence of fumarate, trimethylamine N-oxide (TMAO), dimethyl sulfoxide (DMSO), nitrate, nitrite, and nitric oxide reductase genes. To confirm the annotations for these genes in the V. cambriense genome, a phylogenetic tree was reconstructed with the amino acid sequences of the putative catalytic subunits for the nitrate reductase and DMSO reductase genes. The tree was reconstructed using MEGA5 [56] to produce a maximum-likelihood phylogeny calculated with 100 bootstrap replicates based on the Jones-Taylor-Thornton model of amino acid evolution. All positions containing alignment gaps and missing data were eliminated based on pairwise sequence comparisons (pairwise deletion option).
Analysis of Human Microbiome Project data
The Human Microbiome Project (HMP) [57] hosts QIIME [58] output files for the HMP 16S rRNA Clinical Production Phase I and the HMP 454 Clinical Production Pilot studies (NCBI SRA projects SRP002395 and SRP002012, respectively), which together consist of over 5,700 samples. The 16S rRNA gene variable region 3 to 5 (V35) was sequenced for all samples and the 16S variable region 1 to 3 (V13) was sequenced for a subset of 2,911 samples. The OTU abundance matrices were downloaded for each dataset (V35 and V13) and the abundance of each OTU was calculated as a percent of total reads for each sample. These tables were used to evaluate the relative abundance of Varibaculum across samples and body sites in the HMP data collected for healthy human adults.
Comparative genomics
Although there are no previously sequenced genomes for any member of the genus Varibaculum, several complete and draft genomes are available for members of the family Actinomycetaceae. These genomes, along with the genomes reconstructed from the microbial community of this premature infant, were used in a comparative analysis. Each genome was annotated by finding reciprocal best USEARCH hits between each genome and a subset of the KEGG database containing only prokaryotic protein sequences with KOs (with a minimum bit score of 40 and maximum E-value of 0.01). Metabolic functional potential was compared across genomes by identifying gene sequences associated with specific metabolic functions in each genome (see Additional file 7 for a complete list of these proteins and their associations). These findings were visualized by normalizing the number of genes identified for each function and then using the R pheatmap library to produce a heatmap clustered using the complete linkage method on a Euclidean distance matrix.
Results
Metagenome sequencing, assembly, binning, and annotation
The nine samples collected on days of life 14 through 20 from the infant in our study resulted in 35 gigabase pairs (Gbp) of paired-end Illumina DNA sequences with a length of 101 nucleotides. Filtering out human DNA and quality trimming resulted in 27.8 Gbp of Illumina reads with an average length of 93 bp. The iterative metagenome assembly method used resulted in 89.29% of high quality reads being assembled into 12,184 scaffolds longer than 400 bp (40.8 megabase pairs (Mbp), N50: 13,265 bp, longest scaffold: 608,611 bp). From the scaffolds larger than 400 bp, 46,156 ORFs were predicted (average amino acid length: 254), 94.4% of which had a match to the UniRef90 database with an E-value less than or equal to 0.001.
Scaffolds were clustered based on their time series abundance patterns using an ESOM, resulting in 25 bins (Figure 1, Additional file 2: Table S1 and Additional file 1). These bins represent complete, near-complete, and partial bacterial, plasmid, and viral genomes (Table 1 and Additional file 2: Table S1) and 85.98% of high quality sequencing reads. Six complete (circular) plasmids were assembled along with five putative phage fragments (Table 1 and Additional file 2: Table S4). Plasmid and phage fragments account for only 0.27% and 0.23% of the total sequence data, respectively; however, they account for the majority of the community in terms of relative abundance (41.7% and 16.6%; Table 1 and Additional file 2: Table S4).
Figure 1.
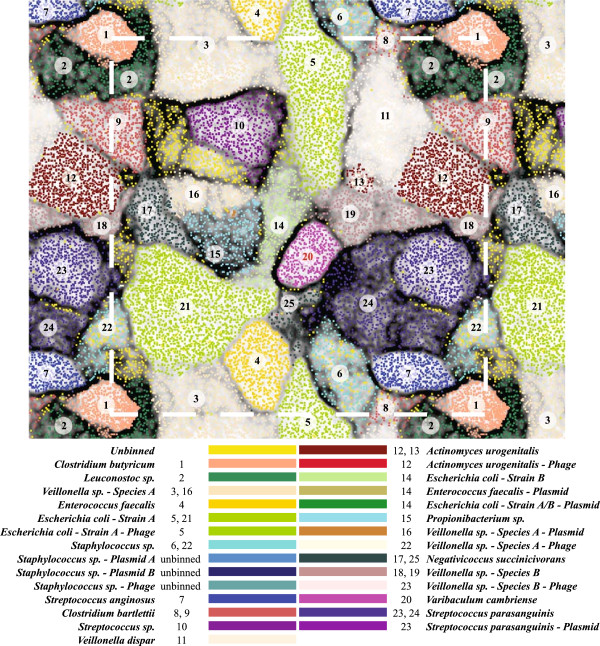
Emergent self-organizing map (ESOM) binning of the metagenome assembly. ESOM showing the clustering and binning of de novo assembled metagenomic data. Each point represents a fragment of an assembled scaffold. Clustering of data points is based on the time series abundance pattern of each assembled scaffold. Dark lines between clusters show definitive separation of genome bins. Colors designate the genome bin for each scaffold fragment.
Table 1.
Assessment of genomes reconstructed from the shotgun-sequenced microbial community: assembly, binning, phylogeny, and genome completeness
| Genome | Near-complete genome | Phylum | Bin | Genome size (bp) | Genome relative abundance (%) | N50 (bp) | Number of ORFs | Predicted% single copy genes |
|---|---|---|---|---|---|---|---|---|
|
Escherichia coli - strain A/B - plasmid |
Yes |
n/a |
14 |
3,225 |
41.67 |
3,225 |
5 |
n/a |
|
Escherichia coli - strain A - phage |
|
n/a |
5 |
3,920 |
16.57 |
3,920 |
4 |
n/a |
|
Enterococcus faecalis - plasmid |
Yes |
n/a |
14 |
5,231 |
5.98 |
5,231 |
5 |
n/a |
|
Streptococcus anginosus |
Yes |
Firmicutes |
7 |
2,108,491 |
5.14 |
537,826 |
2,252 |
100.0 |
|
Escherichia coli - strain A |
|
Proteobacteria |
5, 21 |
5,662,200 |
4.91 |
5,304 |
7,405 |
48.1 |
|
Actinomyces urogenitalis - phage |
|
n/a |
12 |
2,996 |
4.85 |
2,996 |
4 |
n/a |
|
Veillonella dispar |
Yes |
Firmicutes |
11 |
2,445,194 |
3.99 |
53,688 |
2,693 |
100.0 |
|
Veillonella sp. - species A - phage |
|
n/a |
22 |
20,453 |
3.49 |
20,453 |
32 |
n/a |
|
Veillonella sp. - species B - phage |
|
n/a |
23 |
16,533 |
2.43 |
16,533 |
30 |
n/a |
|
Actinomyces urogenitalis |
Yes |
Actinobacteria |
12, 13 |
2,604,957 |
2.28 |
3,337 |
3,035 |
92.6 |
|
Clostridium butyricum |
Yes |
Firmicutes |
1 |
4,350,784 |
2.17 |
103,127 |
4,094 |
100.0 |
|
Veillonella sp. - species A - plasmid |
Yes |
n/a |
16 |
47,631 |
1.20 |
47,631 |
56 |
n/a |
|
Veillonella sp. - species A |
|
Firmicutes |
3, 16 |
2,664,763 |
1.03 |
4,180 |
3,427 |
37.0 |
|
Enterococcus faecalis |
Yes |
Firmicutes |
4 |
2,960,721 |
0.76 |
235,714 |
2,906 |
100.0 |
|
Staphylococcus sp. - plasmid B |
Yes |
n/a |
Unbinned |
2,539 |
0.65 |
2,539 |
2 |
n/a |
|
Staphylococcus sp. - plasmid A |
Yes |
n/a |
Unbinned |
2,556 |
0.54 |
2,556 |
3 |
n/a |
|
Staphylococcus sp. - phage |
|
n/a |
Unbinned |
2,423 |
0.42 |
2,423 |
5 |
n/a |
|
Escherichia coli - strain B |
|
Proteobacteria |
14 |
633,084 |
0.37 |
4,041 |
873 |
3.7 |
|
Varibaculum cambriense
|
Yes |
Actinobacteria |
20 |
2,247,641 |
0.37 |
240,417 |
1,954 |
100.0 |
|
Streptococcus parasanguinis - plasmid |
Yes |
n/a |
23 |
8,975 |
0.31 |
8,975 |
7 |
n/a |
|
Veillonella sp. - species B |
|
Firmicutes |
18, 19 |
639,180 |
0.27 |
2,044 |
879 |
29.6 |
|
Clostridium bartlettii |
Yes |
Firmicutes |
8, 9 |
2,685,446 |
0.18 |
12,095 |
2,633 |
92.6 |
|
Negativicoccus succinicivorans |
Yes |
Negativicoccus |
17, 25 |
1,508,898 |
0.10 |
15,236 |
1,686 |
100.0 |
|
Staphylococcus sp. |
|
Firmicutes |
6, 22 |
1,509,765 |
0.09 |
9,200 |
1,634 |
33.3 |
|
Propionibacterium sp. |
|
Propionibacterium |
15 |
336,576 |
0.07 |
1,117 |
565 |
18.5 |
|
Streptococcus parasanguinis |
Yes |
Firmicutes |
23, 24 |
2,822,032 |
0.06 |
3,494 |
3,647 |
77.8 |
|
Leuconostoc sp. |
|
Firmicutes |
2 |
566,369 |
0.06 |
1,603 |
874 |
25.9 |
| Streptococcus sp. | Firmicutes | 10 | 1,915,777 | 0.05 | 11,008 | 2,114 | 55.6 |
n/a, not available.
Microbial genome identification and curation
Well-defined genomes were binned for Clostridium butyricum, Enterococcus faecalis, Streptococcus anginosus, Streptococcus sp., and Varibaculum cambriense. However, some bins were not clearly delineated owing to the low abundance of the associated species (at the limits of sequencing detection), similar abundance patterns between species, or coverage miscalculations due to strain variation. This was the case for the bins of Actinomyces urogenitalis, Clostridium bartlettii, two Escherichia coli strains, Leuconostoc sp., Negativicoccus succinicivorans, Propionibacterium sp., Staphylococcus sp., Streptococcus parasanguinis, Veillonella dispar, and two additional Veillonella species. Manual curation of these bins resulted in near-complete and partial genome reconstructions for these species, and revealed strain-resolved genomic novelty within the E. coli population (Table 1 and [59]).
We evaluated the completeness of each genome using a list of 26 single copy marker genes (Additional file 3 and [37]), revealing that the genomes for Actinomyces urogenitalis, Clostridium bartlettii, Clostridium butyricum, Enterococcus faecalis, Negativicoccus succinicivorans, Streptococcus anginosus, Streptococcus parasanguinis, Varibaculum cambriense, and Veillonella dispar (9 out of the 17 total genomes) are near-complete (over 75% of marker genes could be identified) (Table 1).
The genome for Varibaculum cambriense was reassembled and manually curated. Before reassembly, the genome was represented by 35 scaffolds with a total length of 2.25 Mbp and an N50 of 240,417 bp. Following reassembly and manual curation, the genome was assembled into three scaffolds, each terminated by a repeat sequence corresponding to a transposase gene. The three scaffolds include completely assembled 16S rRNA and 23S rRNA genes, a total length of 2.28 Mbp, an N50 of 1,648,569 bp, and 105-fold coverage. The V. cambriense genome has a GC-content of 52.5%. Approximately 70% of ORFs could be assigned to a putative function. All three scaffolds are connected to each other but their order cannot be determined; thus, all connections are resolved and we consider this genome to be essentially complete. Furthermore, all of the single copy marker genes used to assess genome completeness could be identified along with all 20 aminoacyl tRNA synthetase genes in the V. cambriense genome (Additional file 3).
The assembled E. coli plasmid has the highest copy number of any assembled plasmid, phage, or bacterial member of the community (Table 1 and Additional file 2: Table S4). The plasmid contains a replication protein distinct from other Enterobacteriaceae plasmids, suggesting that it is novel (Additional file 2: Figure S3). The largest plasmid (47.63 kbp) is associated with Veillonella sp. - species A, and also contains a novel replication protein (Additional file 2: Figure S3), indicating that this plasmid has not been previously studied. Two plasmids are related to known Staphylococcus plasmids and are highly correlated with one another (Pearson coefficient = 0.89), although they are not closely related to one another (based on their replication proteins; Additional file 2: Figure S3). Based on the annotations and abundance patterns for these plasmids, their host is the Staphylococcus sp., for which we reconstructed a near-complete genome (Pearson coefficients of 0.45 and 0.62). Additionally, a complete plasmid genome was assembled and associated with S. parasanguinis (based on protein annotations and time series abundance patterns; Pearson coefficient = 0.99).
Phylogenetic placement of EMIRGE-based 16S rRNA genes
EMIRGE reconstructed 77 candidate 16S rRNA gene sequences, 14 of which could be associated with reconstructed genomes (Table 2 and Additional file 6). The discrepancy between the number of EMIRGE sequences and genomes suggests that EMIRGE is overestimating the number of OTUs. Genes constructed by EMIRGE that could not be assigned to genomes were related to Shigella (probably represented by binned E. coli genomes), Okadaella, Buttiauxella, Brevibacterium, and Citrobacter. Of these, low-abundance ORFs from the community could be assigned to the genus Brevibacterium at greater than 90% amino acid identity, and to Citrobacter, but at less than 90% amino acid identity, suggesting the possible presence of these genera in low abundance in the community. EMIRGE sequences that were not connected with bins were not analyzed further.
Table 2.
Comparison of reconstructed genomes and 16S rRNA gene sequences with reference databases
| Genome | Genome size (bp) | Closest relative with sequenced genome | Closest relative genome size (bp) | % ORFs orthologous to reference | Average% amino acid ID of orthologs | EMIRGE 16S rRNA gene match | EMIRGE 16S% ID |
|---|---|---|---|---|---|---|---|
|
Escherichia coli - strain A/B - plasmid |
3,225 |
Salmonella enterica subsp. enterica serovar Newport str. SL254 plasmid |
684 |
20.0 |
93.9 |
n/a |
n/a |
|
Escherichia coli - strain A - phage |
3,920 |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
Enterococcus faecalis - plasmid |
5,231 |
Enterococcus faecalis 62 plasmid |
1,295 |
100.0 |
100.0 |
n/a |
n/a |
|
Streptococcus anginosus |
2,108,491 |
Streptococcus anginosus 1 2 62CV uid62163 |
1,821,055 |
67.7 |
95.2 |
S. anginosus |
99.8 |
|
Escherichia coli - strain A |
5,662,200 |
Escherichia coli S88 uid62979 |
5,032,268 |
47.7 |
97.5 |
E. coli O83:H1 str. NRG 85 |
99.9 |
|
Actinomyces urogenitalis - phage |
2,996 |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
Veillonella dispar |
2,445,194 |
Veillonella dispar ATCC 17748 |
2,118,767 |
60.0 |
91.6 |
V. dispar |
99.3 |
|
Veillonella sp. - species A - phage |
20,453 |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
Veillonella sp. - species B - phage |
16,533 |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
Actinomyces urogenitalis |
2,604,957 |
Actinomyces urogenitalis DSM 15434 |
2,702,812 |
67.1 |
98.7 |
A. urogenitalis |
99.9 |
|
Clostridium butyricum |
4,350,784 |
Clostridium butyricum 5521 |
4,540,699 |
73.5 |
96.5 |
C. butyricum |
99.3 |
|
Veillonella sp. - species A - plasmid |
47,631 |
Caldicellulosiruptor kristjanssonii 177R1B plasmid |
3,674 |
3.6 |
38.8 |
n/a |
n/a |
|
Veillonella sp. - species A |
2,664,763 |
Veillonella dispar ATCC 17748 |
2,118,767 |
47.1 |
95.6 |
Veillonella sp. oral taxon 158 |
91.2 |
|
Enterococcus faecalis |
2,960,721 |
Enterococcus faecalis OG1RF |
2,739,625 |
79.1 |
98.8 |
E. faecalis OG1RF |
98.7 |
|
Staphylococcus sp. - plasmid B |
2,539 |
Staphylococcus haemolyticus JCSC1435 plasmid |
402 |
100.0 |
99.8 |
n/a |
n/a |
|
Staphylococcus sp. - plasmid A |
2,556 |
Macrococcus caseolyticus JCSC5402 plasmid |
997 |
33.3 |
80.3 |
n/a |
n/a |
|
Staphylococcus sp. - phage |
2,423 |
n/a |
n/a |
n/a |
n/a |
n/a |
n/a |
|
Escherichia coli - strain B |
633,084 |
Escherichia coli S88 uid62979 |
5,032,268 |
52.7 |
82.3 |
n/a |
n/a |
|
Varibaculum cambriense
|
2,247,641 |
Mobiluncus mulieris ATCC 35239
|
2,533,633 |
56.0 |
53.9 |
V. cambriense
|
99.2 |
|
Streptococcus parasanguinis - plasmid |
8,975 |
Enterococcus faecium Aus0004 plasmid |
1,192 |
14.3 |
32.4 |
n/a |
n/a |
|
Veillonella sp. - species B |
639,180 |
Veillonella dispar ATCC 17748 |
2,118,767 |
62.5 |
86.7 |
V. parvula DSM 2008 |
90.3 |
|
Clostridium bartlettii |
2,685,446 |
Clostridium bartlettii DSM 16795 |
2,972,256 |
81.7 |
98.1 |
Clostridium sp. MDA2315 |
99.4 |
|
Negativicoccus succinicivorans |
1,508,898 |
Bacillus coagulans 36D1 |
3,552,226 |
45.0 |
43.4 |
N. succinicivorans |
98.0 |
|
Staphylococcus sp. |
1,509,765 |
Staphylococcus epidermidis ATCC 12228 |
2,499,279 |
78.3 |
98.4 |
S. epidermidis ATCC 12228 |
96.2 |
|
Propionibacterium sp. |
336,576 |
Propionibacterium 5 U 42AFAA |
2,532,807 |
46.4 |
82.4 |
Propionibacterium sp. H456 |
99.4 |
|
Streptococcus parasanguinis |
2,822,032 |
Streptococcus parasanguinis ATCC 15912 |
2,153,652 |
45.0 |
94.3 |
S. parasanguinis |
98.9 |
|
Leuconostoc sp. |
566,369 |
Leuconostoc citreum KM20 |
1,796,284 |
66.7 |
96.0 |
n/a |
n/a |
| Streptococcus sp. | 1,915,777 | Streptococcus M334 | 2,207,013 | 67.6 | 93.9 | n/a | n/a |
Reconstructed genomes were compared with the genomes of isolate strains. Reconstructed 16S rRNA genes were searched against sequences in the RDP and GreenGenes databases to aid classification. n/a, not available.
The 16S rRNA gene sequences reconstructed with EMIRGE were used to build a phylogenetic tree to classify organisms in the community (Additional file 2: Figure S2). The tree shows that many of the reconstructed genomes are very closely related to sequenced genomes, based on their 16S rRNA gene sequences. The tree highlights the lack of reference genomes for Varibaculum, although there are numerous 16S rRNA gene sequences from isolates and from clone libraries. Phylogenetic placement of the EMIRGE sequence for bin 20 confirms that this genome is from a member of the species V. cambriense (99.2% 16S rRNA gene sequence identity).
Comparison of reconstructed genomes to reference genomes
The 25 ESOM bins represent the genomes of 17 unique organisms from the microbial community. Of the nine reconstructed near-complete genomes, seven share over 90% ortholog amino acid identity with reference strains, while five share over 95% ortholog amino acid identity (with at least 60% of ORFs being defined as orthologs) (Table 2). Despite this high-level of similarity, 14% of the ORFs predicted for near-complete reconstructed genomes do not have orthologs within their most closely related reference genomes. At the extreme, 57% of the predicted ORFs for Negativicoccus succinicivorans are not orthologous with genes found in the most closely related reference genome (Table 2). Although the level of genomic divergence observed here does not fall outside of the range previously observed [60], this finding underscores the importance of genome reconstructions, as opposed to 16S rRNA gene sequence analysis, for inferring microbial metabolic potential. Of particular note, the genome for V. cambriense has an average ortholog amino acid sequence identity of 54% with the genome of its closest sequenced relative, Mobiluncus mulieris (Table 2).
Evidence for the importance of anaerobic metabolism and oxygen tolerance
Several genomes encode cytochrome bd oxidase (Table 3 and Figure 2), a high oxygen affinity enzyme indicative of an ability to grow in the presence of low levels of oxygen, either by providing protection from reactive oxygen species or by using oxygen as a terminal electron acceptor during respiration [61,62]. The presence of fumarate, TMAO, DMSO, nitrate, nitrite, and nitric oxide reductase genes supports the notion that members of the community are capable of using several terminal electron acceptors to respire anaerobically (Table 3, Figure 2, and Additional file 2: Figure S4). E. coli was the only organism found to encode heme-copper cytochrome oxidase genes, indicating its ability to use oxygen as an electron acceptor when present. To further assess the oxygen utilization capacity of the community, all binned and unbinned ORFs were searched for cytochrome c oxidase genes, which would indicate aerobic, or possibly aero-tolerant metabolism [62], but none were found. Taken together, we conclude that all organisms in the community are either obligate or facultative anaerobes (Table 3 and Figure 2).
Table 3.
Metabolism of bacterial members of the microbial community
| Genome | Predicted oxygen requirement | NADH: quinone oxidoreductase | Cytochrome bd complex | Fumarate reductase | TMAO reductase | DMSO reductase | Nitrate reductase | Nitrite reductase | Nitric oxide reductase |
|---|---|---|---|---|---|---|---|---|---|
|
Streptococcus anginosus |
Obligate anaerobe |
|
|
|
|
|
|
|
|
|
Escherichia coli - strain A |
Facultative anaerobe |
* |
* |
* |
* |
* |
* |
* |
* |
|
Veillonella dispar |
Obligate anaerobe |
|
* |
* |
|
|
* |
* |
* |
|
Actinomyces urogenitalis |
Facultative anaerobe |
* |
* |
* |
|
* |
* |
* |
|
|
Clostridium butyricum |
Obligate anaerobe |
|
|
|
|
* |
* |
* |
|
|
Veillonella sp. - species A |
Obligate anaerobe |
|
* |
* |
|
|
* |
* |
* |
|
Enterococcus faecalis |
Facultative anaerobe |
|
* |
|
|
|
|
|
|
|
Escherichia coli - strain B |
Facultative anaerobe |
|
|
|
|
|
* |
|
* |
|
Varibaculum cambriense
|
Facultative anaerobe |
* |
* |
* |
|
* |
* |
|
|
|
Veillonella sp. - species B |
Obligate anaerobe |
|
|
|
|
|
* |
|
* |
|
Clostridium bartlettii |
Obligate anaerobe |
|
|
|
|
|
|
|
|
|
Negativicoccus succinicivorans |
Obligate anaerobe |
|
|
|
|
|
* |
|
|
|
Staphylococcus sp. |
Facultative anaerobe |
|
* |
|
|
|
* |
* |
* |
|
Propionibacterium sp. |
Facultative anaerobe |
* |
|
* |
|
|
|
* |
|
|
Streptococcus parasanguinis |
Obligate anaerobe |
|
|
|
|
|
|
|
* |
|
Leuconostoc sp. |
Facultative anaerobe |
|
* |
|
|
|
|
|
|
| Streptococcus sp. | Facultative anaerobe |
Presence (*) and absence of components required for anaerobic respiration and the predicted oxygen requirement of each member of the bacterial community.
Figure 2.
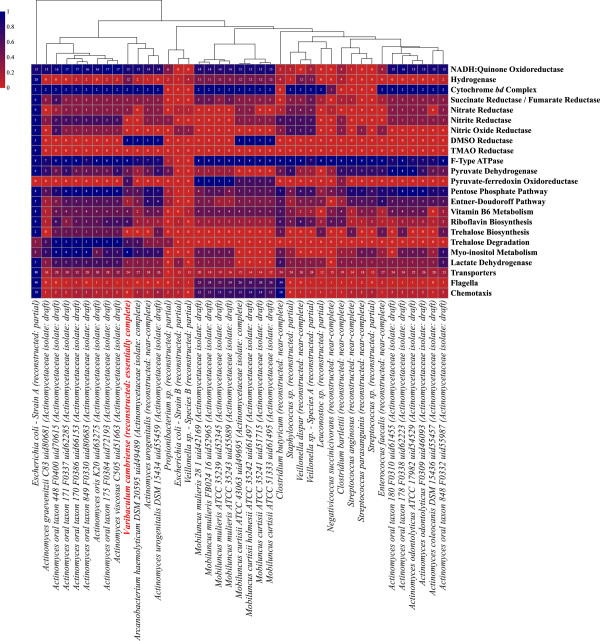
Metabolic analysis of reconstructed community and isolate genomes. Genomes reconstructed from the microbial community were compared with each other and with the genomes of cultured isolates previously sequenced for members of the family Actinomycetaceae. Each genome was annotated with KEGG and the genes that matched specific metabolic features were counted (Additional file 7). The number of genes identified for each group was normalized across genomes to facilitate coloring and clustering. The number of genes identified for each feature in each genome is presented on the heatmap.
Microbial abundance and community shifts
Escherichia coli - strain A accounts for the greatest percentage of high quality sequence data and Propionibacterium sp. accounts for the least amount (33.45% and 0.03%, respectively; Additional file 2: Table S2). Species abundance patterns over the third week of life defined two phases of microbial community composition (Figures 3 and 4). The first phase is observed during DOL 14 to DOL 15, whereas the second covers DOL 18 to DOL 20. The first phase is defined by a dominant E. coli strain (a facultative anaerobe), and the second phase is dominated by obligate anaerobes (Streptococcus anginosus, Clostridium butyricum, and Veillonella dispar). Early in the second phase (first time point on DOL 18) there is a significant increase in the relative abundance of Streptococcus anginosus, followed by a stable abundance afterwards. This is followed by a spike in the abundance of Clostridium butyricum observed on the second time point taken on DOL 18, after which the relative abundance immediately decreases. There is no apparent clinical variable (for example, change in diet or medication) that accounts for the shift between phase one and phase two. The distinct difference in community composition between theses phases corresponds with a shift towards fermentation-based metabolism in the successors of initially dominant E. coli.
Figure 3.
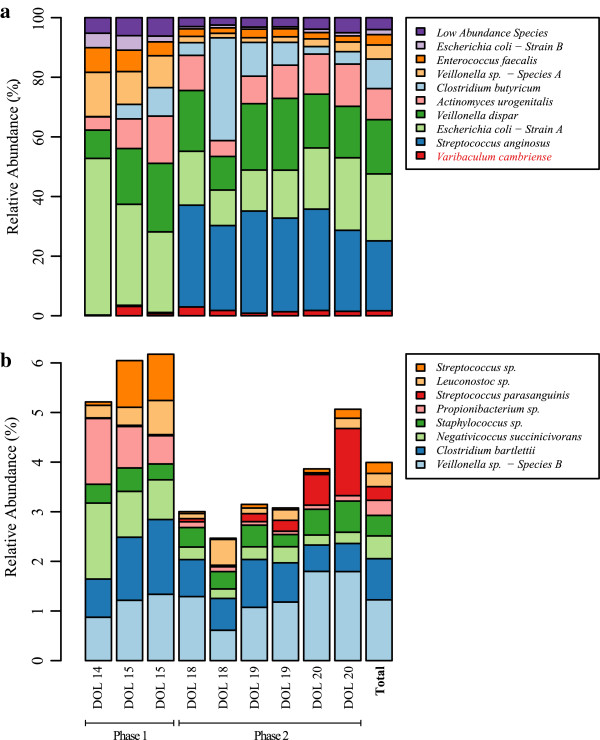
Relative abundance of bacterial species over time. Relative abundances were calculated for bacterial species at nine different time points during the third week of life of a premature male infant. (a) Shows dominant taxa and (b) shows low-abundance species across the time series. During this period, the colonization process is defined by two distinct phases based on the dominance of either facultative (phase 1) or obligate (phase 2) anaerobes.
Figure 4.
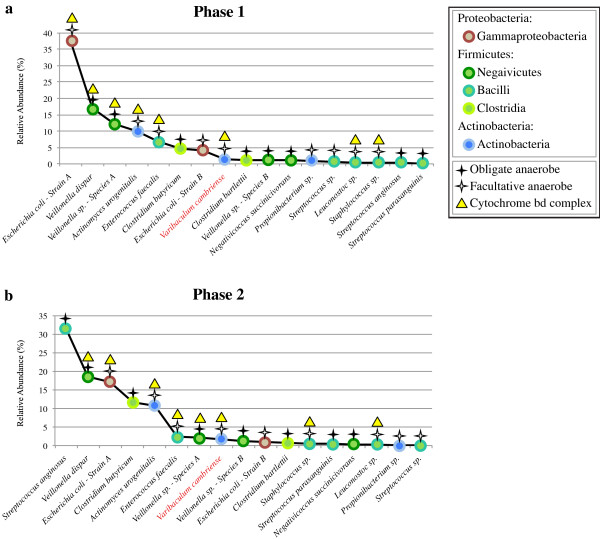
Rank abundance of bacterial species during phases of colonization. Rank abundance was determined from the relative abundance of each species during each phase of microbial colonization. Taxonomic identification and metabolic analysis was completed based on genome reconstructions from the shotgun-sequenced microbial community. The colonization process is broken into two distinct phases defined by the dominance of either (a) facultative anaerobes during phase one or (b) obligate anaerobes during phase two.
V. cambriense is nearly undetectable during the first time point (0.15%) and remains at a low abundance throughout the time series (always ≤3%).
It is interesting to note that Streptococcus, Escherichia, Veillonella, Actinomyces, and Enterococcus dominate the microbial community and that, similar to observations in other premature infants, no Bacteroides, Bifidobacterium, or Lactobacillus were observed throughout the time series [17,18,63].
Metabolism of V. cambriense based on genomic analysis
V. cambriense (strain Dora) (Table 3) became the focus of further analysis for several reasons, including (i) the lack of reference genomes available for Varibaculum (Table 2 and Additional file 2: Figure S2), (ii) the availability of a new, essentially complete genome (Table 1), and (iii) the fact that members of this genus are medically relevant and drastically understudied in the human gut [27,64,65]. Further, Varibaculum have never been studied in the human gut at a species (or genome) level of resolution. Finally, there is some metabolic information in Bergey’s Manual of Systematic Bacteriology for cultured strains of V. cambriense[66].
V. cambriense cell wall and motility
Genome analysis shows that genes involved in the lipopolysaccharide biosynthesis pathway are missing, confirming that V. cambriense does not have a Gram-negative cell envelope. The peptidoglycan biosynthesis pathway containing meso-diaminopimelate is complete. However, the β-lactam resistance pathway for peptidoglycan synthesis is incomplete, suggesting sensitivity to β-lactam antibiotics. No genes for flagella, pili, or chemotaxis were identified, indicating that this strain, like cultured members of this species, is not motile.
V. cambriense transporters and resistance
Twenty different sugar transport ORFs were identified, indicating that V. cambriense has the ability to use many different types of sugars. A putative sialic acid transporter is present, along with genes required for metabolizing N-acetylneuraminate (discussed later), suggesting that this abundant component of both human and non-human cell-surface glycoproteins, human breast milk glycans, and intestinal mucins can be used as a nutrient. Additionally, a glycerol-3-phosphate transporter, a putative phosphotransferase IIA system, a sodium-galactoside symporter, and multiple sugar transport system permease genes were identified. No acetate transporter was found, although several pathways were identified for converting pyruvate into acetate for the bidirectional conversion between acetyl-CoA and acetate. Complete KEGG modules suggest that V. cambriense can transport ribose, phosphate, nickel, lipopolysaccharides, and fructose.
Although no complete antibiotic resistance pathways were identified, a drug resistance transporter (EmrB/QacA subfamily) and a methicillin resistance protein are coded for in the genome. V. cambriense has various resistance mechanisms, including an arsenate resistance pathway, the pathway for glycine betaine biosynthesis (a compound capable of protecting against osmotic stress; [67]), and a P-type ATPase for translocating copper and silver (suggesting Cu2+ tolerance).
The genome contains the enzyme trehalose synthase, which is necessary for trehalose synthesis from β-maltose. The enzymes for synthesizing trehalose from glycogen were also identified, but not the enzymes for degrading trehalose. Trehalose has several biological roles, including that of structural component [68] and stress protector [69]. The pathway for ppGpp is also encoded by the genome; this pathway is known for its role in regulating responses to nutrient or energy starvation and environmental stresses [70].
V. cambriense nutrient sources
Based on the genome, V. cambriense is able to use acetoacetate, ammonia, arabinose, ethanol, fructose, glucose, glycerol, glycogen, lactose, mannose, melibiose, ribose, sialic acid, starch, sucrose, and xylose as nutrient sources. It is interesting to note that cultured representatives of V. cambriense have not been shown to use either starch or xylose [66]. Fructose, glucose, glycerol, glycogen, lactose, mannose, melibiose, starch, sucrose, and xylose can all be directed to glycolysis, for which the complete pathway was identified. Unlike other members of the community, including A. urogenitalis (the other Actinomycetaceae), V. cambriense does not have the enzymes for the Entner-Doudoroff pathway (Figure 2).
The genome encodes several neuraminidases (also known as sialidases), suggesting that V. cambriense is able to cleave various sialic acid species from host-derived substrates. Sialic acids are found terminally bound to cell-surface glycoproteins, human breast milk glycans, and mucins, but are only accessible by microbes once they have been cleaved from their substrate [71-73]. The genome also contains a sialic acid transporter and the enzymes necessary for converting the predominant form of sialic acid found in humans, N-acetylneuraminate, to d-fructose-6-phosphate, which can in turn be fed into glycolysis. Thus, unlike many bacterial species, V. cambriense is probably able both to liberate sialic acids and to make use of them as a nutrient source. However, the genes for the pathway that converts N-acetylneuraminate to CMP-N-acetylneuraminate are not present, making it unlikely that V. cambriense can coat its outer membrane with sialic acids as other species do to evade the host immune system [74].
The pathway for degrading N-acetylglucosamine, a derivative of glucose that is found in chitin, fungal, and prokaryote cell walls, was identified. A near-complete pathway was identified for converting myo-inositol to dihydroxyacetone phosphate, acetyl-CoA, and CO2 (while reducing two NAD+ to NADH). However, no pathways for butyrate or cellulose metabolism could be found, nor genes involved in mucin protein degradation, nor evidence for CO2 fixation (no evidence could be found for the presence of pyruvate formate-lyase).
V. cambriense pentose phosphate pathway
The V. cambriense genome is missing both glucose-6-phosphate dehydrogenase and 6-phosphogluconolactonase, primary components of the oxidative branch of the pentose phosphate pathway. However, as is expected for a facultative anaerobe, the non-oxidative pathway is complete. The presence of a gluconate transporter and ribose transport system suggests that the pentose phosphate pathway could use these precursors to produce D-glyceraldehyde-3-phosphate, which in turn could enter the methylerythritol phosphate pathway and create isopentenyl diphosphate and dimethylallyl diphosphate (fundamental units of isoprenoid biosynthesis), and geranyl diphosphate (a crucial precursor of menaquinone biosynthesis). Likewise, d-fructose-6-phosphate produced in the pentose phosphate pathway can be fed into glycolysis or can participate in the synthesis of UDP-N-acetyl-d-glucosamine, a necessary precursor of cell wall peptidoglycan. Another branch from d-erythrose-4-phosphate in the pentose phosphate pathway could lead to the biosynthesis of chorismate, an important biochemical intermediate. Overall, these pathways indicate the sources of several key metabolic precursors.
V. cambriense fermentation and degradation reactions
The presence of lactate dehydrogenase suggests that pyruvate produced from glycolysis can be fermented to lactate; however, consistent with isolate metabolic data, no pathways were found to consume lactate [66]. We identified that the α, β, and γ subunits of pyruvate-ferredoxin oxidoreductase (EC: 1.2.7.1) are successively encoded on the V. cambriense genome (the δ subunit could not be identified). This enzyme uses an oxidized ferredoxin to ferment pyruvate and produce H+, CO2, and acetyl-CoA, which can subsequently be converted into either acetate or ethanol. Although the directionality of ethanol interconversion is difficult to infer from protein sequences alone, it is possible that the reverse of the ethanol fermentation reaction can occur without additional enzymes, resulting in the formation of acetyl-CoA from ethanol with a gain of two NADH. Additionally, the pathway for acetoacetate degradation through the intermediate acetoacetyl-CoA, which has a net yield of one molecule of acetyl-CoA, also exists. In E. coli, acetoacetate can function as a total source of carbon and energy through this pathway [75], and this may be the case for V. cambriense.
V. cambriense tricarboxylic acid cycle
There is strong evidence for a tricarboxylic acid (TCA) cycle and respiratory capacity in the V. cambriense genome. Two of the three components of the pyruvate dehydrogenase complex are encoded by the genome (the E1 component could not be identified). If functional, V. cambriense could convert pyruvate into acetyl-CoA (which could be used in the TCA cycle) using either this enzyme or pyruvate-ferredoxin oxidoreductase. The genome encodes the enzymes for converting acetyl-CoA into succinate. The TCA cycle can then continue by converting succinate into fumarate using succinate dehydrogenase/fumarate reductase (EC: 1.3.99.1). Four subunits are required for this enzyme, but only three were identified (the iron-sulfur subunit, flavoprotein subunit, and cytochrome b556 subunit are co-localized on the genome). No evidence could be found for the membrane anchor subunit, which may be due to the presence of a small scaffolding gap in this region of the genome, or may indicate the existence of a divergent form of this enzyme. The presence of fumarate lyase provides a way for fumarate to be converted into malate, thus continuing the cycle. The form of malate dehydrogenase that converts malate into oxaloacetate by reducing NAD+ to NADH and H+ is present, but not the form of the enzyme that uses a quinone. Taken together, V. cambriense encodes a complete TCA cycle.
V. cambriense anaerobic respiration
Several components of an anaerobic respiratory chain were identified. The large and small subunits of the hydrogenase enzyme (containing iron-sulfur clusters, EC: 1.12.99.6) and all 14 subunits of NADH dehydrogenase (EC: 1.6.5.3) were identified, indicating that both hydrogen acquired from the environment and NADH produced by glycolysis, the TCA cycle, and substrate degradation reactions (ethanol degradation, for example) can be used as electron donors during anaerobic respiration.
Identification of fumarate reductase (EC: 1.3.99.1), nitrate reductase (EC: 1.7.99.4), and dimethyl sulfoxide (DMSO) reductase (EC: 1.8.5.3), suggests that fumarate, nitrate, and DMSO can all be used as terminal electron acceptors. Phylogenetic analysis of the nitrate reductase and DMSO reductase catalytic subunits supports the functional roles of these genes (Additional file 2: Figure S4). Furthermore, owing to the presence of a TAT signal sequence in the V. cambriense nitrate reductase, the active site is located on the outside of the cytoplasmic membrane, as is common in Archaea [76]. Also, the five signature residues suggested as being involved in nitrite and nitrate binding are conserved [76]. We also identified genes encoding a nitrate-nitrite transporter, but no other reactions that produce or consume nitrate or nitrite (the organism does not fix nitrogen, for example). No reactions for forming DMSO, or transporters for DMSO could be identified. The nitrite resulting from nitrate reduction could be further reduced by several other community members, several of which have the capacity to further reduce nitric oxide to nitrous oxide (Escherichia coli - strain A, Staphylococcus sp., Veillonella dispar, and Veillonella sp. - species A), although no species is predicted to be able to reduce nitrous oxide (Table 3).
As is common in gram-positive bacteria, V. cambriense does not have the genes required for the formation of ubiquinones. However, a near-complete pathway is present for the biosynthesis of menaquinones, electron mediators essential during fumarate, DMSO, and nitrate reduction [77]. All subunits of the F-type H+-transporting ATPase were identified, indicating that V. cambriense is able to produce ATP from the generated proton gradient.
As noted, V. cambriense is not capable of aerobic respiration. However, both subunits of the cytochrome bd complex were identified. This cytochrome along with cysteine synthase and superoxide dismutase (also identified) can protect against oxidative stress and contribute to limited oxygen tolerance [61,78]. Consistent with cultured strains, no evidence was found for catalase production [27,66].
Other metabolic pathways found in V. cambriense
The genome contains complete amino acid biosynthesis and degradation pathways and all 20 aminoacyl tRNA synthetase genes. Complete pathways were found for riboflavin (vitamin B2) and vitamin K2 biosynthesis. The pathway for the synthesis of folate (vitamin B6) is incomplete; however, some crucial and unique enzymes to the pathway were identified, suggesting that this organism may also be able to synthesize this vitamin (approximately 30% of ORFs do not have a predicted function and could be responsible for this and other pathways). The combination of these functions indicates that V. cambriense may exist symbiotically with its human host under certain conditions.
Abundance of Varibaculum in the healthy adult human microbiota
We searched data from the HMP, surveying the V35 region of the 16S rRNA gene in order to assess the abundance and distribution of Varibaculum in the human microbiota (Additional file 2: Figure S5) [57]. Out of the 5,000 samples taken from 235 healthy human subjects, only 90 had hits for Varibaculum (0% oral, 0.31% stool, 1.42% nasal, 3.92% skin, and 10.56% vaginal samples encompassing 24.68% of subjects). In only 29 of these (from 25 different individuals) was the relative abundance of Varibaculum greater than 0.05%, while this genus never represented more than 2.5% of any sample. On average, the most abundant organism in communities studied by the HMP represent 14.42% of the total community (standard deviation of 14.11%), and in communities with Varibaculum, the most abundant organism represented 20.48% (standard deviation 10.96%). Varibaculum was most abundant in samples from the antecubital fossa (skin) and the vagina. Only one stool sample had hits for Varibaculum, where it represented only 0.02% relative abundance. Although Varibaculum is not uncommon, it is never a dominant community member in the large, healthy, adult population surveyed by the HMP.
Comparative genomics of V. cambriense
Comparing the genome for V. cambriense with available genomes for members of the family Actinomycetaceae revealed few unique genes, most of which are phage-associated or are not annotated. Several of these unique genes corresponded with folate, butanoate, and benzoate metabolism (among other pathways), but no complete pathways could be established from this set. However, there is considerable metabolic variation within the Actinomycetaceae (Figure 2). Only members of the genus Mobiluncus are motile, encoding genes for both flagella and chemotaxis. Nitrate reductase is common in the family, but not encoded by Actinomyces coleocanis, Actinomyces graevenitzii, Arcanobacterium haemolyticum, some strains of Mobiluncus curtisii, nor Mobiluncus mulieris, while nitrite reductase and nitric oxide reductase are found only in the Actinomyces. DMSO reductase is found only in Mobiluncus curtisii, Actinomyces urogenitalis, Arcanobacterium haemolyticum, and V. cambriense. Taken together, the Actinomycetaceae rely on several different terminal electron acceptors for anaerobic respiration.
Although genes were identified for riboflavin biosynthesis in V. cambriense, these genes are not common in the Actinomycetaceae. Several species of Actinomyces and members of the microbial community of the premature infant in our study (but not the A. urogenitalis genome reconstructed from the community) encode the genes for trehalose biosynthesis, suggesting a possible interrelationship between these species and V. cambriense, which only encodes the genes for trehalose degradation (Figure 2). In the Actinomycetaceae, pyruvate-ferredoxin oxidoreductase, mentioned previously for its importance in converting pyruvate into acetyl-CoA, is found only in V. cambriense and members of Mobiluncus. Neuraminidases, which are required for cleaving sialic acids from glycoproteins, human breast milk glycans, and intestinal mucins, are distributed throughout the Actinomycetaceae. However, most members of the family are missing transporters for sialic acids, although the presence of the enzymes for their degradation suggests that a currently uncharacterized transporter exists for these species (Additional file 5). Members of the Actinomycetaceae and the microbial community in the gut of this infant engage in diverse metabolisms. Clustering based on select metabolic characters shows that V. cambriense is more metabolically similar to other Actinomycetaceae than to other members identified in the gut of this infant, despite significant metabolic overlap among community members (Figure 2).
Discussion
Genome reconstructions facilitated prediction of the metabolic roles of individual bacterial members in the context of their community. Applied to the gut microbiome of a premature male infant, the time series abundance information also provided by this method revealed strain-specific dynamics during the third week of life. Comparison of reconstructed genomes to the genomes of isolate strains revealed genomic novelty, even among members of this relatively simple microbial community. However, overall similarities between most reconstructed and reference genomes validated the de novo genome binning strategy.
Metabolic analysis revealed a community consisting of facultative anaerobes and obligate (fermentative) anaerobes. The facultative anaerobe Escherichia coli was initially dominant in the time series, but was replaced by obligate anaerobes (Streptococcus anginosus and Clostridium butyricum) during a switch to a community dominated by fermentation-based metabolism. This shift emphasizes the instability of the infant gut in terms of both membership and metabolism, and could be the result of several factors previously observed in the human gut. For example, dominance of species from the family Enterobacteriaceae (including E. coli) has been associated with either high oxygen levels or the availability of nitrate (a natural byproduct of the host immune response) [79]. Thus, this shift could be the result of decreased availability of either nitrate or oxygen, either of which could be depleted by E. coli during respiration. Decreased inflammation could also decrease available nitrate and decrease the competitive advantage of E. coli over obligate fermenters [80]. In terms of the gut environment, succession during early life is driven by the presence of oxygen [81], and replacement of E. coli with obligate fermenters is suggestive of a decrease in oxygen; however, we cannot rule out the hypothesis that this shift in relative abundance is stochastic. Regardless of the mechanism, this represents a dramatic shift in community composition with the potential to affect host metabolism.
Although the abundance of S. anginosus and E. coli appeared to equilibrate by the end of the time series, the drop in abundance of C. butyricum after its initial spike suggests a potential competition between the two obligate anaerobes. Interestingly, S. anginosus and C. butyricum have different clinical presentations. S. anginosus is commonly observed in association with abscess formation [82], while C. butyricum is usually considered a beneficial, butyrate-producing commensal [83]. In this case, the microbe more likely to be beneficial in the gut environment is outcompeted.
To explore the potential role of a species not commonly observed or previously characterized from the human gut, we manually curated and metabolically analyzed the genome of Varibaculum cambriense (strain Dora), resulting in the first genomic sampling of a member of the genus Varibaculum. Strains of V. cambriense isolated from human cerebral and skin abscesses, intrauterine contraceptive devices, and the human vagina have been used to show that it is an anaerobic, catalase-negative, gram-positive, diphtheroid-shaped bacterium [27,66]. However, genome analysis revealed additional insight into the metabolic potential of this organism and informed which substrates V. cambriense may use for anaerobic respiration. V. cambriense is metabolically similar to common gut inhabitants, and is predicted to use various carbon sources, respire anaerobically (using fumarate, nitrate, and DMSO), and produce lactate during fermentation.
Several community members (including V. cambriense) are predicted to use myo-inositol as a nutrient source. This is interesting because myo-inositol plays a role in eukaryotic cell messaging and is found in breast milk and infant formula, although it is generally not found in solutions used for intravenous feeding [84]. Inositol has been shown to benefit premature infants with respiratory ailments [85], suggesting that microbial degradation of this compound would decrease its health benefits. In contrast, the potential for V. cambriense to produce essential vitamins suggests a beneficial contribution by this organism to its human host.
Although this infant was fed fortified breast milk (an abundant source of glycan-bound sialic acids) during the time period studied, only V. cambriense, Streptococcus sp., and Streptococcus parasanguinis, all low-abundance members of the gut community, have neuraminidases (enzymes that cleave sialic acids) and enzymes for sialic acid degradation. Although the E. coli genome does not encode a neuraminidase, it has a sialic acid transporter and degradation machinery. Thus, low-abundance community members may be making sialic acids available to E. coli. It has been shown that some pathogenic species incapable of accessing bound sialic acids are able to make use of sialic acids cleaved by commensal organisms to promote their own growth [86].
The ability of V. cambriense to degrade, but not produce, trehalose suggests a possible dependency on other members of the microbial community able to produce this disaccharide. Furthering community interrelationships, nitrite produced by V. cambriense during anaerobic respiration can by further utilized by community members capable of nitrite and nitric oxide reduction.
Conclusions
This study underlines the higher resolution insight that can be obtained using genome-centric metagenomic approaches. Strain-resolved community dynamics revealed two phases in colonization during the third week of life of a premature infant. The phases were distinguishable based on the dominance of either respiratory or fermentation-based metabolism. Comparison of V. cambriense with other members of the microbial community revealed similarities with traditional gut inhabitants, while comparisons with other members of the family Actinomycetaceae illustrate how the metabolic diversity of this family could mislead species-level functional analysis based on 16S rRNA gene sequencing. Analysis of reconstructed genomes enabled strain-specific metabolic potential to be determined for the microbial community, and suggested potential community interdependencies.
Availability of supporting data
All data have been made publically available and can be accessed through SRA [SRA: SRS470507], DDBJ/EMBL/GenBank [GenBank: AZMA00000000, AZMB00000000, AZMC00000000, AZMD00000000, AZME00000000, AZMF00000000, AZMG00000000, AZMH00000000, AZMI00000000, AZMJ00000000, AZMK00000000, AZML00000000, AZMM00000000], and ggKbase [59]. DDBJ/EMBL/GenBank versions [GenBank: AZMA01000000, AZMB01000000, AZMC01000000, AZMD01000000, AZME01000000, AZMF01000000, AZMG01000000, AZMH01000000, AZMI01000000, AZMJ01000000, AZMK01000000, AZML01000000, AZMM01000000] are described here.
Abbreviations
BLAST: Basic local alignment search tool; DMSO: Dimethyl sulfoxide; DOL: Day of life; ESOM: Emergent self-organizing map; HMP: Human Microbiome Project; ORF: Open reading frame; OTU: Operational taxonomic unit; RDP: Ribosomal database project; TCA: Tricarboxylic acid; TMAO: Trimethylamine N-oxide; TPN: Total parenteral nutrition.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CTB carried out the binning and genome curation, functional, time series abundance and phylogenetic analysis, and drafted the manuscript. IS assembled the sequence data and contributed to the bioinformatics analysis. BCT performed the functional annotation. CJC assisted with biochemical analysis. MJM oversaw sample collection and handled medical aspects of the research. MJM and JFB designed and oversaw the study and data analysis. All authors contributed to manuscript preparation. All authors read and approved the final manuscript.
Supplementary Material
ESOM data.
Supplemental figures and tables.
Single copy genes in reconstructed genomes.
V. cambriense pathway tools data.
KEGG annotations.
EMIRGE reconstructed 16S rRNA gene sequences.
Metabolic features for comparative genomics.
Contributor Information
Christopher T Brown, Email: ctb@berkeley.edu.
Itai Sharon, Email: itaish@berkeley.edu.
Brian C Thomas, Email: bcthomas@berkeley.edu.
Cindy J Castelle, Email: cjcastelle@berkeley.edu.
Michael J Morowitz, Email: michael.morowitz@chp.edu.
Jillian F Banfield, Email: jbanfield@berkeley.edu.
Acknowledgements
This work was supported by NIH Grant 1R01AI092531-01.
References
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1131. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio C-W, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh C-S. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, Casella G, Drew JC, Ilonen J, Knip M, Hyöty H, Veijola R, Simell T, Simell O, Neu J, Wasserfall CH, Schatz D, Atkinson MA, Triplett EW. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE. 2011;6:e25792. doi: 10.1371/journal.pone.0025792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Mshvildadze M, Neu J, Shuster J, Theriaque D, Li N, Mai V. Intestinal microbial ecology in premature infants assessed with non-culture-based techniques. J Pediatr. 2010;156:20–25. doi: 10.1016/j.jpeds.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai V, Young CM, Ukhanova M, Wang X, Sun Y, Casella G, Theriaque D, Li N, Sharma R, Hudak M, Neu J. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS ONE. 2011;6:e20647–e20647. doi: 10.1371/journal.pone.0020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow AL, Lagomarcino AJ, Schibler KR, Taft DH. Early microbial and metabolomic signatures predict later onset of necrotizing enterocolitis in preterm infants. Microbiome. 2013;1:13. doi: 10.1186/2049-2618-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA, Banfield JF. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci USA. 2011;108:1128–1133. doi: 10.1073/pnas.1010992108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 2012;23:111–120. doi: 10.1101/gr.142315.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Chivian D, Brodie EL, Alm EJ, Culley DE, Dehal PS, De Santis TZ, Gihring TM, Lapidus A, Lin LH, Lowry SR, Moser DP, Richardson PM, Southam G, Wanger G, Pratt LM, Andersen GL, Hazen TC, Brockman FJ, Arkin AP, Onstott TC. Environmental genomics reveals a single-species ecosystem deep within earth. Science. 2008;322:275–278. doi: 10.1126/science.1155495. [DOI] [PubMed] [Google Scholar]
- Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, Mackie RI, Pennacchio LA, Tringe SG, Visel A, Woyke T, Wang Z, Rubin EM. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science. 2012;337:1661–1665. doi: 10.1126/science.1224041. [DOI] [PubMed] [Google Scholar]
- Hug LA, Castelle CJ, Wrighton KC, Thomas BC. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome. 2013;1:22. doi: 10.1186/2049-2618-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D, Tringe SG, Singer SW, Eisen JA, Banfield JF. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun. 2013;4:2120. doi: 10.1038/ncomms3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of shiga-toxigenic Escherichia coli O104: H4. JAMA. 2013;309:1502–1510. doi: 10.1001/jama.2013.3231. [DOI] [PubMed] [Google Scholar]
- Sharon I, Battchikova N, Aro E-M, Giglione C, Meinnel T, Glaser F, Pinter RY, Breitbart M, Rohwer F, Béjà O. Comparative metagenomics of microbial traits within oceanic viral communities. ISME J. 2011;5:1178–1190. doi: 10.1038/ismej.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall VV, Collins MDM, Lawson PAP, Hutson RAR, Falsen EE, Inganas EE, Duerden BB. Characterization of some actinomyces-like isolates from human clinical sources: description of Varibaculum cambriensis gen nov, sp nov. J Clin Microbiol. 2003;41:640–644. doi: 10.1128/JCM.41.2.640-644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoetendal EG, Heilig HGHJ, Klaassens ES, Booijink CCGM, Kleerebezem M, Smidt H, De Vos WM. Isolation of DNA from bacterial samples of the human gastrointestinal tract. Nat Protoc. 2006;1:870–873. doi: 10.1038/nprot.2006.142. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultsch A, Mörchen F. ESOM-Maps: Tools For Clustering, Visualization, And Classification With Emergent SOM. Technical Report No. 46. Marburg: Dept. of Mathematics and Computer Science, University of Marburg, Germany; 2005. [Google Scholar]
- Altschul S, Gish W, Miller W, Myers E. Basic local alignment search tool. J Mol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40(Database issue):D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes J, Korbel JO, Lercher MJ, Mering Von C, Bork P. Prediction of effective genome size in metagenomic samples. Genome Biol. 2007;8:R10. doi: 10.1186/gb-2007-8-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wixon J, Kell D. The Kyoto encyclopedia of genes and genomes–KEGG. Yeast. 2000;17:48–55. doi: 10.1002/(SICI)1097-0061(200004)17:1<48::AID-YEA2>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinforma. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18:S225–S232. doi: 10.1093/bioinformatics/18.suppl_1.S225. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. Proceedings 20th IEEE International Parallel & Distributed Processing Symposium. Washington: IEEE; 2006. Phylogenetic models of rate heterogeneity: a high performance computing perspective. doi: 10.1109/IPDPS.2006.1639535. [Google Scholar]
- Shimodaira H. Multiple comparisons of log-likelihoods and combining nonnested models with applications to phylogenetic tree selection. Commun Stat Theory Methods. 2001;30:1751–1772. doi: 10.1081/STA-100105696. [DOI] [Google Scholar]
- FigTree. [ http://tree.bio.ed.ac.uk/software/figtree/]
- The R Project for Statistical Computing. [ http://www.r-project.org/]
- Miller CS, Baker BJ, Thomas BC WSS, Banfield JF. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011;12:R44. doi: 10.1186/gb-2011-12-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giongo A, Davis-Richardson AG, Crabb DB, Triplett EW. TaxCollector: modifying current 16S rRNA databases for the rapid classification at six taxonomic levels. Diversity. 2010;2:1015–1025. doi: 10.3390/d2071015. [DOI] [Google Scholar]
- Maidak BL, Olsen GJ, Larsen N. The RDP (Ribosomal Database Project) Nucleic Acids. 1997;25:109–111. doi: 10.1093/nar/25.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. Some mathematical questions in biology: DNA sequence analysis. Rhode Island: American Mathematical Society; 1986. [Miura RM (Series Editor): Lectures on Mathematics in the Life Sciences, vol. 17.] [Google Scholar]
- Tamura KK, Peterson DD, Peterson NN, Stecher GG, Nei MM, Kumar SS. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J. The NIH HMP Working Group et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Biotechnology (NY) 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ggKbase: DORA. [ http://ggkbase.berkeley.edu/DORA/organisms]
- Konstantinidis KT, Tiedje JM. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005;187:6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Silaghi-Dumitrescu R, Ljungdahl LG, Kurtz DM. Cytochrome bd oxidase, oxidative stress, and dioxygen tolerance of the strictly anaerobic bacterium Moorella thermoacetica. J Bacteriol. 2005;187:2020–2029. doi: 10.1128/JB.187.6.2020-2029.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Schmidt TM. Shallow breathing: bacterial life at low O2. Nat Rev Microbiol. 2013;11:205–212. doi: 10.1038/nrmicro2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan MS. Probiotic and prebiotic supplementation for the prevention of neonatal necrotizing enterocolitis. J Perinatol. 2009;29(Suppl 2):S2–6. doi: 10.1038/jp.2009.21. [DOI] [PubMed] [Google Scholar]
- Hall V. Actinomyces - Gathering evidence of human colonization and infection. Anaerobe. 2008;14:1–7. doi: 10.1016/j.anaerobe.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Chu YW, Wong CH, Chu MY, Cheung CPF, Cheung TKM, Tse C, Luk WK, Lo JYC. Varibaculum cambriense infections in Hong Kong, China, 2006. Emerging Infect Dis. 2009;15:1137–1139. doi: 10.3201/eid1507.081291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman WB, Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-I, Ludwig W. Bergey’s Manual of Systematic Bacteriology: Volume 5: The Actinobacteria. New York, NY: Springer New York; 2012. pp. 139–140. [Google Scholar]
- Boch J, Nau-Wagner G, Kneip S, Bremer E. Glycine betaine aldehyde dehydrogenase from Bacillus subtilis: characterization of an enzyme required for the synthesis of the osmoprotectant glycine betaine. Arch Microbiol. 1997;168:282–289. doi: 10.1007/s002030050500. [DOI] [PubMed] [Google Scholar]
- Leslie SB, Teter SA, Crowe LM, Crowe JH. Trehalose lowers membrane phase transitions in dry yeast cells. Biochim Biophys Acta. 1994;1192:7–13. doi: 10.1016/0005-2736(94)90136-8. [DOI] [PubMed] [Google Scholar]
- Strøm AR, Kaasen I. Trehalose metabolism in Escherichia coli: stress protection and stress regulation of gene expression. Mol Microbiol. 1993;8:205–210. doi: 10.1111/j.1365-2958.1993.tb01564.x. [DOI] [PubMed] [Google Scholar]
- Traxler MF, Summers SM, Nguyen H-T, Zacharia VM, Hightower GA, Smith JT, Conway T. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol. 2008;68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AL, Lewis WG. Host sialoglycans and bacterial sialidases: a mucosal perspective. Cell Microbiol. 2012;14:1174–1182. doi: 10.1111/j.1462-5822.2012.01807.x. [DOI] [PubMed] [Google Scholar]
- David CD. Protein-linked glycan degradation in infants fed human milk. J Glycomics Lipidomics. 2012;S1:002. doi: 10.4172/2153-0637.S1-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimr ER. Unified theory of bacterial sialometabolism: how and why bacteria metabolize host sialic acids. ISRN Microbiol. 2013;2013:816713. doi: 10.1155/2013/816713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severi E, Hood DW, Thomas GH. Sialic acid utilization by bacterial pathogens. Microbiology. 2007;153:2817–2822. doi: 10.1099/mic.0.2007/009480-0. [DOI] [PubMed] [Google Scholar]
- Pauli G, Overath P. ato operon: a highly inducible system for acetoacetate and butyrate degradation in Escherichia coli. Eur J Biochem. 1972;29:553–562. doi: 10.1111/j.1432-1033.1972.tb02021.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Espinosa RM, Dridge EJ, Bonete MJ, Butt JN, Butler CS, Sargent F, Richardson DJ. Look on the positive side! The orientation, identification and bioenergetics of Archaeal™ membrane-bound nitrate reductases. FEMS Microbiol Lett. 2007;276:129–139. doi: 10.1111/j.1574-6968.2007.00887.x. [DOI] [PubMed] [Google Scholar]
- Wissenbach U, Kröger A, Unden G. The specific functions of menaquinone and demethylmenaquinone in anaerobic respiration with fumarate, dimethylsulfoxide, trimethylamine N-oxide and nitrate by Escherichia coli. Arch Microbiol. 1990;154:60–66. doi: 10.1007/BF00249179. [DOI] [PubMed] [Google Scholar]
- Rolfe RDR, Hentges DJD, Campbell BJB, Barrett JTJ. Factors related to the oxygen tolerance of anaerobic bacteria. Appl Environ Microbiol. 1978;36:306–313. doi: 10.1128/aem.36.2.306-313.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Lopez CA, Bäumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013;14:319–327. doi: 10.1038/embor.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, Popova IE, Parikh SJ, Adams LG, Tsolis RM, Stewart VJ, Bäumler AJ. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PBP, Bik EME, Bernstein CNC, Purdom EE, Dethlefsen Les L, Sargent MM, Gill SRS, Nelson KEK, Relman DAD. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Yoshida A, Nagata E, Hoshino T, Oho T, Awano S, Takehara T, Ansai T. Streptococcus anginosusl-cysteine desulfhydrase gene expression is associated with abscess formation in BALB/c mice. Mol Oral Microbiol. 2011;26:221–227. doi: 10.1111/j.2041-1014.2010.00599.x. [DOI] [PubMed] [Google Scholar]
- Woo TDHT, Oka KK, Takahashi MM, Hojo FF, Osaki TT, Hanawa TT, Kurata SS, Yonezawa HH, Kamiya SS. Inhibition of the cytotoxic effect of Clostridium difficile in vitro by Clostridium butyricum MIYAIRI 588 strain. J Med Microbiol. 2011;60:1617–1625. doi: 10.1099/jmm.0.033423-0. [DOI] [PubMed] [Google Scholar]
- Pereira GR, Baker L, Egler J, Corcoran L, Chiavacci R. Serum myoinositol concentrations in premature infants fed human milk, formula for infants, and parenteral nutrition. Am J Clin Nutr. 1990;51:589–593. doi: 10.1093/ajcn/51.4.589. [DOI] [PubMed] [Google Scholar]
- Hallman M, Bry K, Hoppu K, Lappi M, Pohjavuori M. Inositol supplementation in premature infants with respiratory distress syndrome. N Engl J Med. 1992;326:1233–1239. doi: 10.1056/NEJM199205073261901. [DOI] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ESOM data.
Supplemental figures and tables.
Single copy genes in reconstructed genomes.
V. cambriense pathway tools data.
KEGG annotations.
EMIRGE reconstructed 16S rRNA gene sequences.
Metabolic features for comparative genomics.