

ABSTRACT
Murine polyomavirus small t antigen (PyST) regulates cell cycle, cell survival, apoptosis, and differentiation and cooperates with middle T antigen (MT) to transform primary cells in vitro and in vivo. Like all polyomavirus T antigens, PyST functions largely via its interactions with host cell proteins. Here, we show that PyST binds both Yes-associated protein 1 (YAP1) and YAP2, integral parts of the Hippo signaling pathway, which is a subject of increasing interest in human cancer. The transcription factor TEAD, which is a known target of YAP, is also found in PyST complexes. PyST enhanced YAP association with protein phosphatase 2A (PP2A), leading to decreased YAP phosphorylation. PyST increased YAP levels by decreasing its degradation. This effect was mediated by a reduction in YAP association with β-transducin repeat protein (βTRCP), which is known to regulate YAP turnover in a phosphorylation-dependent manner. Genetic analysis has identified PyST mutants defective in YAP binding. These mutants demonstrated that YAP binding is important for PyST to block myoblast differentiation and to synergize with the phosphodiesterase inhibitor isobutylmethylxanthine (IBMX) to promote cell death in 3T3-L1 preadipocytes placed under differentiation conditions. In addition to YAP binding, both of these phenotypes require PyST binding to PP2A.
IMPORTANCE The Hippo/YAP pathway is a highly conserved cascade important for tissue development and homeostasis. Defects in this pathway are increasingly being associated with cancer. Polyomavirus small t antigen is a viral oncogene that cooperates with middle T antigen in transformation. On its own, small t antigen controls cell survival and differentiation. By binding YAP, small t antigen brings it together with protein phosphatase 2A. This work shows how this association of small t antigen with YAP is important for its effects on cell phenotype. It also suggests that PyST can be used to characterize cellular processes that are regulated by YAP.
INTRODUCTION
Studies of murine polyomavirus have been instrumental in the identification and establishment of several key signaling pathways, including tyrosine phosphorylation (1), regulation of c-src (2), and phosphoinositide-3-kinase (3). Small t antigen (PyST), which can complement middle T antigen (MT) for transformation (4, 5), has provided insights into cellular control mechanisms for differentiation and survival. PyST can promote either cell survival or cell death, depending on the circumstances (6). It is a “professional killer,” causing cell death via mitotic catastrophe even at low levels of expression. PyST blocks differentiation to adipocytes, myocytes, or osteoblasts (7, 8). From a virology perspective, it is interesting that these effects are not generally observed with simian virus 40 (SV40) small t antigen (SV40ST).
Studies on proteins that bind t antigens have been enormously fruitful in explaining their biological functions in normal and transformed cells. Work on the biology of PyST and SV40ST focused attention on protein phosphatase 2A (PP2A). A comparison of SV40 to polyomavirus showed that only PyST strongly associated with the Aβ scaffold subunit of the PP2A family of enzymes (7). Subsequent work showed the importance of the Aβ isoform in regulation of survival and differentiation, at least in part because of its effects on Akt (8). Earlier work suggested that all three T antigens, including PyST, could bind to TAZ (9), an interaction affecting the host range of the virus.
TAZ belongs to a conserved family of transcriptional coactivators that includes Yes-associated protein (YAP) and Yorkie (10–12). These proteins are part of the Hippo signaling system (13–15). Upon Hippo pathway activation, a phosphorylation cascade ensues: mammalian sterile 20-like kinase (MST) kinase phosphorylates large tumor suppressor kinase (LATS), which in turn phosphorylates YAP. The transcriptional activity of YAP is modulated by its phosphorylation status (15, 16). YAP appears to have other roles, such as controlling microRNA (miRNA) production and β-catenin function (17, 18). YAP, particularly, is a candidate oncogene in several cancers (19–21). Overexpression of YAP can lead to transformation of cells in vitro (21), can induce tumors in vivo (22, 23), and is a predictor of prognosis for patients with colorectal cancer (24). YAP is required for Kras-mediated transformation (25). While oncogenic signals arising from YAP promote excessive proliferation (22, 23, 26), YAP can also promote cell death via a p73-dependent pathway (27–30). YAP also has been shown to play a role in myoblast (31) and neuronal stem cell (32, 33) differentiation. Additionally, recent evidence suggests that YAP is involved in cell survival in Wnt- and β-catenin-dependent colorectal cancers (34). Clearly, YAP and PyST are both pleiotropic in their actions, with considerable overlap in their biology.
In the current work, we used a proteomic screen to identify YAP as a PyST binding protein. We found that by bringing together PP2A and YAP, PyST directs the dephosphorylation of YAP. Genetic analysis shows that this interaction is important for PyST effects on differentiation and survival.
MATERIALS AND METHODS
Cells and growth medium.
293T cells were used for transient overexpression to permit an assessment of YAP binding. 3T3 cells were used because they are a traditional, well-studied substrate for polyomavirus. 3T3-L1 preadipocytes and C2C12 myoblasts are well-characterized differentiation systems used previously in our PyST experiments (7, 8). MCF10A cells were used only for immunofluorescence experiments, because as seen from the literature (16), these cells are where YAP localization gives the clearest results. Serum (Dulbecco's modified Eagle's medium [DMEM]) supplemented with 4 mM l-glutamine (catalog no. 25030081; Invitrogen), 1 unit of penicillin-streptomycin (15140122; Invitrogen), 2.25 g/liter NaHCO3, and either 10% fetal bovine serum (FBS) (16000044; Invitrogen) or 10% calf serum (CS) (26170043; Invitrogen). Cells that were grown in 10% FBS medium include C2C12 cells, 3T3-L1 cells, 293T cells, and Phoenix Ampho cells, while NIH 3T3 cells were grown in medium with 10% CS. MCF10A cells were grown in DMEM/F-12 medium supplemented with 0.5% FBS, 0.5 mg/ml hydrocortisone, 10 ng/ml human epidermal growth factor (hEGF), 10 mg/ml insulin, and 10 mg/ml cholera toxin.
Retrovirus infections and generation of cell lines.
Phoenix Ampho cells were transfected with 10 μg of pBABE retrovirus constructs. Forty-eight hours later, the supernatants were removed and replaced with fresh medium. The filtered viral supernatant mixed with fresh medium at a 1:1 ratio was placed on target cells with 2 μg/ml of Polybrene (Sigma AL-118) for 4 h twice. The next day, the process was repeated twice. Cells were selected the following day with puromycin (2 mg/ml). The cell lines were maintained in DMEM with 10% FBS and the respective drug for up to 1 month or until experiments were performed. At this point, the cell lines were generated with newly thawed cells.
Cell lysates.
Harvested cells washed once in cold PBS were suspended in extraction buffer (TEB) (137 mM NaCl, 20 mM Tris, 0.92 mM Ca2+, 0.49 mM Mg2+, 1% NP-40, and 10% glycerol adjusted to pH 7) and incubated for 30 min at 4°C in the presence of protease inhibitors, Pepstatin, aprotinin at 1 μg/ml, 1 mM phenylmethanesulfonyl fluoride (PMSF), and phosphatase inhibitors II and III (1:100 dilution, Sigma) for 30 min. The cleared supernatant was boiled with SDS loading buffer.
PEI transfection.
The polyethyleneimine (PEI) transfection method was used in retrovirus generation of cell lines. This method of transfection has a high efficiency in a large number of cells, allowing maximal production of the intended protein products. However, PEI transfection can stress the cells (J. H. Hwang, unpublished data). To begin, 10-cm plates containing cells that were 60 to 80% confluent were supplied with fresh DMEM containing 10% FBS and supplements for 4 h. Three micrograms of PEI and at least 7 μg of the plasmid DNA were mixed in 1 ml of Opti-MEM. The mixture was incubated for 5 min and slowly distributed into the medium. The medium was changed the next day. Forty-eight hours was usually needed for optimal expression.
BES transfection.
Luciferase assays measuring ATF2 promoter activity were done exclusively through BES transfection (35), for this method appears to be less toxic to the cell. Briefly, 10-cm plates of NIH 3T3 or HEK 293T cells were split a day before transfection, with the goal of having them approximately 40% to 60% confluent the next day. A total of about 6 to 10 μg of the DNA was mixed with 450 μl water and then 500 μl of 2× N,N-bis(hydroxyethyl)-2-aminoethanesulfonic acid (BES) buffer (0.05 M BES [pH 6.95], 0.28 M NaCl, 0.015 M Na2HPO4). Finally, 50 μl of 2.5 M CaCl2 was added dropwise, and the precipitate was allowed to form for 20 min. The precipitate was then added dropwise on the plates and incubated overnight at 35°. The plates were washed the next day with PBS, and then fresh medium was added. At 48 h posttransfection, the cells were harvested and the extracts were used for the assays.
Immunoprecipitation (IP).
Cell extracts were added to antibody premixed with either protein A (catalog no. L00210; Genscript) or protein G beads (L00209; Genscript) for 1 to 2 h at 4°C. Each of the samples was subsequently washed with TEB 4 times, boiled in loading buffer, and analyzed by Western blotting.
Reporter assays.
The BES-calcium phosphate method was used for the initial transfections as described above with the combination of expression and luciferase constructs for each assay. The medium was changed the next day, and at 48 h posttransfection, cells were harvested. Luciferase activity was assayed after mixing the lysate with luciferase buffer (20 mM Tricine, 1.07 mM Mg(CO3)Mg(OH)2 5H2O, 2.07 mM MgSO4, 0.1 mM EDTA, 33.3 mM dithiothreitol [DTT], 270 μM coenzyme A (CoA), 470 mM Luciferin, and 530 mM ATP in water). To normalize the luciferase values, the same lysates were also measured for β-galactosidase activity (60 μl of 100× Mg solution [0.1 M MgCl2 and 4.5 M β-mercaptoethanol], 1.32 ml of 1× o-nitrophenyl-β-d-galactopyranoside [ONPG], 4.02 ml of 0.1 M NaPO4).
Retroviral plasmids.
PyST and SV40ST pBABE puro vector (36) (obtained from Addgene) were inserted at EcoRI restriction sites. In the pBABE vector, STs were tagged with a tandem FLAG and hemagglutinin (HA) tag at the C terminus (ST-FLAG-HA). Mutants of PyST were generated in the pBABE backbone. HA-tagged pBABE myr-Akt and myc-tagged Akt were gracious gifts from Phillip Tsichlis.
C2C12 cell differentiation to myocytes.
C2C12 cell lines were allowed to grow to confluence in DMEM containing 10% FBS. The medium was then replaced with DMEM containing 2% horse serum (HS) every 2 days. The observation of an elongated spindle-like cell morphology represents the formation of myocytes.
3T3-L1 apoptosis.
Two days after 3T3-L1 cells were confluent, 0.5 mM isobutylmethylxanthine (IBMX) was added to the cells for 24 h.
Knockdown of YAP.
Lentivirus pLKO short hairpins against mouse YAP (TRCN0000095866) was obtained from the Broad Institute (target sequence, CGGTTGAAACAACAGGAATTA). This hairpin as well as a scramble control at 3 μg were transfected into 293T cells along with packaging vectors PLP1 (4 μg), PLP2 (2.5 μg), and vesicular stomatitis virus glycoprotein 3 (VSVG3; 3 μg). At 48 h posttransfection, the supernatant was collected, filtered, and added to target cells at amounts that yielded similar amounts of infection based on equalizing puromycin-resistant expression. Twenty-four hours later, the infection medium was changed. At 48 to 72 h postinfection, cells were assayed by Western blotting for YAP knockdown.
Site-directed mutagenesis.
Primers were designed to extend 10 bp beyond the site of mutagenesis, and the reverse primer was a simple reverse complement of the forward primer. The obtained primers and the target plasmid were used in a PCR and transformed into XL1 cells with a QuikChange II XL site-directed mutagenesis kit (Agilent 200522). The resulting clones were subsequently expanded and isolated for plasmid DNA with QIAprep Spin Miniprep kit (Qiagen 27106) and then sequenced for the mutation using the appropriate primers at the Tufts University Core Facility.
Antibodies and reagents.
MF20 antibody to myosin heavy chain (MHC) was from the Developmental Studies Hybridoma Bank. Monoclonal pAB419 and pN116 recognize the N terminus of SV40ST and PyST, respectively (7). Antibodies from Cell Signaling Technologies were Akt (9272), pS473 Akt (XP 4060), caspase 3 (9664), YAP (12395), YAP pS127 (4911), and YAP pS381 (13619). HA11 (MMS-101P) was from Covance, and EE antibody (AB3788) was from Millipore. YAP(H-125) was from Santa Cruz. FLAG M2 (F1804) and actin (A2066) were ordered from Sigma. MK2206 was a gift from Phillip Tsichlis. Polyclonal anti-ST antibody was raised in rabbits against recombinant PyST expressed in Escherichia coli.
Immunostaining.
Cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Fixed cells were permeabilized with 0.3% Triton X-100 for 10 min and blocked with 5% goat serum in PBS for 1 h at room temperature. For staining, cells were incubated overnight with the appropriate primary antibodies at 4°C. HA tag antibody from Cell Signaling (catalog no. 2367) was used instead of HA11 antibody for these experiments. Following incubation with primary antibodies, cells were washed three times with PBS and incubated with fluorescent secondary antibodies (Alexa Fluor antibodies). Cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI) and mounted on glass slides using Prolong Gold mounting medium. Cells were imaged using a Zeiss epifluorescence microscope.
RESULTS
PyST interacts with YAP.
To identify cellular proteins that interact with PyST, human U2OS cells that expressed PyST under the control of doxycycline were prepared. Mass spectrometry was performed with PyST complexes isolated by tandem affinity purification from these cells after induction. These experiments identified Yes-associated protein, YAP, as a PyST binding protein. YAP is a mammalian ortholog of Drosophila WW domain-containing transcription factor Yorkie. This observation drew our attention, since PyST has already been reported (9) to interact with TAZ, another family member. To confirm the mass spectrometry result, PyST was immunoprecipitated from extracts of murine 3T3 cells expressing PyST. Western blotting showed that YAP was coimmunoprecipitated with PyST (Fig. 1A). Comparison of the PyST and YAP levels in whole-cell lysates with their levels in immunoprecipitates indicated that only a small fraction of total YAP is coming down with PyST. YAP is well known to interact with the TEAD transcription factor (37). TEAD was also found in the mass spectrometry data and coimmunoprecipitated with PyST (Fig. 1A).
FIG 1.
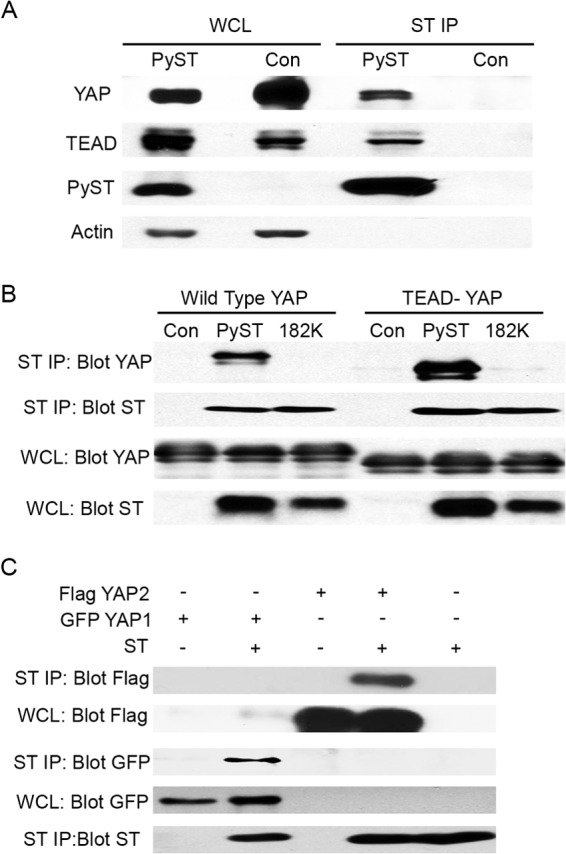
PyST binds to YAP. (A) NIH 3T3 were infected with lentivirus to express PyST or green fluorescent protein (GFP) control (Con) after induction with doxycycline. Whole-cell lysates (WCL) were collected after the induction by 300 ng/ml of doxycycline for 48 h. PyST was immunoprecipitated with ST1 antibody (ST IP). Western blotting with the same extracts was utilized to determine interactions with ST by blotting for YAP, TEAD, and ST (using PN116 antibody). Actin was used as a loading control. (B) 293T cells were cotransfected with combinations of HA-tagged wild-type PyST or YAP binding mutant PyST 182K along with either Flag-tagged wild-type YAP or S94A, a YAP mutant unable to bind TEAD (37). The whole-cell lysate was collected 48 h after transfection and immunoprecipitated with HA antibody. The immunoprecipitates were separated by SDS-PAGE and blotted for YAP or PyST, respectively. (C) 293T cells were transfected with the indicated combinations of PyST, GFP-tagged YAP1, or FLAG-tagged YAP2. Immunoprecipitation was performed with anti-ST antibody 48 h posttransfection. The PyST/YAP interaction from the immunoprecipitates was illustrated by Western blotting against YAP1, YAP2, and PyST (using PN116 antibody).
Since YAP and TEAD were both found in PyST immunoprecipitates, and YAP and TEAD are found in a complex, we wanted to determine whether the interaction was with YAP or TEAD. A point YAP mutant (S94A) (37) that is defective in TEAD binding was used to answer this question. This YAP mutant bound PyST (Fig. 1B), indicating that the interaction with PyST was not via TEAD. Comparison of the whole-cell lysates with the immunoprecipitates shows that PyST actually bound the TEAD-negative (TEAD−) YAP even better than the wild type.
YAP exists in two isoforms, YAP1 and YAP2 (11). To determine whether PyST interacts with both of them, tagged YAP constructs were cotransfected along with PyST. After PyST immunoprecipitation, blotting of the precipitates for each tag confirmed the association of PyST with both YAP isoforms (Fig. 1C).
Genetic analysis of PyST binding to YAP.
A set of PyST mutants was screened for binding to YAP. As shown in Fig. 2A, both R103A and D182K are defective in the ability to bind YAP as measured in cotransfection/IP experiments with 293T cells. To confirm that the binding was not a result of overexpression, C2C12 cell lines expressing wild-type and mutant small t antigens were derived. Figure 2B shows that D182K PyST is also defective in YAP binding in C2C12 cell lines. A similar result was obtained with 3T3-L1 cells used in the adipocyte apoptosis experiments (not shown).
FIG 2.

Genetics of PyST interactions with YAP and PP2A. (A) 293T cells were transfected with control (Con), HA-tagged PyST, as well as YAP binding mutants (103A, 182K). At 48 h after transfection, whole-cell lysate was collected and ST was immunoprecipitated using an HA antibody. The immunoprecipitates were used to quantify ST/YAP and ST/PP2A interaction by Western blotting against YAP and the PP2A A subunit. (B) Whole-cell lysates (WCL) were collected from C2C12 cells expressing PyST, YAP binding mutant PyST 182K, or GFP. The lysate was immunoprecipitated with a PyST antibody (IP PyST), and the relative YAP interaction was analyzed through Western blotting. The figure shows both short (YAP short) and longer (YAP long) film exposures. (C) NIH 3T3 cells were cotransfected with TKGal4 luciferase, GAL4-ATF2, β-galactosidase, empty vector control (Con), PyST, YAP binding mutants 103A and 182K, and PP2A binding mutants 135E, 142Y, and 157S. Cell lysates were collected 48 h posttransfection. Lysates were assayed (luciferase relative light units × 10−3/β-galactosidase activity), and the average from each experiment is plotted relative to the control. PyST had an average activation of about 8-fold. The standard error is indicated. (D) 293T cells were cotransfected with HA-PP2A Aα, Flag-YAP, and PyST PP2A binding mutants (157S, 142Y, and 135E). After 48 h posttransfection, whole-cell lysates (WCL) were collected. Immunoprecipitation (IP) of HA-PP2A Aα and Flag-YAP was performed from the same WCL. Western blotting against ST demonstrates YAP interaction relative to results for wild-type ST and the YAP binding mutant 182K. (E) Structural model of the complex formed between PyST (white), PP2A A (green), and C subunits (red). Residues leading to defects in YAP/TAZ binding (103 and 182) are highlighted in yellow. Residues required for PP2A binding (135, 142, and 157) are in red. 3DW8 (Protein Data Bank identifier) (70) was used for the structure of the PP2A holoenzyme; this included PP2A A, B, and C subunits. 2PF4 (71) was used for the SV40 ST and Aα interaction. These two structures were completely superimposed. The B subunit from 3DW8 was removed from this structure. Using the nuclear magnetic resonance (NMR) structure of murine polyomavirus, the J domain (amino acids 1 to 75) of murine polyomavirus was then superimposed in place of the SV40 J domain. Subsequently, the PyST unique domain sequence (amino acids 76 to 182) was put in place of the SV40 ST unique domain through homology modeling by Swiss-Model. (F) 293T cells were cotransfected with Flag-YAP, PyST, or YAP binding-deficient PyST (182K). At 48 h after transfection, whole-cell lysates (WCL) were collected and immunoprecipitated (IP) with anti-Flag antibody. The YAP/ST and YAP/PP2A interactions were assessed by Western blotting using an anti-PP2A A, anti-FLAG, or PN116 anti-T antibody.
Since PyST binds to YAP and to PP2A, experiments were performed to probe the relationship of these interactions. Figure 2A shows that mutants defective in YAP binding retain the ability to bind PP2A, a major target of PyST. PyST is known to activate ATF2 in a manner dependent on PP2A binding (6). Mutations that affect YAP binding do not affect the ability of PyST to act on PP2A, since R103A and D182K are wild type in activating ATF2 in reporter assays (Fig. 2C). For PyMT, binding PP2A is a prerequisite for c-src binding (38). To see whether PP2A might be needed for PyST to bind YAP, mutants bc1075 (C142Y) (39), K135E, and W157S (7), all known to be defective in PP2A binding, were examined. Coimmunoprecipitation experiments show that they retain the ability to bind YAP despite their PP2A binding defect (Fig. 2D). Taken together with the PP2A binding data for YAP mutants in Fig. 2A and D, these results show that YAP and PP2A bind PyST independently. This is reasonable, since the mutations affecting YAP binding and those affecting PP2A binding are located in different places in structural models of PyST (Fig. 2E). A final question is whether PyST affects the interaction of YAP with PP2A. As shown in Fig. 2F, cotransfection of YAP and PP2A gave only limited coimmunoprecipitation in the absence of PyST. This interaction was significantly enhanced by wild-type PyST but not by the YAP binding-defective D182K allele. This result demonstrated that PyST facilitates the interaction of YAP and PP2A.
PyST affects the phosphorylation state of YAP.
YAP is phosphorylated in a complicated fashion. In addition to LATS and possible Akt phosphorylation (29, 40), YAP is phosphorylated by casein kinase 1 (41), on three sites by cdk1 (42), and on tyrosine by src family kinases as well (43). Phosphorylation often slows the electrophoretic mobility of proteins. Figure 3A shows that wild-type but not 182 mutant PyST causes YAP to migrate more rapidly. It is evident that PyST affects a substantial fraction of YAP. Comparison of the shift seen here with the limited extent of coimmunoprecipitation seen in Fig. 1 and 2 suggests that PyST seems to be acting in a catalytic fashion rather than affecting only the YAP associated with it at a given moment. We do not favor, but cannot exclude, the alternative possibility that only a small fraction of the YAP/PyST complex survives immunoprecipitation. Extended electrophoresis on 12.5% gels results in a broad series of YAP bands (Fig. 3B). Mutants 135 and 157, which are defective in association with PP2A, failed to shift the mobility, supporting the argument that dephosphorylation is responsible for the faster migration. There are phosphospecific antibodies for some of the YAP phosphorylation sites. Phosphoserine 127 is associated with Hippo signaling to YAP via LATS. The center panel of Fig. 3C shows that S127 phosphorylation is associated with slower-migrating forms. Quantitation of these blots indicates that the ratio of phospho signal to total signal is 5.5 times lower in the PyST sample than in the control in this experiment. S381 (S397 in human YAP) is a phosphorylation site that regulates YAP stability (41). Phosphospecific antibody blotting indicates that pS381 is reduced 8- to 9-fold by PyST. Figure 3D summarizes the data from three independent experiments. Dephosphorylation of both sites depended on the ability of PyST to bind YAP, since the 182K mutant looked like the control. The simple explanation for these results is that PyST serves to bring YAP together with PP2A; this idea is supported by immunoprecipitation experiments from Fig. 2F.
FIG 3.
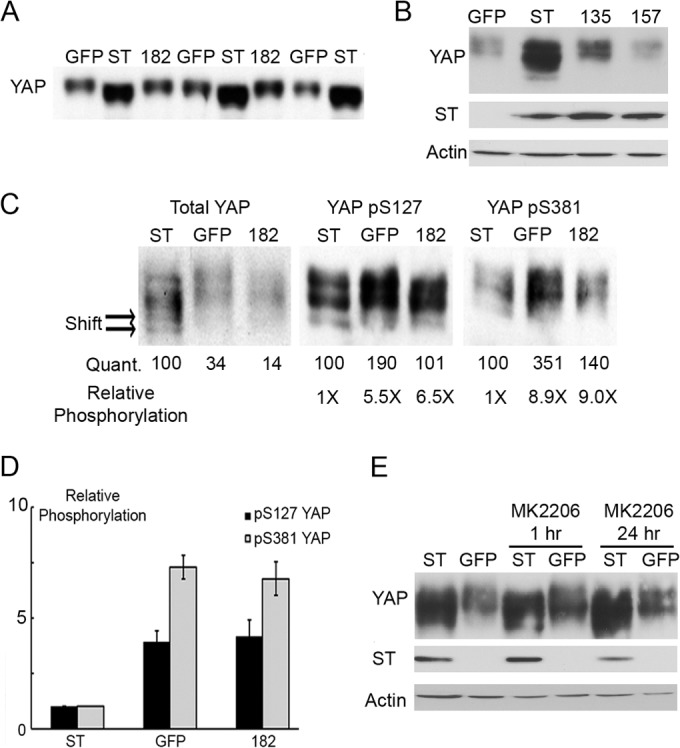
ST expression dephosphorylated YAP in cells. (A) Cell lysates were collected from fully confluent C2C12 cells expressing PyST (ST), the GFP control, or YAP binding mutant 182K (182). Multiple identical samples were analyzed by Western blotting for YAP after extensive gel electrophoresis on a 10% acrylamide gel to show the effect of wild-type PyST on YAP mobility. (B) Cell lysates were collected from fully confluent C2C12 cells expressing PyST (ST), the GFP control, or PP2A binding mutants 135E and 157S. Differences in YAP mobility were analyzed by Western blotting for YAP after extensive electrophoresis (24 h) on a 12.5% gel. (C) Samples from panel A were further analyzed by Western blotting after extensive gel electrophoresis on a 12.5% acrylamide gel for mobility shifts in total YAP, as well as YAP phosphorylation on S127 and S381. The extensive mobility shift induced by PyST expression is labeled with arrows (Shift). The intensity of each band was quantified by ImageJ and compared to the value of ST, which is set at 100 (Quant.). These values were used to calculate the relative intensity of YAP phosphorylation at S127 and S381 (Relative Phosphorylation). (D) The quantitation of relative phosphorylation from panel C and that of two additional biological repeats were averaged. The phosphorylation at S127 and S381 was plotted for cells expressing wild-type PyST, GFP, or the 182K mutant. (E) Akt inhibitor MK2206 was added to C2C12 cells expressing PyST (ST) or the GFP control for the indicated times. Western blotting was used to analyze YAP mobility, while actin was used as a loading control.
YAP phosphorylation is known to be regulated by Akt phosphorylation (29). Although PyST affects Akt activity (6, 7), the YAP mobility shift was not affected by inhibition of Akt by MK2206 (Fig. 3E), suggesting that PyST does not regulate YAP phosphorylation through Akt.
PyST stabilizes YAP expression by inhibiting interaction with βTRCP.
As shown for C2C12 cells growing toward confluence, the level of YAP increases when PyST is expressed (Fig. 4A). Even after induction of differentiation, the level of YAP remains higher in PyST cells. Figure 4B shows the same result for 3T3-L1 cells used here for both differentiation and apoptosis assays. The figure also shows that the effect of PyST on YAP requires binding, since neither 182 nor 103 PyST mutants elevated YAP levels. Protein turnover experiments were carried out by the addition of cycloheximide. As shown in Fig. 4C, YAP is much more stable when wild-type PyST is present than in control cells. Binding of YAP is required for this process, as it is for effects on YAP phosphorylation. The D182K mutant had no stabilizing effect on YAP. YAP is known to interact with the ubiquitin ligase β-transducin repeat protein (βTRCP) (41). βTRCP has attracted increasing attention for a possible role in tumorigenesis (44). Figure 5A shows that immunoprecipitation of βTRCP brought down YAP associating with it. When PyST was also expressed, this interaction was reduced. Figure 5B shows that the interaction was not affected by the D182K mutant, again suggesting that the physical interaction of PyST and YAP is important. PP2A mutant 135 was also unable to disrupt the interaction of YAP with βTRCP1. These results are consistent with the observed decrease in S381 phosphorylation in PyST-expressing cells.
FIG 4.
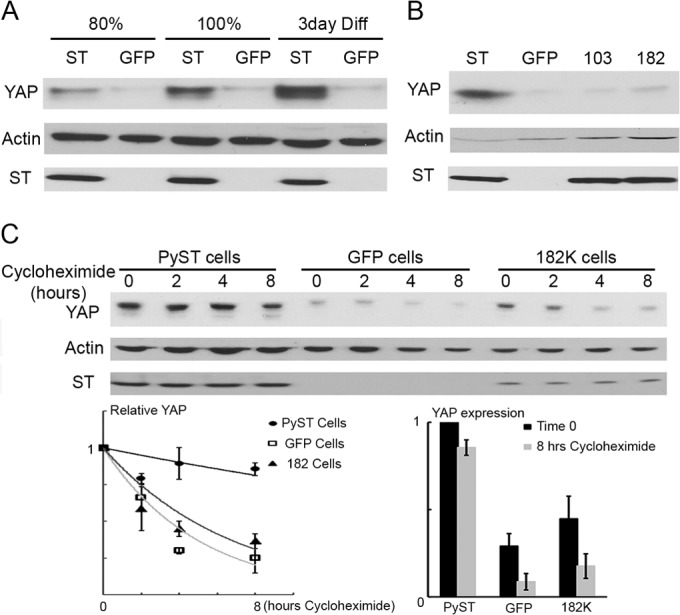
ST stabilizes expression of YAP. (A) YAP expression was analyzed by Western blotting from lysates collected from C2C12 cells expressing PyST (ST) or the GFP control at the following conditions: growing (80% confluent), confluent (100%), or after 3 days of differentiation (Diff). Actin is used as a loading control in each case. (B) YAP expression was analyzed as in panel A in 3T3-L1 cells induced by isobutylmethylxanthine (IBMX) for 24 h. As shown in Fig. 8, these are conditions under which PyST affects survival through YAP. These cells expressed PyST (ST), the GFP control, or YAP binding mutant 103A (103) or 182K (182). (C) Postconfluent C2C12 cells were analyzed for YAP expression by Western blotting at the indicated time points after the addition of 50 μg/ml cycloheximide. After quantification by ImageJ, the relative expression (YAP expression divided by actin expression) was analyzed. The relative rate of degradation was plotted along with the standard error from three experiments (graph at left). The overall change between 0 and 8 h is shown on the right with the standard error indicated.
FIG 5.
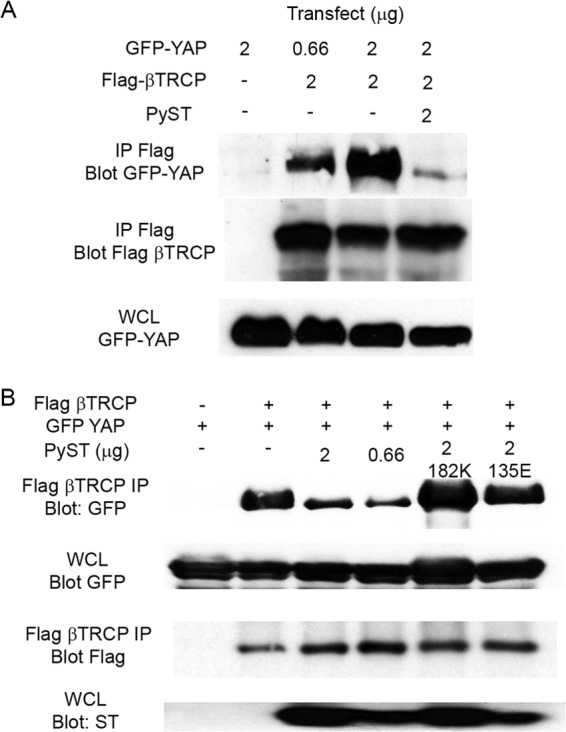
PyST inhibits βTRCP interaction with YAP. (A) 293T cells were cotransfected with the indicated amounts (in micrograms) of GFP-YAP, FLAG-βTRCP, and PyST. The whole-cell lysates (WCL) were collected and immunoprecipitated with Flag antibody. The interaction between YAP and βTRCP is determined by Western blotting against YAP and βTRCP. (B) An experiment similar to that in panel A was performed with the additional cotransfection of YAP binding mutant 182K and PP2A binding mutant 135E. Flag-βTRCP was immunoprecipitated and analyzed for YAP interaction by Western blotting.
PyST enhances nuclear localization of YAP.
YAP is found in both the cytoplasm and the nucleus. MCF10A breast cells have been quite useful in looking at changes in YAP localization (16). MCF10As were infected with retrovirus expressing PyST. In the cells that did not express PyST, YAP is dispersed throughout the cell (Fig. 6, bottom four cells indicated by arrows). However, when PyST is expressed, YAP is highly concentrated in the nucleus, as shown by the top two arrows.
FIG 6.
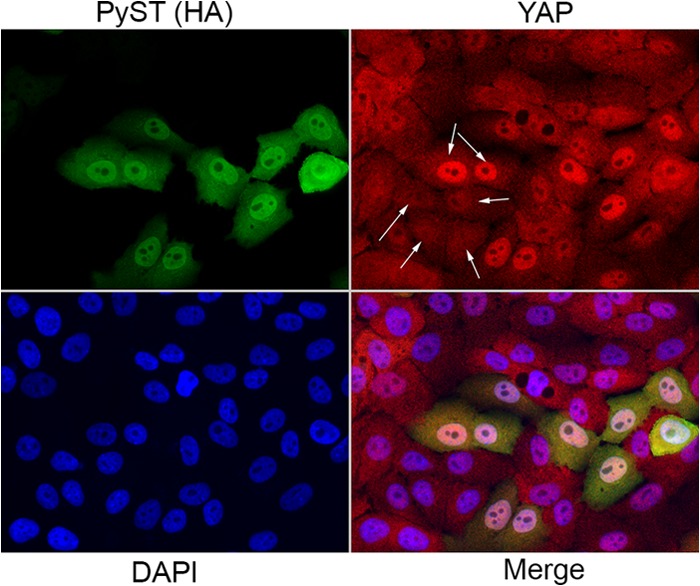
PyST promotes nuclear localization of YAP. (A) YAP localization in the presence of PyST was analyzed by immunofluorescence. MCF10a cells were infected with retroviruses expressing PyST. Fixed cells were stained with YAP antibody (shown in red; the top two arrows show highly concentrated YAP in the nucleus when PyST is expressed, and the bottom four arrows indicate that YAP is dispersed throughout the cell in cells that did not express PyST). PyST expression was detected with an HA tag antibody (shown in green). DAPI was used to stain the nucleus (shown in blue). A panel showing all three channels (Merge) is also shown.
PyST binding to YAP is needed to fully block differentiation of C2C12 myoblasts.
The important question is how YAP binding contributes to PyST phenotypes. A requirement for PP2A interaction has been demonstrated for a variety of PyST functions (6, 7, 45), including C2C12 myoblast differentiation (7, 8). To test the role of YAP binding in a PyST differentiation block, C2C12 cells expressing wild-type or mutant PySTs were prepared and allowed to differentiate for 9 days. We then estimated the extent of differentiation by immunoblotting of myosin heavy chain (MHC) (Fig. 7A). As shown previously, wild-type PyST completely blocked differentiation and the PP2A mutant W157S was completely ineffective. Both the 103A and 182K YAP binding mutants that bind PP2A, as well as the PP2A binding-defective 135E mutant that binds YAP, showed only very minor decreases in differentiation. These results show that binding of YAP by PyST contributes to its ability to inhibit differentiation. The 135E mutant was also defective even though it shows high levels of YAP binding. Thus, YAP binding by itself is not sufficient to recreate the wild-type phenotype. PP2A is also required.
FIG 7.
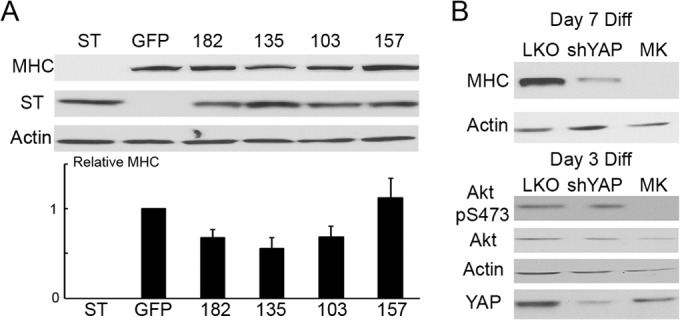
PyST requires YAP binding to block differentiation, and YAP is required for differentiation. (A) C2C12 cells expressing ST, the GFP control, or the indicated mutants were differentiated for 9 days. The extent of differentiation was first analyzed by Western blotting against myosin heavy chain (MHC). Actin was used as a loading control. This representative experiment was quantitated with ImageJ, and the value for GFP was set to 1. The numerical values were subsequently averaged with those of 5 other biological repeats, and relative MHC (numerical quantity of MHC divided by respective expression of actin in the same sample) is plotted below. Error bars represent standard errors. (B) C2C12 cells were infected with a lentivirus containing a short hairpin RNA (shRNA) against YAP (shYAP) or a scrambled control (LKO) prior to differentiation. Differentiation was also performed in the presence of 2 μM Akt inhibitor MK2206. The extent of differentiation after 7 days was quantitated by Western blotting against MHC using actin as a control. At day 3, the knockdown of YAP was determined by Western blotting measuring total YAP, while the effect of MK2206 was verified by detecting Akt phosphorylation at S473. Actin was also used as a loading control.
The role of YAP signaling in myoblast differentiation seems to be complex. While it was clear that PyST action on YAP is needed for its effect on differentiation, it could either inhibit or activate YAP. YAP is a coactivator for TEAD, which is required for C2C12 differentiation (46). Consistent with this, Fig. 7B shows that knockdown of YAP reduced differentiation as measured by the expression of MHC.
Akt is also required for myoblast differentiation (47–49). Its activation is observed earlier in the differentiation process, before most MHC expression. Figure 7B confirms this by showing that the Akt inhibitor MK2206 completely blocks differentiation. The ability of PyST to block differentiation has also been strongly connected to inhibition of Akt activity (8). Knockdown of YAP by itself (Fig. 7B) seems to block differentiation without affecting Akt activation as measured by S473 phosphorylation. This indicates a role for YAP in differentiation apart from Akt. In contrast to the need for YAP activity for differentiation to proceed, constitutive activation using a nonphosphorylatable S127A YAP blocks differentiation (31). Given the effect of PyST on YAP as shown in Fig. 3, this seems a likely mechanism.
PyST requires YAP to induce IBMX-dependent apoptosis in preadipocytes.
Differentiation of preadipocyte 3T3-L1 cells to adipocytes is achieved using a set of stimulators that includes a phosphodiesterase inhibitor, isobutylmethylxanthine (IBMX). IBMX elevates both cyclic AMP (cAMP) and cGMP. PyST blocks adipocyte differentiation but also causes cell death in this system. Addition of IBMX alone to confluent 3T3-L1 cells allows PyST to kill. This can be seen in the morphologies of the treated cells in Fig. 8A and the induction of activated caspase 3 in Fig. 8B. The ability of PyST to induce apoptosis was fully dependent on YAP binding, as demonstrated by the lack of apoptosis in YAP binding PyST mutants 103A and 182K (Fig. 8A). The YAP binding-defective mutants also failed to activate caspase 3, in contrast to wild-type PyST (Fig. 8B). Both the 135 and 157 mutants, which are defective in PP2A but retain YAP binding, showed reduced killing and caspase activation. Figure 8C demonstrates that YAP knockdown blocks caspase 3 activation by PyST. This supports a role for YAP in this death. In other situations, PyST-induced cell death involves Akt (6, 7). The death seen here appears to be independent of Akt. The addition of Akt inhibitors MK2206 and IBMX to control cells did not synergize to induce death nor did MK2206 enhance caspase 3 induction by PyST (Fig. 8D).
FIG 8.
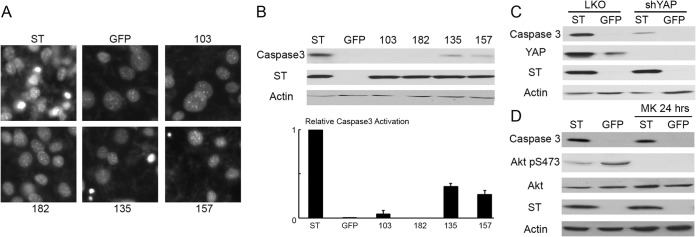
ST induces caspase-dependent apoptosis when IBMX is added. (A) Postconfluent 3T3-L1 cells expressing ST, the GFP control, YAP binding mutants (103A and 182K), or PP2A binding mutants (135E and 157S) were stimulated with 0.5 mM IBMX. Live-cell nuclei were stained with Hoechst 33342 for 30 min. These were observed at ×20 magnification, and pictures were taken. Cell rounding and detachment were observed in culture, and representative pictures were taken. (B) Lysates from the set of cells in panel A were also collected after 24 h of IBMX stimulation. The apoptosis was analyzed by Western blotting illustrating the relative expression of active caspase 3. Actin was used as a loading control in this experiment. The figure was quantitated with ImageJ, and the relative amount of active caspase 3 (numerical value of caspase 3 divided by actin in the same sample) was plotted. Error bars represent standard errors. (C) YAP was suppressed in 3T3-L1 cells with a lentivirus expressing shRNA against YAP and compared to a scrambled control. After the cells reached confluence, they were stimulated with 0.5 mM IBMX for 24 h. The relative amount of apoptosis was quantitated by Western blotting to detect activated caspase 3. Actin was used as a control. (D) Akt inhibitor MK2206 (2 μM) was added to confluent cells expressing PyST (ST) and GFP along with IBMX for 24 h. The relative amount of apoptosis is illustrated by Western blotting using actin as a control.
DISCUSSION
This work demonstrates that PyST binds to YAP1 and YAP2. The YAP partner TEAD is also found in the YAP-PyST complex. Genetic analysis has identified a region on PyST required for this binding. Since this same sequence is also present in PyMT, it is not surprising that PyMT is also a YAP binding protein (not shown). Binding of YAP to PyST results in its dephosphorylation at key residues. Neither PyST mutants defective in YAP binding nor mutants with a defect in PP2A binding cause YAP dephosphorylation. At any given moment, the fraction of YAP bound to PyST as measured by coimmunoprecipitation is small. However, a significant fraction of YAP shows a phosphorylation-related mobility shift, and the amount of phosphorylation at specific sites, such as S127 and S381, decreases 5- to 9-fold (Fig. 3B). Thus, the PyST-YAP interaction seems rather like an enzyme, in this case PP2A with a PyST subunit, interacting with a substrate, YAP. Since YAP is degraded through a pathway involving binding of its phosphodegron to βTRCP (41), binding to PyST results in its stabilization.
Two clear examples of the contribution of YAP binding to PyST biology are documented. Previously, we have shown that PyST blocks differentiation of preadipocytes to adipocytes and myoblasts to myocytes and osteoblasts (8). Mutants that do not bind YAP are not as effective in blocking myoblast differentiation as the wild type, although they still have some effect. From one point of view, this is not surprising. It is known that unphosphorylatable S127A YAP, often viewed as constitutively active, blocks differentiation (31). However, we also find that YAP knockdown interferes with differentiation. This may mean that YAP activity makes a positive contribution at some stage of differentiation and then its suppression is also required for differentiation. Previously, we have shown that PyST affects Akt activity as part of the differentiation block (8). The role of YAP appears to be independent of Akt, since YAP knockdown reduces differentiation without affecting Akt (Fig. 7B).
The second pathway specifically affected by PyST interaction with YAP is a survival pathway. PyST effects on cell survival are complex. In 3T3 cells, acute expression of PyST leads to cell death when cells are grown in normal serum-containing medium, but PyST can protect against cell death promoted by the absence of serum (6). The acute cell death caused by PyST expression results from cell cycle arrest in the G2/M phases of the cell cycle, followed by mitotic catastrophe (50). This kind of killing does not seem to require YAP binding, since YAP binding mutants remain active in these assays (not shown). Since the phosphorylation of YAP is limited in growing cells (16), perhaps PyST does not need to target YAP under those conditions. 3T3-L1 cells differentiate starting from confluence by induction with a cocktail of activators (insulin, dexamethasone, IBMX, troglitazone). PyST induces cell death when differentiation is induced under these conditions. Examination of the activators showed that IBMX was necessary and sufficient to induce death. The requirement for IBMX to induce killing strongly suggests a role for cyclic nucleotides in the killing phenotype. There is already significant interest in the role of cyclic nucleotides in cancer (see reference 51 for a review). Since forskolin, an inducer of cAMP, does not substitute for IBMX in triggering cell death, it is quite likely that cGMP is involved. Because PyST could be shown to block differentiation even when IBMX was omitted, the death and the block to differentiation are clearly distinct phenotypes.
Death induced in the presence of IBMX required that PyST bind to YAP. Since YAP is ordinarily suppressed by phosphorylation at high cell densities, perhaps it is the activation of YAP by PyST decreasing its phosphorylation that facilitates cell death. In support of this idea, knockdown of YAP prevented killing by PyST. The involvement of YAP in support of apoptosis is in contrast with the usual role of Hippo signaling through YAP, which is most often thought to promote proliferation and survival. However, YAP can also promote cell death via a p73-dependent pathway, mainly via Bax induction (27–30). Nevertheless, this does not explain our observations, since we do not see an increase in Bax protein levels during cell death (data not shown). This suggests that there might be a novel Bax-independent mode of YAP-mediated cell death.
This work stresses the need for a detailed examination of YAP function as it relates to PyST. YAP is phosphorylated in a very complex fashion. In addition to tyrosine phosphorylation by src family members, YAP is phosphorylated on Ser/Thr by LATS in the canonical Hippo pathway, as well as by cdk1 (42), casein kinase 1 (41), Akt (29), and other kinases (40, 43, 52, 53). We have shown that S127 and S381 are dephosphorylated by PyST, but does PyST affect all, or only some, of these phosphorylations? How do these effects, dependent on PP2A, compare to regulation by PP1 (54)? The YAP/TAZ family is well known for its effects on transcription (10, 15, 55). The TEAD/TEF family may be the most familiar target/partner of YAP (37, 56). However, the YAP/TAZ family is known to interact with TBX-5 (57), Runx (58, 59), Smad (60, 61), and p73 (62), among others. In these systems, it is the dephosphorylated form of YAP that is thought to be active, with phosphorylated YAP sequestered in the cytosol by association with the 14-3-3 proteins (56, 63). Here, we observe that PyST increases nuclear localization of YAP. It is possible that the difference in YAP's targets arises from the specifics of the phosphorylation control of YAP. YAP mutant analysis should eventually test this idea. There is also cross talk between YAP and Wnt signaling on different levels, including beta catenin (34) and Disheveled (64; for a review, see reference 65). In these situations, it is the phosphorylated YAP that is active. It will be of interest to see which of these transcriptional activities PyST modulates. Recently, attention has focused on YAP and microRNA production (17, 18). MicroRNAs are generally downregulated in tumors (66). In some cases, miRNA can be used as an antitumor therapeutic (67). Contact inhibition, which is associated with YAP inactivation, activates microRNA biosynthesis (68). YAP regulates miRNAs by binding p72 (DDX17) (18). Does PyST regulate the amount or spectrum of miRNAs? At a different level, YAP is involved in mechanical-sensory regulation relating to architecture and environment (see reference 69 for a review). By studying the interactions of t antigens with host proteins, it has often been possible to gain insights into convoluted control mechanisms at work in the host cell. Future work will determine how PyST modulates all of these functions and hopefully shed light on their regulation in the absence of PyST.
ACKNOWLEDGMENTS
This work was supported by NIH grants CA50661 (W.C.H., T.M.R., and B.S.S.), CA30002 (T.M.R.), and CA34722 (B.S.S.).
We thank Celia Harrison for constructing the structural model in Fig. 2E. We also thank Yanan Chen for technical assistance.
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1. Eckhart W, Hutchinson MA, Hunter T. 1979. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18:925–933. 10.1016/0092-8674(79)90205-8 [DOI] [PubMed] [Google Scholar]
- 2. Cartwright CA, Hutchinson MA, Eckhart W. 1985. Structural and functional modification of pp60c-src associated with polyoma middle tumor antigen from infected or transformed cells. Mol. Cell. Biol. 5:2647–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. 1985. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239–242. 10.1038/315239a0 [DOI] [PubMed] [Google Scholar]
- 4. Asselin C, Gelinas C, Branton PE, Bastin M. 1984. Polyoma middle T antigen requires cooperation from another gene to express the malignant phenotype in vivo. Mol. Cell. Biol. 4:755–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Shea CC, Fried M. 2005. Modulation of the ARF-p53 pathway by the small DNA tumor viruses. Cell Cycle 4:449–452. 10.4161/cc.4.3.1555 [DOI] [PubMed] [Google Scholar]
- 6. Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B. 2007. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 104:19011–19016. 10.1073/pnas.0706696104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andrabi S, Hwang JH, Choe JK, Roberts TM, Schaffhausen BS. 2011. Comparisons between murine polyomavirus and Simian virus 40 show significant differences in small T antigen function. J. Virol. 85:10649–10658. 10.1128/JVI.05034-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hwang JH, Jiang T, Kulkarni S, Faure N, Schaffhausen BS. 2013. Protein phosphatase 2A isoforms utilizing Abeta scaffolds regulate differentiation through control of Akt protein. J. Biol. Chem. 288:32064–32073. 10.1074/jbc.M113.497644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tian Y, Li D, Dahl J, You J, Benjamin T. 2004. Identification of TAZ as a binding partner of the polyomavirus T antigens. J. Virol. 78:12657–12664. 10.1128/JVI.78.22.12657-12664.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang K, Degerny C, Xu M, Yang XJ. 2009. YAP, TAZ, and Yorkie: a conserved family of signal-responsive transcriptional coregulators in animal development and human disease. Biochem. Cell Biol. 87:77–91. 10.1139/O08-114 [DOI] [PubMed] [Google Scholar]
- 11. Sudol M, Shields DC, Farooq A. 2012. Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin. Cell Dev. Biol. 23:827–833. 10.1016/j.semcdb.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu C, Huang W, Lei Q. 2011. Regulation and function of the TAZ transcription co-activator. Int. J. Biochem. Mol. Biol. 2:247–256 [PMC free article] [PubMed] [Google Scholar]
- 13. Varelas X. 2014. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141:1614–1626. 10.1242/dev.102376 [DOI] [PubMed] [Google Scholar]
- 14. Zhao B, Li L, Guan KL. 2010. Hippo signaling at a glance. J. Cell Sci. 123:4001–4006. 10.1242/jcs.069070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishio M, Otsubo K, Maehama T, Mimori K, Suzuki A. 2013. Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci. 104:1271–1277. 10.1111/cas.12227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21:2747–2761. 10.1101/gad.1602907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chaulk SG, Lattanzi VJ, Hiemer SE, Fahlman RP, Varelas X. 2014. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 289:1886–1891. 10.1074/jbc.C113.529362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI. 2014. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 156:893–906. 10.1016/j.cell.2013.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu MZ, Yao TJ, Lee NP, Ng IO, Chan YT, Zender L, Lowe SW, Poon RT, Luk JM. 2009. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 115:4576–4585. 10.1002/cncr.24495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW. 2006. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125:1253–1267. 10.1016/j.cell.2006.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. 2006. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. U. S. A. 103:12405–12410. 10.1073/pnas.0605579103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. 2007. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17:2054–2060. 10.1016/j.cub.2007.10.039 [DOI] [PubMed] [Google Scholar]
- 23. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133. 10.1016/j.cell.2007.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A, Wen W, Zhu Q. 2013. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS One 8:e65539. 10.1371/journal.pone.0065539 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T, Hahn WC. 2014. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158:171–184. 10.1016/j.cell.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD. 2011. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144:782–795. 10.1016/j.cell.2011.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levy D, Adamovich Y, Reuven N, Shaul Y. 2007. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 14:743–751. 10.1038/sj.cdd.4402063 [DOI] [PubMed] [Google Scholar]
- 28. Strano S, Monti O, Pediconi N, Baccarini A, Fontemaggi G, Lapi E, Mantovani F, Damalas A, Citro G, Sacchi A, Del Sal G, Levrero M, Blandino G. 2005. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol. Cell 18:447–459. 10.1016/j.molcel.2005.04.008 [DOI] [PubMed] [Google Scholar]
- 29. Basu S, Totty NF, Irwin MS, Sudol M, Downward J. 2003. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 11:11–23. 10.1016/S1097-2765(02)00776-1 [DOI] [PubMed] [Google Scholar]
- 30. Lee KK, Yonehara S. 2012. Identification of mechanism that couples multisite phosphorylation of Yes-associated protein (YAP) with transcriptional coactivation and regulation of apoptosis. J. Biol. Chem. 287:9568–9578. 10.1074/jbc.M111.296954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watt KI, Judson R, Medlow P, Reid K, Kurth TB, Burniston JG, Ratkevicius A, De Bari C, Wackerhage H. 2010. Yap is a novel regulator of C2C12 myogenesis. Biochem. Biophys. Res. Commun. 393:619–624. 10.1016/j.bbrc.2010.02.034 [DOI] [PubMed] [Google Scholar]
- 32. Zhang H, Deo M, Thompson RC, Uhler MD, Turner DL. 2012. Negative regulation of Yap during neuronal differentiation. Dev. Biol. 361:103–115. 10.1016/j.ydbio.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin YT, Ding JY, Li MY, Yeh TS, Wang TW, Yu JY. 2012. YAP regulates neuronal differentiation through Sonic hedgehog signaling pathway. Exp. Cell Res. 318:1877–1888. 10.1016/j.yexcr.2012.05.005 [DOI] [PubMed] [Google Scholar]
- 34. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, Vazquez F, Cowley GS, Root DE, Mesirov JP, Beroukhim R, Kuo CJ, Goessling W, Hahn WC. 2012. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151:1457–1473. 10.1016/j.cell.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen CA, Okayama H. 1988. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques 6:632–638 [PubMed] [Google Scholar]
- 36. Morgenstern JP, Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596. 10.1093/nar/18.12.3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao B, Ye X, Yu JD, Li L, Li WQ, Li SM, Yu JJ, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22:1962–1971. 10.1101/gad.1664408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fluck MM, Schaffhausen BS. 2009. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol. Mol. Biol. Rev. 73:542–563. 10.1128/MMBR.00009-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martens I, Nilsson SA, Linder S, Magnusson G. 1989. Mutational analysis of polyomavirus small-T-antigen functions in productive infection and in transformation. J. Virol. 63:2126–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hao Y, Chun A, Cheung K, Rashidi B, Yang X. 2008. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283:5496–5509. 10.1074/jbc.M709037200 [DOI] [PubMed] [Google Scholar]
- 41. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. 2010. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24:72–85. 10.1101/gad.1843810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. 2013. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 73:6722–6733. 10.1158/0008-5472.CAN-13-2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Levy D, Adamovich Y, Reuven N, Shaul Y. 2008. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 29:350–361. 10.1016/j.molcel.2007.12.022 [DOI] [PubMed] [Google Scholar]
- 44. Lau AW, Fukushima H, Wei W. 2012. The Fbw7 and betaTRCP E3 ubiquitin ligases and their roles in tumorigenesis. Front. Biosci. 17:2197–2212. 10.2741/4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mullane KP, Ratnofsky M, Cullere X, Schaffhausen B. 1998. Signaling from polyomavirus middle T and small T defines different roles for protein phosphatase 2A. Mol. Cell. Biol. 18:7556–7564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Benhaddou A, Keime C, Ye T, Morlon A, Michel I, Jost B, Mengus G, Davidson I. 2012. Transcription factor TEAD4 regulates expression of myogenin and the unfolded protein response genes during C2C12 cell differentiation. Cell Death Differ. 19:220–231. 10.1038/cdd.2011.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilson EM, Tureckova J, Rotwein P. 2004. Permissive roles of phosphatidyl inositol 3-kinase and Akt in skeletal myocyte maturation. Mol. Biol. Cell 15:497–505. 10.1091/mbc.E03-05-0351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gonzalez I, Tripathi G, Carter EJ, Cobb LJ, Salih DA, Lovett FA, Holding C, Pell JM. 2004. Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways in myogenesis. Mol. Cell. Biol. 24:3607–3622. 10.1128/MCB.24.9.3607-3622.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Q, Wu Z. 2000. The insulin-like growth factor-phosphatidylinositol 3-kinase-Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma-derived RD cells. J. Biol. Chem. 275:36750–36757. 10.1074/jbc.M005030200 [DOI] [PubMed] [Google Scholar]
- 50. Pores Fernando AT, Andrabi S, Cizmecioglu O, Zhu C, Livingston DM, Higgins JM, Schaffhausen BS, Roberts TM. 7 July 2014. Polyoma small T antigen triggers cell death via mitotic catastrophe. Oncogene, in press. 10.1038/onc.2014.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fajardo AM, Piazza GA, Tinsley HN. 2014. The role of cyclic nucleotide signaling pathways in cancer: targets for prevention and treatment. Cancers (Basel) 6:436–458. 10.3390/cancers6010436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, Rosato A, Piccolo S, Del Sal G. 2014. Metabolic control of YAP and TAZ by the mevalonate pathway. Nature Cell Biol. 16:357–366. 10.1038/ncb2936 [DOI] [PubMed] [Google Scholar]
- 53. Huang J, Wu S, Barrera J, Matthews K, Pan D. 2005. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122:421–434. 10.1016/j.cell.2005.06.007 [DOI] [PubMed] [Google Scholar]
- 54. Wang P, Bai Y, Song B, Wang Y, Liu D, Lai Y, Bi X, Yuan Z. 2011. PP1A-mediated dephosphorylation positively regulates YAP2 activity. PLoS One 6:e24288. 10.1371/journal.pone.0024288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hong W, Guan KL. 2012. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 23:785–793. 10.1016/j.semcdb.2012.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. 2001. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 15:1229–1241. 10.1101/gad.888601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murakami M, Nakagawa M, Olson EN, Nakagawa O. 2005. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. U. S. A. 102:18034–18039. 10.1073/pnas.0509109102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zaidi SK, Sullivan AJ, Medina R, Ito Y, van Wijnen AJ, Stein JL, Lian JB, Stein GS. 2004. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23:790–799. 10.1038/sj.emboj.7600073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cui CB, Cooper LF, Yang X, Karsenty G, Aukhil I. 2003. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol. Cell. Biol. 23:1004–1013. 10.1128/MCB.23.3.1004-1013.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW, Wrana JL. 2008. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nature Cell Biol. 10:837–848. 10.1038/ncb1748 [DOI] [PubMed] [Google Scholar]
- 61. Lee MJ, Ran Byun M, Furutani-Seiki M, Hong JH, Jung HS. 2014. YAP and TAZ regulate skin wound healing. J. Invest. Dermatol. 134:518–525. 10.1038/jid.2013.339 [DOI] [PubMed] [Google Scholar]
- 62. Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G. 2001. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 276:15164–15173. 10.1074/jbc.M010484200 [DOI] [PubMed] [Google Scholar]
- 63. Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, Hisaminato A, Fujiwara T, Ito Y, Cantley LC, Yaffe MB. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19:6778–6791. 10.1093/emboj/19.24.6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, Kuo CJ, Camargo FD. 2013. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493:106–110. 10.1038/nature11693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Konsavage WM, Jr, Yochum GS. 2013. Intersection of Hippo/YAP and Wnt/beta-catenin signaling pathways. Acta Biochim. Biophys. Sin. (Shanghai) 45:71–79. 10.1093/abbs/gms084 [DOI] [PubMed] [Google Scholar]
- 66. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. 2005. MicroRNA expression profiles classify human cancers. Nature 435:834–838. 10.1038/nature03702 [DOI] [PubMed] [Google Scholar]
- 67. Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT. 2009. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137:1005–1017. 10.1016/j.cell.2009.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hwang HW, Wentzel EA, Mendell JT. 2009. Cell-cell contact globally activates microRNA biogenesis. Proc. Natl. Acad. Sci. U. S. A. 106:7016–7021. 10.1073/pnas.0811523106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hao J, Zhang Y, Wang Y, Ye R, Qiu J, Zhao Z, Li J. 2014. Role of extracellular matrix and YAP/TAZ in cell fate determination. Cell. Signal. 26:186–191. 10.1016/j.cellsig.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 70. Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. 2008. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol. Cell 31:873–885. 10.1016/j.molcel.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cho US, Morrone S, Sablina AA, Arroyo JD, Hahn WC, Xu W. 2007. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 5:e202. 10.1371/journal.pbio.0050202 [DOI] [PMC free article] [PubMed] [Google Scholar]