

Abstract
Vibrio harveyi is a marine bacterial pathogen responsible for episodic epidemics generally associated with massive mortalities in many marine organisms, including the European abalone Haliotis tuberculata. The aim of this study was to identify the portal of entry and the dynamics of infection of V. harveyi in the European abalone. The results indicate that the duration of contact between V. harveyi and the European abalone influences the mortality rate and precocity. Immediately after contact, the epithelial and mucosal area situated between the gills and the hypobranchial gland was colonized by V. harveyi. Real-time PCR analyses and culture quantification of a green fluorescent protein-tagged strain of V. harveyi in abalone tissues revealed a high density of bacteria adhering to and then penetrating the whole gill-hypobranchial gland tissue after 1 h of contact. V. harveyi was also detected in the hemolymph of a significant number of European abalones after 3 h of contact. In conclusion, this article shows that a TaqMan real-time PCR assay is a powerful and useful technique for the detection of a marine pathogen such as V. harveyi in mollusk tissue and for the study of its infection dynamics. Thus, we have revealed that the adhesion and then the penetration of V. harveyi in European abalone organs begin in the first hours of contact. We also hypothesize that the portal of entry of V. harveyi in the European abalone is the area situated between the gills and the hypobranchial gland.
INTRODUCTION
The halophilic Gram-negative bacterium Vibrio harveyi is a common pathogen of many marine vertebrate and invertebrate species (1). V. harveyi is known to induce gastroenteritis, inflammation of the circulatory system or eye, and skin lesions in various species of fishes, crustaceans, and mollusks (2). In the European abalone Haliotis tuberculata, V. harveyi is responsible for the massive mortalities that occur during the summer spawning period both in natural populations and in farmed stocks (3, 4). Vibriosis of the European abalone is characterized by the presence of white spots on the foot and inflammation of the pericardial tissue, resulting in impaired mobility and septicemia (5).
Schematically, the bacterial infection process can be summarized to occur in three stages: (i) colonization, adhesion of the pathogen to the host surface, and initial multiplication and then penetration of the pathogen into the body through one or several portals, (ii) invasion of the host organs and/or the circulatory system concurrently with the expression of virulence factors by the pathogen, and (iii) exit of the pathogen and transmission of the disease (6, 7). Invasion and multiplication of V. harveyi inside the circulatory system, the hemolymph, seem to take place after 24 h of contact with the European abalone (5), suggesting that the adhesion and penetration of V. harveyi must occur in the first 24 h of contact. The portal of entry and the precise timing of the earlier stages of V. harveyi infection in the European abalone remain uncharacterized.
Many studies which have focused on the infection dynamics of fish pathogens have revealed a wide variety of modes of infection among Vibrio species. For example, V. anguillarum seems to penetrate preferentially through the gastrointestinal tract in the turbot Scophthalmus maximus (8), while both the skin and gills are the portal of entry in the Atlantic salmon (Salmo salar) (9) and in the rainbow trout (Salmo gairdneri) (10). In marine mollusks, the organic matrix of the shell may be a putative portal of entry of Vibrio pathogens, as observed with V. tapetis, which is known to invade Ruditapes philippinarum clams after adhesion on the periostracal lamina shell and the extrapallial fluid (11, 12). Considering the different modes of infection in pathogenic Vibrio species, the objectives of this study were to localize and verify the existence of a portal of entry of V. harveyi in the European abalone and to determine the infection dynamics occurring during the early stages of vibriosis.
MATERIALS AND METHODS
Abalones and bacterial strains.
Sexually mature abalones were transported in containers filled with macroalgae (Palmaria palmata) from the France Haliotis Hatchery, Plouguerneau, France, to our laboratory at the European Institute for Marine Studies, Brest, France, in June 2011. The abalones were used in the experiments if they did not present any tissue injuries or pedal muscle pustules and were capable of moving and adhering to the tank surface. The abalones were placed in 5-liter tanks of seawater, which was replaced daily with seawater pumped from the Bay of Brest (dissolved oxygen concentration, >7.5 mg · liter−1; salinity, 35‰) and filtered through a 1-μm-pore-size filter. The seawater temperature was increased by 1°C per day until it reached 19°C, and the animals were then maintained at this temperature for 1 week before beginning the experiments.
Two virulent strains of V. harveyi were used in this study. The first strain, ORM4, had been isolated from diseased H. tuberculata abalones in Normandy, France, during an episode of massive mortalities in 1999 and was affiliated with V. carchariae, the synonym species of V. harveyi, after 16S rRNA gene sequencing and phylogenetic analysis (3, 13). The second strain was a green fluorescent protein (GFP)-tagged and kanamycin-resistant bacterium constructed from the ORM4 strain by Travers et al. (5). The initial ORM4 strain and the GFP-tagged ORM4 strain have similar growth rates and virulences, and the GFP-labeled plasmid is stable in the GFP-tagged ORM4 strain for 8 days (5). Bacteria were grown in Luria-Bertani broth (LB; Sigma) supplemented with salt (LBS; final concentration, 20 g · liter−1) in a temperature-controlled shaker at 28°C for 16 h before challenge. Prior to use in experiments, bacterial cultures were washed and resuspended in sterile seawater.
Experiment 1: effect of contact duration on abalone mortality.
Five triplicate groups of 15 sexually mature abalones (n = 270, 2.5 years old, 38.8 ± 2.1 mm) were challenged with a suspension of strain ORM4 at a final concentration, estimated by the spectrophotometric method (5), of 105 CFU · ml−1 per tank, the concentration usually used to reproduce European abalone vibriosis under controlled conditions (4), for 1 h, 3 h, 6 h, 9 h, and 24 h. A control group not exposed to bacteria was also included. Dead abalones were counted and removed daily until mortality rate stabilization. The mortality of an abalone was attributed to vibriosis if the ORM4 strain was detected in its hemolymph. Comparison of the mortality curves between different challenge durations was performed with a Kaplan-Meier analysis followed by a log-rank test (14).
Experiment 2: microscopic observations.
Two noninfected abalones were removed from their shells and visualized using a Lumar stereomicroscope (Carl Zeiss Group, Oberkochen, Germany) to identify autofluorescent organs prior to adding a 100-μl GFP-tagged V. harveyi ORM4 strain suspension at a concentration of 105 bacteria/ml at 19°C. Microscopic observation was immediately done to detect a putative preferential tropism of V. harveyi for one or several organs.
Experiment 3: quantification of V. harveyi in European abalone organs during the first hours after contact. (i) Bacterial challenge and animal sampling.
Two triplicate groups of 10 sexually mature abalones (n = 60, 2 years old, 34.6 ± 1.9 mm) were placed in tanks. The first group contained animals challenged with the GFP-tagged strain at a final concentration of 106 CFU · ml−1, and the second group contained animals immersed in filtered seawater (control group). A high bacterial concentration was used in this experiment to avoid interindividual variability in the response to pathogen virulence. Ten animals were removed from the tanks prior to infection and were used as time zero (t0) samples. Five control and five challenged individuals were randomly sampled from the tanks at 1 h, 3 h, 6 h, 9 h, and 24 h postinfection.
(ii) Hemolymph and tissue sampling.
The hemolymph of each abalone was collected from the pedal sinus using a 2.5-ml sterile syringe mounted with a 23-gauge needle. One milliliter of hemolymph was reserved for bacterial quantification by plating on LBS agar dishes and then kept on ice until use, and 1 ml was saved for real-time quantitative PCR (qPCR) analysis and stored at −20°C until DNA extraction. After hemolymph sampling, all animals were dissected under sterile conditions. The following tissues were dissected for further analysis: (i) the two gills and attached hypobranchial gland, (ii) the central portion of the foot muscle, (ii) a section of the gonad-digestive gland, (iv) the mouth, and (v) epipodia. All organs were cut into two equal portions. The first portion was immediately washed in a 70% ethanol bath to remove external bacteria and then rinsed with sterile seawater. The second portion was briefly plunged in a sterile seawater bath to detect both adhered and penetrated bacteria and was considered untreated tissue. Each organ piece was weighed and homogenized in 1 ml sterile seawater using a mechanical disruptor. Two hundred microliters of homogenate was reserved for V. harveyi quantification by plating and kept on ice until use, and 800 μl was stored at −20°C until DNA extraction.
(iii) Bacterial quantification by plating.
The bacterial quantification of abalone samples collected after 1 h, 3 h, and 6 h of contact with V. harveyi was done by plating tissue homogenates. Two series of 10-fold dilutions (10−1 and 10−2) of tissue homogenates and hemolymph samples were made with sterile seawater. One hundred microliters of the initial and diluted tissue homogenates was plated onto LBS agar dishes containing 100 μg · ml−1 kanamycin, to ensure the selection of GFP-tagged bacteria. The dishes were incubated overnight at 28°C, and the number of colonies fluorescing under UV light was counted. The number of colonies per 100 mg of tissue (density) or 1 ml of hemolymph (concentration) is reported.
(iv) Bacterial quantification by real-time quantitative PCR.
(a) DNA extraction procedure. DNA extraction was done on hemolymph and untreated tissue samples. Five hundred microliters of hemolymph or homogenates of untreated tissue were diluted in 500 μl of extract salted buffer (100 mM Tris HCl, 100 mM NaCl, 50 mM EDTA, pH 8). Total DNA was extracted using a traditional phenol-chloroform-isoamyl alcohol method (15), after treatment with RNase A (10 mg/ml), lysozyme (10 mg/ml), 10% (vol/vol) SDS, 10% (vol/vol) Sarkosyl, and proteinase K (25 mg/ml). Finally, DNA was suspended in 50 μl of DNase-free water. The concentration and quality of the nucleic acids were determined using a spectrometer (NanoDrop ND-1000; Thermo Fisher Scientific, USA).
The standard samples used for qPCR were obtained after DNA extraction from 500 μl of hemolymph or 100-mg homogenates of organs dissected from noninfected abalones and supplemented with 10-fold serial dilutions of V. harveyi from 108 to 0 CFU. Viable bacteria were counted by plating and optical microscopy.
(b) Real-time quantitative PCR conditions and analysis. The concentration of V. harveyi in hemolymph was measured by qPCR using a 7500 Fast real-time PCR system and TaqMan Universal master mix (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA, USA). Amplification was done with V. harveyi toxR gene-specific primers: forward primer CCA-CTG-CTG-AGA-CAA-AAG-CA and reverse primer GTG-ATT-CTG-CAG-GGT-TGG-TT. Fluorescent visualization of amplification was done using a toxR probe (CAG-CCG-TCG-AAC-AAG-CAC-CG) dually labeled with a 5′ reporter dye (Texas Red) and a downstream 3′ quencher dye (black hole quencher 2) (16). After optimization of the primer and TaqMan probe concentrations, each reaction was run in duplicate with a final volume of 20 μl containing 4 μl of DNA sample, a 1× concentration of TaqMan master mix, 300 nM each primer, and 200 nM probe. The reactions were initiated by activation of the Thermo-Start DNA polymerase for 10 min at 95°C, followed by 45 amplification cycles (denaturation at 95°C for 15 s, annealing at 60°C for 1 min). The threshold cycle (CT) was defined as the first PCR cycle in which the probe reporter fluorescence was detectable above a baseline signal and was inversely proportional to the logarithm of the initial bacterial density or concentration. Thus, CT values, obtained for each sample using 7500 software (v2.0.3), were compared with those on standard curves to determine the initial V. harveyi density or concentration.
The sensitivity of the amplification was estimated for each abalone organ from three separate PCR assays. The primer efficiency (E) was determined from the slope of the standard curves using the method described by Yuan et al.: E = 10(−1/slope) (17).
The influence of time on bacterial density or concentration was assessed for the different organs using a nonparametric Wilcoxon test. Differences in bacterial concentrations or densities between two infection intervals were detected by a Wilcoxon signed-rank test (18). Statistical tests were done using JMP (v10.0.0) software (SAS Institute Inc.).
RESULTS
Effect of contact duration on abalone mortality.
European abalone mortality due to vibriosis was assessed over a 12-day period after different durations of contact with V. harveyi (Fig. 1; Table 1). The cause of mortality was assessed each day by detection of V. harveyi in abalone hemolymph. The cumulative mortality rate of infected abalones increased, while the lethal time period (LT50), which was the time period over which 50% of the abalones died, decreased throughout the different contact durations (Table 1). Kaplan-Meier analyses inferred that mortality curves were different between 1 h and 6 h of contact (P = 0.0114) and between 6 h and 24 h of contact (P = 0.042). Contact with V. harveyi for 1 h was sufficient to induce 80% and 77% mortality in two tanks, whereas only one abalone died in the last one after 11 days of the experiment. Abalone mortality reached 72% and 80% when abalones were in contact with V. harveyi for 6 h and 24 h, respectively, after 8 days of the experiment. No mortality was reported in the control groups.
FIG 1.
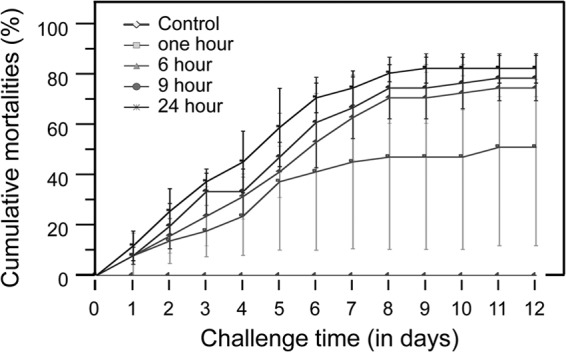
Influence of contact duration with Vibrio harveyi on mortality kinetics in the European abalone. Animals were exposed to challenge with 105 bacteria/ml by immersion for 1 h, 6 h, 9 h, and 24 h at 19°C. Results correspond to the means from 3 experimental replicates with 15 abalones per tank. Bars indicate standard error rates. Control animals were immersed only in filtered seawater. Comparison of mortality curves was performed with a Kaplan-Meier analysis (14).
TABLE 1.
Cumulative mortality in the European abalone after 12 days of infection and LT50 for each contact duration with V. harveyi
| Contact duration (h) | % cumulative mortality after 12 days of infectiona |
LT50b (days) | |
|---|---|---|---|
| Mean | SD | ||
| 1 | 57.8 | 44.4 | 6.8 |
| 6 | 84.4 | 10.2 | 5.3 |
| 9 | 88.9 | 3.8 | 4.5 |
| 24 | 93.3 | 6.7 | 3.7 |
The results correspond to the means of 3 experimental replicates with 15 animals.
LT50, lethal time period, that is, the time period over which 50% of abalones died.
Portal of entry. (i) Microscopic observations.
Fluorescent bacteria were observed on the surface of gill filaments immediately after contact with the GFP-tagged V. harveyi strain (data not shown). After 6 h of contact, fluorescent bacteria were aggregated on the epithelial and mucosal area between the gills and the hypobranchial gland (data not shown).
(ii) Quantification measures.
Quantification by plating revealed that V. harveyi densities increased exponentially in all organs during the first few hours after contact and throughout the different contact times (Fig. 2). The density of the adhered GFP-tagged bacteria was the highest on the surface of the gills, being 5-fold greater than that in the digestive gland and 12-fold greater than that on the surface of the foot muscle after 6 h of contact (Fig. 2A). Penetrated bacteria were also 10-fold more numerous in the gills than in the digestive gland after 6 h of contact (Fig. 2B).
FIG 2.
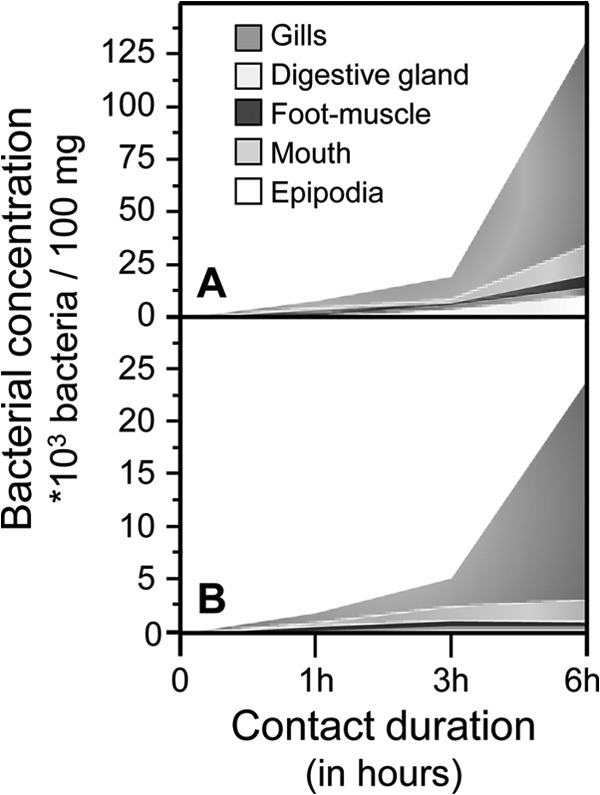
Vibrio harveyi densities in European abalone organs during the first hours of contact. Densities are expressed as the number of bacteria per 100 mg of tissue. Quantification of the GFP-tagged and kanamycin-resistant V. harveyi ORM4 strain was done after plating of tissue homogenate on kanamycin-LBS agar dishes after overnight incubation at 28°C. (A) Quantification of adhered and penetrated bacteria in organs that were not treated with ethanol; (B) quantification of only bacteria that penetrated organs that were washed in an ethanol bath.
Efficiency and sensitivity of qPCR for V. harveyi quantification.
The sensitivity and efficiency of V. harveyi qPCR quantification via toxR gene amplification were assessed with samples from the gills, digestive gland, foot muscle, and hemolymph (see Fig. S1 in the supplemental material). The sensitivity threshold was estimated to be 102 bacteria/100 mg of tissue for the gills, 103 bacteria/100 mg for the digestive gland, 101 bacteria/100 mg for the foot muscle, and 102 bacteria/ml for hemolymph. The equation of the regression line was then used to calculate the bacterial concentration or density from the qPCR data that were obtained. The average amplification efficiencies were 95% in the gills, 84% in the digestive gland, and 105% in the foot muscle and hemolymph.
Infection kinetics.
The V. harveyi concentration depended on the contact duration in all abalone compartments (P < 0.01 by the Wilcoxon test). In hemolymph, the bacterial concentration increased with contact duration, except in samples in which the V. harveyi concentration was measured by the qPCR quantification method between 3 h or 6 h and 9 h of contact (Fig. 3; Table 2). In the gills, the density of adhered and penetrated vibrios also increased significantly with duration until 6 h of contact for both of the quantification methods used (Table 2). Moreover, the density of penetrated bacteria in the gills surpassed 103 CFU/100 mg after the first hour of contact, while that of the penetrated bacteria in the digestive gland and muscle did not reach the threshold of 102 CFU/100 mg until 6 h of contact (Fig. 3). For the digestive gland, quantification by plating revealed a significant increase in the density of either adhered or penetrated bacteria during infection. However, qPCR measures showed a stabilization of the V. harveyi densities in the digestive gland after 1 h, except that a difference was detected between 3 h and 6 h of contact (Fig. 3; Table 2). In the foot muscle, the V. harveyi density measured by plating increased significantly during the first hour and after 6 h of contact (Fig. 3; Table 2). Meanwhile, qPCR analysis of foot muscle tissue showed significant differences between controls and infected animals until 9 h of contact (Table 2).
FIG 3.
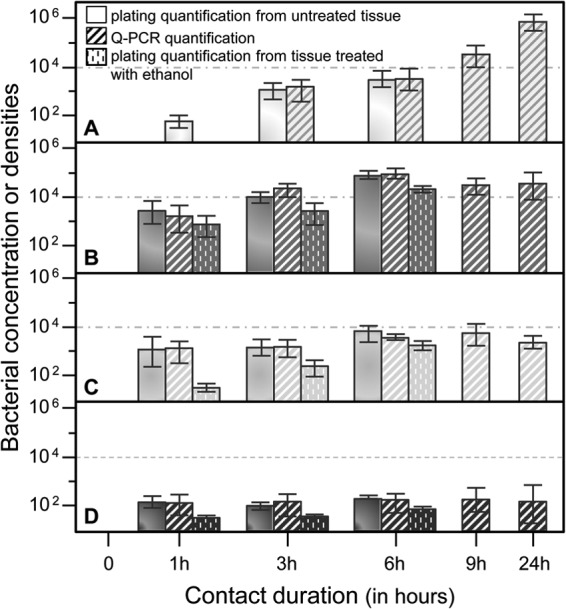
Infection dynamics of Vibrio harveyi in European abalone tissues. Concentrations or densities (bacterial count per ml of hemolymph or per 100 mg of tissue) in hemolymph (A), gills (B), digestive gland (C), and foot muscle (D) were estimated during the first hours of contact. Results are given as the average concentrations or densities of five biological replicates, estimated by plating tissues from organs treated or not treated with ethanol on kanamycin-LBS agar dishes and by qPCR using specific primers and a TaqMan probe. Bars indicate standard errors. No bacterium was detected in samples obtained at t0 or in control individuals immersed in filtered seawater without V. harveyi in suspension.
TABLE 2.
Statistical significance of V. harveyi concentrations or densities in European abalone organs after the first hours of contact
| Organ | Timea (h) | Statistical significance at the indicated times by the following quantification methodsc: |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plating without ethanol treatment |
qPCR |
Plating with ethanol treatment |
||||||||||
| 1 h | 3 h | 6 h | 1 h | 3 h | 6 h | 9 h | 24 h | 1 h | 3 h | 6 h | ||
| HLb | 0 | ** | ** | ** | ND | ** | ** | ** | ** | |||
| 1 | ** | ** | ** | ** | ** | ** | ||||||
| 3 | ** | ** | NS | ** | ||||||||
| 6 | NS | ** | ||||||||||
| 9 | ** | |||||||||||
| Gills | 0 | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** |
| 1 | ** | ** | ** | ** | ** | ** | NS | ** | ||||
| 3 | ** | ** | ** | ** | ** | |||||||
| 6 | * | NS | ||||||||||
| 9 | NS | |||||||||||
| Digestive gland | 0 | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** | ** |
| 1 | NS | ** | NS | NS | NS | NS | ** | ** | ||||
| 3 | ** | * | NS | NS | ** | |||||||
| 6 | NS | NS | ||||||||||
| 9 | NS | |||||||||||
| Muscle | 0 | ** | ** | ** | ** | ** | ** | ** | NS | ** | ** | ** |
| 1 | NS | NS | NS | NS | NS | NS | NS | ** | ||||
| 3 | ** | NS | NS | NS | * | |||||||
| 6 | NS | NS | ||||||||||
| 9 | NS | |||||||||||
Duration of contact between the European abalone and V. harveyi.
HL, hemolymph.
Differences were detected by the Wilcoxon signed-rank test (18). *, a statistically significant difference at P < 0.1; **, a statistically significant difference at P < 0.05; NS, not significant; ND, not detected.
For the majority of the measures, the bacterial count determined by qPCR analysis was slightly higher than the count determined by plating. No bacteria were counted either in samples obtained at t0 or in samples from the controls by plating or by qPCR analysis.
DISCUSSION
The duration of contact with the pathogen V. harveyi influenced the mortality rate and infection kinetics in European abalones (Fig. 1). Immediately after contact, a large number of bacteria could be observed in the gills and hypobranchial gland. Moreover, after 6 h of contact, the density of V. harveyi was at least 5-fold greater in the gills and in the hypobranchial gland than in the other tissues tested (Fig. 2). Finally, V. harveyi was detected in hemolymph after 3 h of contact (Fig. 3).
Our results show that the duration of contact between V. harveyi and the European abalones was correlated with the mortality rate. Moreover, 1 h of exposure was sufficient to induce mortality. This observation suggests that V. harveyi is able to adhere to and then penetrate abalone tissues during the first hour of contact. The relative rapidity of adhesion and penetration in the host has been described for many Vibrio species and for V. harveyi and may contribute to their symbiotic process. For example, in the association between the luminous bacterium V. fischeri and the sepiolid squid Euprymna scolopes, the symbiont is able to adhere to the ciliated epithelium of juvenile squid light organs after only 3 h (19). In the kuruma prawn Penaeus japonicus, the study of Vibrio sp. infection dynamics after oral inoculation showed that the pathogen can be detected in the stomach and hemolymph after 3 h and in all other sampled organs after 6 h (20). In the giant tiger prawn Penaeus monodon, modulation of immune gene expression and of the synthesis of protein involved in immune function was observed in hemocytes after 6 h of immersion with a pathogenic strain of V. harveyi, confirming its adhesion, penetration, and invasion capacities during the first hours of contact (21, 22).
The quantification of adhered or penetrated V. harveyi by plating showed a high density of V. harveyi on the surface and then within gill filaments and the hypobranchial gland after 1 h of contact. Microscopic observation confirmed a tropism of V. harveyi for the epithelial and mucosal region situated between the gills and the hypobranchial gland in the European abalone. Previous studies showed that gills seem to be an ideal place of adhesion and, hence, are a relevant entry point for Vibrio pathogens in marine mollusks. In the case of the challenge with a GFP-tagged virulent strain of V. parahaemolyticus, the density of the bacteria was maintained in the gills of Tiostrea chilensis oysters for 48 h after contact (23). Sawabe et al. also revealed a tropism of a GFP-tagged virulent strain of V. harveyi for gills in the abalone Haliotis discus hannai (24). Our results add another degree of clarification and functional complexity regarding the observations made by Travers et al., who visualized an accumulation of V. harveyi on the surface of the gills in infected animals (5). This region may constitute the portal of entry of V. harveyi in the European abalone. The proximity of this targeted region to the heart and the anterior aorta may allow the pathogen to quickly invade the circulatory system of the European abalone (25, 26). This hypothesis of an entry route of V. harveyi via the epithelial region between the gills and the hypobranchial gland could be confirmed using some additional advanced electron microscopy techniques, which may also specify the strategy of V. harveyi entry into gill tissue via an intra- or extracellular route.
In the current study, we have used and tested a TaqMan probe developed by Schikorski et al. (2013) for qPCR assays of V. harveyi (16). Measurements obtained by TaqMan qPCRs following a scheme of serial dilution of a pure culture of V. harveyi in samples of hemolymph or tissues showed an excellent linear correlation, and TaqMan qPCRs have been proven to be powerful and sensitive techniques for V. harveyi detection and quantification in European abalones. TaqMan qPCRs have previously been useful for the detection of pathogenic strains of V. vulnificus, V. parahaemolyticus, and V. aestuarianus in marine mollusks (27–31). Nevertheless, in our measurements we observed that the counts obtained by qPCR were higher than the counts obtained by the plating method. This observation may be due to the quantification of dead or uncultured bacteria by qPCR.
Quantification of the adhered and penetrated V. harveyi organisms in abalone organs (i.e., gills-hypobranchial gland, digestive gland, and foot muscle) and in hemolymph has elucidated the infection dynamics. After 1 h of contact, V. harveyi was already detected by plating and by qPCR on the surface of all the abalone tissue analyzed, hence inferring that V. harveyi is able to adhere to abalones during the first hour of contact. The concentration of V. harveyi in the hemolymph increased progressively between 3 h and 24 h after contact. This observation suggests that the concentration of V. harveyi in hemolymph may be a proxy for the development of infection in the European abalones and that the stage of multiplication and invasion may occur during the first hours after contact. The relative rapidity of Vibrio invasion was also observed in the oyster Crassostrea gigas, in which virulent strains of V. splendidus and V. aestuarianus were detected by qPCR in hemolymph after 2 h of contact, and the concentrations of both pathogens were maximal after 6 h (32). On observation of all the organs analyzed here, the density of V. harveyi increased significantly with contact duration in the gills, in which the density was maximal after 6 h of contact. This increase in pathogen density may be due to the local multiplication of V. harveyi on the surface of the gills and the hypobranchial gland, which supports the hypothesis of a putative portal of entry of V. harveyi in this region.
In conclusion, V. harveyi seems to have the capacity to adhere to and penetrate the European abalone during the first hours of contact. We hypothesize that the portal of entry of V. harveyi is the epithelial region situated between the gills and the hypobranchial gland. Further observations need to be conducted to verify the putative portal of entry of V. harveyi in the European abalone and localize it precisely. The invasion of V. harveyi in abalone organs is fast and progressive during the first hours of contact and may be characterized by quantification in the hemolymph. In this study, we have also successfully tested a TaqMan qPCR assay for V. harveyi quantification in European abalone tissue which may be useful for pathogen detection in the natural environment and in farmed stocks. This article demonstrates the importance of pathogen concentration measures during the first hours of contact with the host, improving our knowledge and understanding of the infection dynamics of Vibrio pathogens in marine mollusks.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Pole Mer Bretagne and the Université de Bretagne Occidentale.
We are also grateful to Flavia Nunes from the Laboratoire des Sciences de l'Environnement MARin-LEMAR and Sarah Culloty from the University College Cork for language support and to Isabelle Calves for her experimental help.
Footnotes
Published ahead of print 8 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01036-14.
REFERENCES
- 1.Austin B, Zhang XH. 2006. Vibrio harveyi: a significant pathogen of marine vertebrates and invertebrates. Lett. Appl. Microbiol. 43:119–124. 10.1111/j.1472-765X.2006.01989.x [DOI] [PubMed] [Google Scholar]
- 2.Austin B. 2010. Vibrios as causal agents of zoonoses. Vet. Microbiol. 140:310–317. 10.1016/j.vetmic.2009.03.015 [DOI] [PubMed] [Google Scholar]
- 3.Nicolas JL, Basuyaux O, Mazurie J, Thebault A. 2002. Vibrio carchariae, a pathogen of the abalone Haliotis tuberculata. Dis. Aquat. Organ. 50:35–43. 10.3354/dao050035 [DOI] [PubMed] [Google Scholar]
- 4.Travers MA, Basuyaux O, Le Goic N, Huchette S, Nicolas JL, Koken M, Paillard C. 2009. Influence of temperature and spawning effort on Haliotis tuberculata mortalities caused by Vibrio harveyi: an example of emerging vibriosis linked to global warming. Glob. Chang. Biol. 15:1365–1376. 10.1111/j.1365-2486.2008.01764.x [DOI] [Google Scholar]
- 5.Travers MA, Barbou A, Le Goic N, Huchette S, Paillard C, Koken M. 2008. Construction of a stable GFP-tagged Vibrio harveyi strain for bacterial dynamics analysis of abalone infection. FEMS Microbiol. Lett. 289:34–40. 10.1111/j.1574-6968.2008.01367.x [DOI] [PubMed] [Google Scholar]
- 6.Mims C, Nash A, Stephen J, Fitzgerald R. 2002. Mims' pathogenesis of infectious disease. Academic Press, London, United Kingdom [Google Scholar]
- 7.Finlay BB, Falkow S. 1997. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 61:136–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsson JC, Joborn A, Westerdahl A, Blomberg L, Kjelleberg S, Conway PL. 1996. Is the turbot, Scophthalmus maximus L., intestine a portal of entry for the fish pathogen Vibrio anguillarum? J. Fish Dis. 19:225–234 [Google Scholar]
- 9.Svendsen YS, Bogwald J. 1997. Influence of artificial wound and non-intact mucus layer on mortality of Atlantic salmon Salmo salar L. following a bath challenge with Vibrio anguillarum and Aeromonas salmonicida. Fish Shellfish Immunol. 7:317–325 [Google Scholar]
- 10.Laurencin FB, Germon E. 1987. Experimental infection of rainbow trout, Salmo gairdneri R., by dipping in suspensions of Vibrio anguillarum: ways of bacterial penetration; influence of temperature and salinity. Aquaculture 67:203–205. 10.1016/0044-8486(87)90028-7 [DOI] [Google Scholar]
- 11.Paillard C, Maes P. 1995. The brown ring disease in the Manila clam, Ruditapes philippinarum. I. Ultrastructural alterations of the periostracal lamina. J. Invertebr. Pathol. 65:91–100. 10.1006/jipa.1995.1015 [DOI] [Google Scholar]
- 12.Allam B, Paillard C, Ford SE. 2002. Pathogenicity of Vibrio tapetis, the etiological agent of brown ring disease in clams. Dis. Aquat. Organ. 48:221–231. 10.3354/dao048221 [DOI] [PubMed] [Google Scholar]
- 13.Gauger EJ, Gómez-Chiarri M. 2002. 16S ribosomal DNA sequencing confirms the synonymy of Vibrio harveyi and V. carchariae. Dis. Aquat. Organ. 52:39–46. 10.3354/dao052039 [DOI] [PubMed] [Google Scholar]
- 14.Kaplan EL, Meier P. 1958. Nonparametric-estimation from incomplete observations. J. Am. Stat. Assoc. 53:457–481. 10.1080/01621459.1958.10501452 [DOI] [Google Scholar]
- 15.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 16.Schikorski D, Renault T, Paillard C, Bidault-Toffin A, Tourbiez D, Saulnier D. 2013. Development of TaqMan real-time PCR assay targeting both the marine bacteria Vibrio harveyi, a pathogen of European abalone Haliotis tuberculata, and plasmidic DNA harbored by virulent strains. Aquaculture 392-395:106–112. 10.1016/j.aquaculture.2013.02.005 [DOI] [Google Scholar]
- 17.Yuan JS, Reed A, Chen F, Stewart CN. 2006. Statistical analysis of real-time PCR data. BMC Bioinformatics 7:85. 10.1186/1471-2105-7-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilcoxon F. 1945. Individual comparisons by ranking methods. Biometrics Bull. 1:80–83. 10.2307/3001968 [DOI] [Google Scholar]
- 19.Altura MA, Heath-Heckman EAC, Gillette A, Kremer N, Krachler AM. 2013. The first engagement of partners in the Euprymna scolopes-Vibrio fischeri symbiosis is a two-step process initiated by a few environmental symbiont cells. Environ. Microbiol. 15:2937–2950. 10.1111/1462-2920.12179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delapena LD, Nakai T, Muroga K. 1995. Dynamics of Vibrio sp in organs of orally infected kuruma prawn Penaeus japonicus. Fish Pathol. 30:39–45. 10.3147/jsfp.30.39 [DOI] [Google Scholar]
- 21.Somboonwiwat K, Supungul P, Rimphanitchayakit V, Aoki T, Hirono I, Tassanakajon A. 2006. Differentially expressed genes in hemocytes of Vibrio harveyi-challenged shrimp Penaeus monodon. J. Biochem. Mol. Biol. 39:26–36. 10.5483/BMBRep.2006.39.1.026 [DOI] [PubMed] [Google Scholar]
- 22.Somboonwiwat K, Chaikeeratisak V, Wang HC, Lo CF, Tassanakajon A. 2010. Proteomic analysis of differentially expressed proteins in Penaeus monodon hemocytes after Vibrio harveyi infection. Proteome Sci. 8:39. 10.1186/1477-5956-8-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabello AE, Espejo RT, Romero J. 2005. Tracing Vibrio parahaemolyticus in oysters. Tiostrea chilensis using a green fluorescent protein tag. J. Exp. Mar. Biol. Ecol. 327:157–166. 10.1016/j.jembe.2005.06.009 [DOI] [Google Scholar]
- 24.Sawabe T, Inoue S, Fukui Y, Yoshie K, Nishihara Y, Miura H. 2007. Mass mortality of Japanese abalone Haliotis discus hannai caused by Vibrio harveyi infection. Microbes Environ. 22:300–308. 10.1264/jsme2.22.300 [DOI] [Google Scholar]
- 25.Crofts DR. 1929. Haliotis. Memoirs on typical British marine plants and animals. 29 University Press of Liverpool, Liverpool, United Kingdom [Google Scholar]
- 26.Manganaro M, Laura R, Guerrera MC, Lanteri G, Zaccone D, Marino F. 2012. The morphology of gills of Haliotis tuberculata (Linnaeus, 1758). Acta Zool. 93:436–443. 10.1111/j.1463-6395.2011.00518.x [DOI] [Google Scholar]
- 27.Panicker G, Bej AK. 2005. Real-time PCR detection of Vibrio vulnificus in oysters: comparison of oligonucleotide primers and probes targeting vvhA. Appl. Environ. Microbiol. 71:5702–5709. 10.1128/AEM.71.10.5702-5709.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordon KV, Vickery MC, DePaola A, Staley C, Harwood VJ. 2008. Real-time PCR assays for quantification and differentiation of Vibrio vulnificus strains in oysters and water. Appl. Environ. Microbiol. 74:1704–1709. 10.1128/AEM.01100-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanco-Abad V, Ansede-Bermejo J, Rodriguez-Castro A, Martinez-Urtaza J. 2009. Evaluation of different procedures for the optimized detection of Vibrio parahaemolyticus in mussels and environmental samples. Int. J. Food Microbiol. 129:229–236. 10.1016/j.ijfoodmicro.2008.11.028 [DOI] [PubMed] [Google Scholar]
- 30.Saulnier D, De Decker S, Haffner P. 2009. Real-time PCR assay for rapid detection and quantification of Vibrio aestuarianus in oyster and seawater: a useful tool for epidemiologic studies. J. Microbiol. Methods 77:191–197. 10.1016/j.mimet.2009.01.021 [DOI] [PubMed] [Google Scholar]
- 31.Baker-Austin C, Gore A, Oliver JD, Rangdale R, McArthur JV, Lees DN. 2010. Rapid in situ detection of virulent Vibrio vulnificus strains in raw oyster matrices using real-time PCR. Environ. Microbiol. Rep. 2:76–80. 10.1111/j.1758-2229.2009.00092.x [DOI] [PubMed] [Google Scholar]
- 32.De Decker S, Saulnier D. 2011. Vibriosis induced by experimental cohabitation in Crassostrea gigas: evidence of early infection and down-expression of immune-related genes. Fish Shellfish Immunol. 30:691–699. 10.1016/j.fsi.2010.12.017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.