

ABSTRACT
Few drugs targeting picornaviruses are available, making the discovery of antivirals a high priority. Here, we identified and characterized three compounds from a library of kinase inhibitors that block replication of poliovirus, coxsackievirus B3, and encephalomyocarditis virus. Using an in vitro translation-replication system, we showed that these drugs inhibit different stages of the poliovirus life cycle. A4(1) inhibited both the formation and functioning of the replication complexes, while E5(1) and E7(2) were most effective during the formation but not the functioning step. Neither of the compounds significantly inhibited VPg uridylylation. Poliovirus resistant to E7(2) had a G5318A mutation in the 3A protein. This mutation was previously found to confer resistance to enviroxime-like compounds, which target a phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ)-dependent step in viral replication. Analysis of host protein recruitment showed that E7(2) reduced the amount of GBF1 on the replication complexes; however, the level of PI4KIIIβ remained intact. E7(2) as well as another enviroxime-like compound, GW5074, interfered with viral polyprotein processing affecting both 3C- and 2A-dependent cleavages, and the resistant G5318A mutation partially rescued this defect. Moreover, E7(2) induced abnormal recruitment to membranes of the viral proteins; thus, enviroxime-like compounds likely severely compromise the interaction of the viral polyprotein with membranes. A4(1) demonstrated partial protection from paralysis in a murine model of poliomyelitis. Multiple attempts to isolate resistant mutants in the presence of A4(1) or E5(1) were unsuccessful, showing that effective broad-spectrum antivirals could be developed on the basis of these compounds.
IMPORTANCE Diverse picornaviruses can trigger multiple human maladies, yet currently, only hepatitis A virus and poliovirus can be controlled with vaccination. The development of antipicornavirus therapeutics is also facing significant difficulties because these viruses readily generate resistance to compounds targeting either viral or cellular factors. Here, we describe three novel compounds that effectively block replication of distantly related picornaviruses with minimal toxicity to cells. The compounds prevent viral RNA replication after the synthesis of the uridylylated VPg primer. Importantly, two of the inhibitors are strongly refractory to the emergence of resistant mutants, making them promising candidates for further broad-spectrum therapeutic development. Evaluation of one of the compounds in an in vivo model of poliomyelitis demonstrated partial protection from the onset of paralysis.
INTRODUCTION
Picornaviruses are a family of positive-strand RNA viruses that infect diverse human and animal hosts. Many members of this group, such as polioviruses, rhinoviruses, foot-and-mouth disease viruses, and others, can cause serious diseases associated with a significant public health burden and high economic costs. Currently, only hepatitis A virus and poliovirus can be effectively controlled by vaccination, while for most picornavirus-induced pathologies, modern medicine can offer nothing more than supportive therapies. The major obstacle in vaccine development is the broad antigenic diversity of viruses associated with specific diseases, which in many cases makes the vaccination approach impractical. For example, rhinoviruses, the major cause of the common cold, resulting in multibillion-dollar losses annually due to loss of productivity and cost of treatment (1, 2), comprise more than a hundred known individual serotypes, and the number is growing (3). Similarly, other serious human conditions, such as type I diabetes and myocarditis, may be associated with diverse viruses from the Enterovirus genus of the Picornaviridae family (4–6), making the development of comprehensive vaccines problematic. Thus, antiviral therapies would be highly desirable for many picornavirus-associated pathological conditions that are impossible to control by vaccination. Even for poliovirus, which has almost been eliminated via massive vaccination campaigns in the course of the WHO polio eradication initiative, antiviral drugs could play an important role in treating chronically infected individuals and preventing them from shedding virulent viruses into the environment. Maintaining stockpiles of antipoliovirus drugs could also mitigate risks of polio reemergence after circulation of wild and vaccine-derived polioviruses has been stopped (7).
Traditionally, antiviral drugs are designed to target virus-specific proteins. This approach holds the advantage of minimizing host toxicity, since the drug is expected to specifically interact with only the viral protein and ideally not interfere with cellular metabolism. At the same time, therapeutics targeting virus-specific proteins are inevitably effective against only very closely related viruses with minimal divergence of protein sequences. An alternative approach is to inhibit host-specific proteins involved in the viral replication cycle. Since related viruses are expected to share basic mechanisms of replication, targeting of one host factor may potentially generate a broad-spectrum antiviral effective against all viruses that rely on this host protein. Host factors as antiviral targets came into focus relatively recently, largely because very few such factors are still known but also because targeting of a host protein bears a higher risk of inducing toxicity.
The daunting problem for designing effective antiviral therapeutics is the emergence of resistant mutants. Positive-strand RNA viruses, including picornaviruses, are notorious for their ability to escape therapeutic pressure. The low fidelity of the viral RNA-dependent RNA polymerase results in an accumulation of a range of mutants that coexist in viral populations (quasispecies), providing a source for rapid selection of variants resistant to antiviral drugs. Poliovirus readily generates resistance to compounds targeting either viral proteins or cellular factors involved in the viral life cycle (8–10), imposing significant restrictions on the repertoire of targets for drug development. Resistance to the prospective antipicornavirus compounds pleconaril and V-073 is well documented, restricting their use to cases of life-threatening infections and public health emergencies (11). Currently, the emergence of resistant mutants is accepted as an unavoidable pitfall. However, it does not mean that it is impossible to find drugs that will not easily induce resistant virus. Inhibition of at least one cellular protein, the chaperone Hsp-90, involved in the folding of poliovirus capsid proteins, was shown to be refractory to the emergence of resistant mutants in vitro and in vivo (12).
Poliovirus is the prototype member of the Picornaviridae family and is among the best-studied animal viruses. The arsenal of available research tools makes poliovirus a perfect model for the discovery of novel antiviral approaches, especially those targeting picornaviruses and in particular members of the Enterovirus genus. The poliovirus genome RNA of ∼7,500 nucleotides (nt) is directly translated into one polyprotein that is cleaved in cis and in trans by viral proteases into structural proteins that make up the capsid and nonstructural proteins involved in the replication of viral RNA (Fig. 1A). Viral RNA replication is performed by RNA-dependent RNA polymerase 3D, which also uridylylates the small viral protein VPg (3B), which serves as a primer for the synthesis of both negative and positive RNA strands. Other viral nonstructural proteins facilitate the replication process, but their precise role is not well established. Replication of poliovirus RNA is associated with membranous replication organelles where viral and host factors involved in replication are assembled (13). The complex process of viral genome expression and replication apparently depends on the coordinated activity and multiple interactions of cellular and viral proteins, providing potentially countless targets for antiviral therapeutics.
FIG 1.

Effect of kinase inhibitors on poliovirus replication. (A) Scheme of the poliovirus genome and the replicon construct coding for Renilla luciferase. (B and C) Representative experiments showing poliovirus replicon replication in the presence of 10 μM kinase inhibitors from library I (B) and library II (C) and 100 μM z-VAD-fmk. HeLa cells grown on a 96-well plate were transfected with poliovirus replicon RNA with a Renilla luciferase gene. After transfection, each inhibitor was added to one well of a 96-well plate (x-axis labels). The cells were incubated for 16 h in the presence of a cell-permeable Renilla luciferase substrate. The total integrated luciferase signal measured each hour over 16 h of observation is plotted. Data are normalized to values for control replication in the presence of DMSO (solvent); BFA served as a negative control. Toxicity was measured in the same wells of a 96-well plate after the replication experiment was finished; data are normalized to control values (DMSO).
Here, we report the identification of three novel compounds from a screen of cell-permeable kinase inhibitors that effectively blocked replication of poliovirus at different stages of the replication cycle: A4(1) (Akt inhibitor IV), E5(1) (platelet-derived growth factor [PDGF] receptor tyrosine kinase inhibitor III), and E7(2) (indirubin derivative E804). All three compounds were effective against poliovirus, the related enterovirus coxsackievirus B3 (CVB3), as well as the much more distantly related encephalomyocarditis virus (EMCV). Separate assessments of individual steps in viral replication showed that A4(1) blocked both the formation and functioning of the poliovirus replication complexes. E5(1) and E7(2) prevented the assembly of the functional replication complexes but could not interfere with the replication reaction. Replication complexes assembled in the presence of the compounds were fully competent in the synthesis of a uridylylated VPg primer but could not support effective RNA replication. The antiviral effect of these compounds is not likely to be related to their annotated cellular targets, since other inhibitors of the same kinases did not interfere with viral replication. A resistant poliovirus mutant selected upon propagation in the presence of E7(2) contained a mutation in viral protein 3A that was previously reported to confer resistance to a group of so-called enviroxime-like compounds, strongly suggesting that E7(2) has a similar mechanism of action (10, 14). Analyses of the effects of E7(2) on poliovirus polyprotein processing and recruitment of the viral and cellular proteins to replication complexes strongly suggest that enviroxime-like compounds compromise interactions of the viral polyprotein with membranes but not the recruitment of viral polymerase 3D, as was suggested previously (15). Evaluation of A4(1) in vivo in a murine model of poliomyelitis showed that while this compound had significant toxicity, it delayed the development of disease. Multiple attempts to select mutants resistant to compounds A4(1) and E5(1) were unsuccessful, showing that these drugs may be developed into superior antiviral therapeutics.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were grown in high-glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS). Poliovirus type 1 strain Mahoney, coxsackievirus B3, and encephalomyocarditis virus were propagated on HeLa cells. Titers were determined by standard plaque assays.
Kinase inhibitors.
Two libraries of cell-permeable kinase inhibitors were purchased from Calbiochem. A list of the compounds is available in Tables S1 and S2 in the supplemental material. Wortmannin was obtained from Sigma-Aldrich; LY294002 was obtained from Cell Signaling Technology.
Plasmids.
Plasmid pXpA-RenR, coding for a poliovirus replicon in which the P1 structural region is replaced with a gene encoding Renilla luciferase, was described previously (16). The plasmid coding for a similar coxsackievirus B3 replicon was a kind gift from Frank van Kuppeveld (University of Utrecht, The Netherlands). Plasmid pXpA-P2P3 contains poliovirus cDNA coding for nonstructural proteins under the control of a T7 RNA polymerase promoter. pXpA plasmids contain the ribozyme sequence that generates the authentic 5′-UU sequence of poliovirus RNA upon T7 RNA polymerase transcription (17). Plasmid pPVΔP1 was a gift from Natalya Teterina (NIH). It contains poliovirus cDNA coding for the nonstructural proteins (P2P3) under the control of a T7 RNA polymerase promoter but does not contain the ribozyme sequence resulting in a 5′-GG sequence on the RNA generated by T7 RNA polymerase. Poliovirus RNAs were generated by using a MEGAscript T7 transcription kit (Ambion) and purified as described previously (18).
Antibodies.
Rabbit polyclonal anti-GBF1 antibodies were gifts from Nihal Altan-Bonnet (NIH). Rabbit polyclonal antibodies against phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ) were obtained from Millipore. Mouse monoclonal antibodies against calnexin were obtained from Sigma-Aldrich. Rabbit polyclonal antipoliovirus 3D antibodies were produced by Chemicon, using full-length recombinant 3D as an antigen. Antipoliovirus 2C and antipoliovirus 3A mouse monoclonal antibodies were gifts from Kurt Bienz (Basel University).
Poliovirus replicon assays.
Poliovirus RNA replication assays were performed essentially as described previously (16). HeLa cells were grown on a 96-well plate overnight. Transfection mix with purified Renilla luciferase poliovirus replicon RNA (10 ng RNA/well) was prepared with mRNA TransIt transfection reagent (Mirus) according to the manufacturer's protocol. After 5 min of incubation at room temperature, the transfection mix was combined with normal cell growth medium supplemented with the cell-permeable Endu-Ren Renilla luciferase substrate (Promega). For the initial library screening experiments, medium on the 96-well plate was replaced with this master mix (75 μl/well), and every inhibitor was added to a single well of a 96-well plate (1 μl of a 75× solution in dimethyl sulfoxide [DMSO]). DMSO (positive control) and 10 μM brefeldin A (BFA) (negative control) were added to 8 wells (one column on the 96-well plate each), thus also providing a measure of the reproducibility of the signals from individual wells. After the addition of compounds, the plate was incubated on a shaker for 2 min for mixing. For characterization of the selected inhibitors, the transfection master mix was divided into aliquots sufficient for transfection of the required number of wells (at least 16 wells per sample), and the inhibitors (or the corresponding amount of DMSO for controls) were added directly to the master mix before transfection. After the transfection medium was added to the cells, the plates were sealed with optical clear film and incubated directly in an M5 (Molecular Devices) or Infinite M1000 (Tecan) plate reader equipped with heated cameras at 37°C, and the luciferase signal was measured every hour for 16 h. Results were plotted by using GraphPad Prism statistical software, with error bars showing standard deviations. Total replication was calculated based on the integrated luciferase signal (area under the curve).
In vitro translation and replication assays.
HeLa S10 cell extracts were prepared and translation-replication reactions were performed essentially as previously described (19). Translation mixtures contained 2 mM guanidine-HCl to prevent replication. An aliquot of the translation reaction mixture was mixed with 1 μl of EasyTag Express 35S protein labeling mix (PerkinElmer) to detect newly synthesized proteins. Translation reaction mixtures were incubated at 34°C for 3.5 h and then centrifuged at 15,000 rpm to collect the membrane-associated poliovirus replication complexes. The pellet was resuspended in a replication buffer containing 32P-labeled αCTP (PerkinElmer), without guanidine-HCl. Replication reactions were carried out for 1 h at 37°C. Total RNA was isolated and purified with the RNeasy minikit (Qiagen). Proteins from 35S-labeled translation reactions were resolved by SDS-PAGE. RNA was denatured for 45 min in a glyoxal buffer at 65°C and resolved on a glyoxal-containing denaturing agarose gel. The gel was stained with ethidium bromide to assess the total RNA content, dried, and exposed to a radiographic film to reveal the newly synthesized poliovirus RNA. Image density quantitation was performed with ImageJ software (NIH).
Analysis of proteins associated with poliovirus replication complexes.
In vitro translation was performed as described above, and the membranous pellets containing replication complexes were collected by centrifugation and resuspended in 50 μl of 1× protein sample buffer. Aliquots of 20 μl were run on 10% or 15% SDS-PAGE gels, and proteins were analyzed by Western blotting.
VPg uridylylation assay.
The VPg uridylylation assay was performed essentially as described previously (20), with minor modifications. In vitro translation was performed as described above. Following translation, replication complexes were collected via centrifugation at 15,000 rpm and resuspended in a replication buffer without guanidine-HCl containing 32P-labeled αUTP (PerkinElmer). For the negative control, 2 mM guanidine-HCl was added to the replication buffer. Replication reactions were carried out for 1 h at 37°C. Following replication, the replication complexes were spun down at 15,000 rpm and resuspended in 1× protein sample buffer. Samples were fractionated on a polyacrylamide-Tris-Tricine gel. Image density quantitation was performed with ImageJ software (NIH).
Cell viability assay.
Cell viability was measured with the CellTiter-Glo luminescent cell viability assay kit (Promega).
Serial passages.
For the first passage, a HeLa cell monolayer grown on a 6-cm plate was infected with poliovirus at 10 PFU/cell. After virus attachment, medium containing 10% FBS and the indicated concentrations of inhibitors were added. The samples were frozen at 6 h postinfection (p.i.). After three cycles of freeze-thawing to release the intracellular virus, 1/10 of the viral material was used for subsequent passages, and the rest was stored for further analysis. Every five passages, standard plaque assays were performed to determine virus titers.
Identification of resistant mutations.
Resistant viruses were picked from individual plaques, and viral RNA was isolated with the QIAamp viral RNA minikit (Qiagen) according to the Spin protocol. RNA was reverse transcribed into cDNA with the MonsterScript first-strand cDNA synthesis kit (Illumina). Three overlapping cDNA fragments covering the entire polyprotein coding region were amplified by PCR using a Phusion PCR kit (New England BioLabs) and were commercially sequenced (Genewiz Inc.). Primers for PCR and sequencing are available upon request.
Solubilization of inhibitors in water using molecular containers M1 and M2.
The abilities of molecular containers M1 and M2 to increase the solubility of water-insoluble compounds A4(1), E5(1), and E7(2) were determined according to a methodology described previously (21). The concentrations of compounds A4(1), E5(1), and E7(2) in the resulting solutions were determined by proton nuclear magnetic resonance spectroscopy by comparing the integrals for protons of the inhibitors with the integral of the singlet at 8.3 ppm for 1,3,5-benzene tricarboxylic acid, which was used as an internal standard.
Murine model of poliomyelitis.
TgPVR21 mice expressing poliovirus receptor (22, 23) were randomized into three groups (each containing 5 males and 5 females) to be infected with poliovirus and treated with (i) compound A4(1), solubilized by using molecular container M2 dissolved in phosphate-buffered saline (PBS); (ii) PBS; and (iii) empty molecular container M2 dissolved in PBS. Groups of two mice (one male and one female) were treated with the same substances but mock infected with the virus. Mice received the first intraperitoneal injection of the A4(1) solution (10 mg/kg of body weight) (or the corresponding amount of control solutions) 3 h before intramuscular challenge with 10 50% protective doses (PD50s) of poliovirus type 1 strain Mahoney. The animals received the second injection 24 h after viral challenge (day 2) and were not treated after that, since mice treated with A4(1) showed signs of toxicity. The mice were observed for signs of paresis/paralysis for 4 days, until all animals were paralyzed. The animal care protocol was approved by the Institutional Animal Care and Use Committees (IACUCs) of the FDA Center for Biologics Evaluation and Research.
RESULTS
Identification of small-molecule inhibitors that block enterovirus replication.
We first screened 192 cell-permeable kinase inhibitors for their effects on poliovirus replication. HeLa cells were transfected with poliovirus replicon RNA that contains the Renilla luciferase gene instead of the P1 structural proteins (RenR) (Fig. 1A), and replication was monitored in live cells incubated in the presence of 10 μM inhibitors for 16 h. Samples incubated in the presence of the corresponding amount of DMSO (solvent for the inhibitors) served as positive controls, and those incubated with brefeldin A, a known strong suppressor of poliovirus replication (24, 25), served as negative controls. Some screening experiments were performed in the presence of 100 μM z-VAD-fmk, a cell-permeable caspase inhibitor, to prevent cell death from apoptosis that may be induced by kinase inhibitors. z-VAD-fmk was shown previously not to interfere with poliovirus replication (26). After replication, the effects of the inhibitors on cell survival were evaluated. In our screen, we identified several compounds that effectively inhibited poliovirus replication without significant cell toxicity (Fig. 1B). Among others, we identified Flt3 inhibitor II (Fig. 1B, library I, position C5), which was previously reported to inhibit poliovirus replication (27), thus validating our screening approach. We repeated the screen using a similar replicon construct based on CVB3 (another enterovirus) RNA. The compounds that suppressed poliovirus replication were equally effective against CVB3 (not shown), highlighting the similarity of the replication processes of the two viruses and the possibility of the development of broad-spectrum antienterovirus therapeutics.
Next, we determined if the compounds showing significant inhibition of replicon replication could block normal poliovirus infection in HeLa cells. Interestingly, many of the inhibitors that were effective in the replicon assay did not significantly affect virus propagation, suggesting that their effect was mediated by an inhibition of the RNA transfection process rather than actual replication (see Table S1 in the supplemental material). Three compounds, Akt inhibitor IV, henceforth called A4(1) (position A4 in the library I plate); PDGF receptor tyrosine kinase inhibitor III [E5(1)]; and indirubin derivative E804 [E7(2)], which considerably reduced poliovirus replicon replication in the initial screen, effectively suppressed virus infection in cell culture, and did not show significant cellular toxicity, were chosen for further characterization (Fig. 1B).
The selected inhibitors suppress poliovirus replication in a dose-dependent manner, but their antiviral effect is likely not related to their annotated cellular targets.
To determine the 50% inhibitory concentrations (IC50s) for the selected inhibitors, we performed the poliovirus replicon replication assay in the presence of different concentrations of the compounds. Cellular toxicity of treatment was assessed after replication (∼18 h of inhibitor treatment). The most effective treatment was A4(1), with an IC50 of 3.2 μM, followed by E5(1) and E7(2), with IC50s of 12 and 16 μM, respectively. We observed noticeable cytotoxicity with treatment with A4(1) at a concentration of 50 μM; however, lower concentrations of this inhibitor as well as all treatment conditions for the other two inhibitors were well tolerated by the cells (Fig. 2A to C). In the presence of E7(2), especially at higher concentrations, the luciferase signal was noticeably decreasing toward the end of the experiment (Fig. 2C), likely reflecting the death of cells harboring the poliovirus replicon in the presence of this compound. Activation of the cellular apoptotic program under conditions of suppressed poliovirus infection was described previously for inhibitors of translation and replication (26).
FIG 2.

The kinase inhibitors interfere with poliovirus replication in a dose-dependent manner. The molecular structures of the inhibitors tested are shown. HeLa cells grown on 96-well plates were transfected with poliovirus replicon RNA with a Renilla luciferase gene. Inhibitors were added after transfection, the cells were incubated for 16 h in the presence of a cell-permeable Renilla luciferase substrate, and the signal was measured every hour for 16 h posttransfection. Replication kinetics and total replication (integrated Renilla luciferase signal) are shown. An integrated signal plot was used to determine the IC50s of the inhibitors. Cell toxicity was measured in the same plate after the replication experiments were finished. (A) Akt inhibitor IV [A4(1)]. (B) PDGF receptor tyrosine kinase inhibitor III [E5(1)]. (C) Indirubin derivative E804 [E7(2)]. RLU, relative luciferase units.
A4(1) inhibits PI3K-dependent activation of Akt signaling by inhibiting a kinase upstream of Akt but downstream of PI3K (28). However, Akt inhibitor V (triciribine), which prevents activation of all Akt isoforms (29), as well as Akt inhibitor X had no significant effect on poliovirus replicon replication (Fig. 1B, positions A5 and A7, and data not shown). This controversy prompted us to further test the importance of the Akt pathway for poliovirus replication. We pretreated cells for 2 h before replicon RNA transfection with 25 μM wortmannin and LY294002, well-known strong inhibitors of PI3K-dependent Akt activation (30). After this, the replication assay was also performed in the presence of 25 μM these inhibitors. These treatment conditions had minimal effects on the efficiency of replication compared to that of A4(1) (not shown), arguing that inhibition of the PI3K-Akt pathway is not detrimental to poliovirus RNA replication, at least in cell culture, and that the antiviral activity of A4(1) is unrelated to this pathway.
Similarly, the antiviral effects of the other two selected inhibitors seem to not be related to their annotated cellular targets. E5(1) targets the PDGF receptor family of tyrosine kinases (31); however, other PDGF receptor tyrosine kinase inhibitors, PDGF receptor inhibitors II and IV, had no significant effect on replication (Fig. 1B, positions E4 and E6, and data not shown).
E7(2) inhibits cyclin-dependent kinases as well as Src kinase activity, resulting in inhibition of the Stat3 signaling pathway (32). Other inhibitors targeting Src kinase (SU6656) and cyclin-dependent kinases (kenpaullone, aminopurvalanol A, SB 218078, and SU9516) had minimal or no effects on replicon replication (Fig. 1C, positions E2, F5, A9, H4, and H10, and data not shown).
We conclude that the observed antiviral action of the small-molecule inhibitors A4(1), E5(1), and E7(2) is due to their effect on as-yet-unknown cellular or viral targets.
A4(1), E5(1), and E7(2) block propagation of diverse picornaviruses.
To characterize the antiviral activities of A4(1), E5(1), and E7(2) in cell culture, we infected HeLa cells with 10 PFU/cell of poliovirus and incubated the cells with 10 μM, 25 μM, or 50 μM each inhibitor after virus attachment. The cells were frozen at 6 h postinfection and subjected to three cycles of freeze-thawing to release the intracellular virus. The virus yield in each sample was determined by a plaque assay. All three inhibitors reduced the titer of poliovirus in a dose-dependent manner (Fig. 3A). Accordingly, poliovirus-infected cells incubated in the presence of 25 μM A4(1) and E5(1) did not show signs of viral cytopathic effects (CPE), at least up to 8 h postinfection (Fig. 3B). The protective effect of the compounds correlated directly with the inhibition of accumulation of the viral proteins (Fig. 3C). In spite of the strong suppression of viral propagation, E7(2) was not as protective to infected cells as the other two inhibitors, although the development of CPE was still delayed compared to the control sample (Fig. 3B). Mock-infected cells incubated with any of the inhibitors for 8 h were essentially indistinguishable from the DMSO (solvent) control (not shown). In order to determine the effect of the inhibitors on other members of the Picornaviridae family, we infected HeLa cells with 10 PFU/cell of CVB3 or EMCV in the presence of 25 μM each inhibitor. EMCV belongs to the Cardiovirus genus of the Picornaviridae family and is significantly different from poliovirus and CVB3, which are both members of the Enterovirus genus. All inhibitors reduced propagation of these viruses, similar to their effect on poliovirus. E7(2) reduced the EMCV yield by ∼2 logs, while A4(1) and E5(1) reduced the virus yield by 3 logs (Fig. 3C). A4(1) and E7(2) reduced the CVB3 yield by 2 logs, and E5(1) reduced the yield by >4 logs (Fig. 3C). This shows that the three inhibitors have a broad-spectrum effect on picornavirus replication, suggesting that they target replication components conserved even among distantly related viruses.
FIG 3.

A4(1), E5(1), and E7(2) inhibit diverse picornaviruses. (A) HeLa cells were infected with 10 PFU/cell of poliovirus and incubated in the presence of the inhibitors for 6 h p.i. Virus yield was determined by a plaque assay. (B) HeLa cells infected with 10 PFU/cell of poliovirus were incubated for 8 h p.i. in the presence of 25 μM the inhibitors (or DMSO as a control). (C) Accumulation of viral protein 2C in HeLa cells infected with 10 PFU/cell of poliovirus in the presence of 25 μM the inhibitors (or DMSO as a control) at 4 h p.i. (D) HeLa cells were infected with 10 PFU/cell of EMCV or CVB3 and incubated for 8 h p.i. in the presence of 25 μM the inhibitors. Virus yield was determined by a plaque assay.
Inhibitors block different steps of the poliovirus replication cycle.
Since the inhibitors were selected in the replicon assay, which bypasses early steps of infection such as virion-receptor interactions and RNA release, the inhibitors must be acting on RNA translation and/or replication. To determine the step(s) in the replication life cycle affected by the inhibitors, we analyzed them in an in vitro poliovirus translation-replication system based on a crude HeLa cell extract, which allows separate investigations of RNA translation, polyprotein processing, and RNA replication (6, 33). During the first step (translation), poliovirus RNA is translated in the extract in the presence of 2 mM guanidine-HCl, which does not interfere with viral polyprotein expression and processing but specifically blocks poliovirus RNA replication. During the second step (replication), the membranes with the stalled replication complexes are collected by centrifugation and resuspended in a guanidine-HCl-free replication buffer supplemented with a labeled nucleotide, thus allowing a synchronous start of viral RNA replication and visualization of the newly synthesized RNA (Fig. 4A).
FIG 4.

A4(1), E5(1), and E7(2) inhibit formation and/or functioning of poliovirus replication complexes. (A) Scheme of the two-step in vitro translation-replication system. The inhibitors were added during either the translation step (formation of the replication complexes) or the replication step (functioning of the replication complexes). DMSO (solvent) was added as a control where indicated. An aliquot of the in vitro translation mixture was labeled with [35S]methionine to measure viral polyprotein synthesis and processing. Replication was performed in the presence of [α-32P]CTP to label the newly synthesized RNA. (B) A4(1). (C) E5(1). (D) E7(2). (E) Effect of E7(2) on P3 processing. The scheme summarizes the results of these and other translation experiments performed in the presence of E7(2) and shows the most affected proteolytic sites. Arrowheads indicate 3C-dependent cleavages, and arrows indicate 2A-dependent cleavages.
First, we assessed the effects of the inhibitors on the replication of fully replication-competent poliovirus RNA that codes for the replication polyprotein P2P3 without capsid region P1. Since poliovirus capsid proteins are not involved in replication, and their input is nonessential for the antiviral activity of the selected inhibitors, the absence of the P1 region simplifies the system and facilitates interpretation of the data. We added the inhibitors at concentrations of 10 μM, 25 μM, or 50 μM to either the translation or the replication reaction mixture. An equivalent amount of DMSO (solvent) was added to the samples incubated without the inhibitors. A4(1) inhibited RNA replication in a dose-dependent manner when added to either the translation or replication reaction mixture but had a more pronounced effect if present during the replication step, suggesting that it can directly block proper functioning of the replication complexes. This compound had no effect on the efficiency of RNA translation or polyprotein processing (Fig. 4B). E5(1) also had no apparent effect on polyprotein expression and processing, and if the inhibitor was present during the translation step, subsequent replication was inhibited in a dose-dependent manner. If the inhibitor was added only during the replication step, it had no effect on RNA replication. Thus, E5(1) prevents formation of functional replication complexes but does not interfere with RNA replication once the complexes are assembled (Fig. 4C). E7(2), like E5(1), was effective only if it was added during the translation step, showing that it interferes with the formation of the replication complexes but not their functioning. In contrast to the other two inhibitors, E7(2) had a strong effect on polyprotein processing. It resulted in a significant accumulation of the unprocessed P2P3 polyprotein, a decrease of the 3CD signal and a concomitant increase of the 3D signal, as well as a decrease of 2C accumulation (Fig. 4D, arrowheads). These cleavages are mediated by viral protease 3C. E7(2) also inhibited the accumulation of 2A-dependent cleavage fragments 3C′ and 3D′ (Fig. 4D, arrows). The strong effect of E7(2) on 3C-dependent processing was confirmed in an experiment where only the P3 (3ABCD) fragment was expressed in the presence or in the absence of E7(2). As in the case of full-length P2P3, processing of P3 into 3BCD and 3CD was significantly suppressed in the presence of the compound (Fig. 4E). Thus, A4(1) blocks both the formation and functioning of the replication complexes, while the effects of E5(1) and E7(2) are restricted to the assembly of functional replication complexes.
The inhibitors do not block VPg uridylylation but inhibit minus-strand RNA synthesis.
To understand the steps of viral RNA replication affected by the inhibitors, we examined the formation of uridylylated VPg, which serves as a primer for initiation of synthesis of both strands of poliovirus RNA. To assess VPg uridylylation, poliovirus RNA was translated in a HeLa cell extract in the presence of guanidine-HCl and 50 μM the inhibitors. The membrane-associated preinitiation complexes were collected by centrifugation and resuspended in a buffer without the inhibitors and guanidine-HCl. The replication buffer contained [α-32P]UTP to monitor the formation of uridylylated VPg. Guanidine-HCl (2 mM), which strongly suppresses VPg uridylylation, was added to the negative-control reaction mixture (Fig. 5A). None of the inhibitors had a significant effect on VPg uridylylation; in the case of A4(1), the signal was even stronger than that in the control sample (Fig. 5B). This shows that the compounds block viral RNA replication at a downstream step(s). The next step in poliovirus replication is the copying of the genome RNA into the negative RNA template that will be used for the generation of progeny positive-strand genomes. T7 RNA polymerase generates transcripts with the 5′-GG sequence. If these G's are present at the 5′ end of poliovirus RNA, which has an authentic 5′-UU sequence, they do not interfere with RNA translation, and such an RNA is copied by the replication machinery to the minus strand, but the subsequent step of plus-strand RNA synthesis is blocked (34). We programmed the translation-replication reaction with poliovirus RNA generated from a plasmid, pPV1ΔP1, that does not encode a ribozyme sequence, and thus, the T7 RNA polymerase transcripts retain extra G's at the 5′ end. The inhibitors were present at a concentration of 50 μM during the translation step, when the replication complexes are formed. A4(1) was the strongest inhibitor of minus-strand RNA synthesis, while E5(1) and E7(2) showed more modest inhibition (Fig. 5C). Thus, the poliovirus replication complexes formed in the presence of the compounds are fully competent in the generation of the uridylylated VPg primer but cannot support effective synthesis of RNA.
FIG 5.

A4(1), E5(1), and E7(2) inhibit poliovirus RNA replication but not VPg uridylylation. (A) Scheme of the VPg uridylylation experiment. (B) Effect of 50 μM the inhibitors on VPg uridylylation. The inhibitors were present during the RNA translation step (formation of replication complexes). 32P-uridylylated VPg is shown. Guanidine-HCl was added as a negative control. An aliquot of the in vitro translation mixture was labeled with [35S]methionine to measure viral polyprotein synthesis and processing. The asterisk indicates accumulation of unprocessed P2P3 polyprotein in the presence of E7(2). (C) Effect of 50 μM the inhibitors on negative-RNA-strand synthesis. The inhibitors were present during the translation step (formation of replication complexes). Replication of the poliovirus template with extra 5′-GG sequence (allowing only negative-strand synthesis) was performed in the presence of [α-32P]CTP to label the newly synthesized RNA. An aliquot of the in vitro translation mixture was labeled with [35S]methionine to measure viral polyprotein synthesis and processing. The asterisk indicates accumulation of unprocessed P2P3 polyprotein in the presence of E7(2).
E7(2) affects binding of cellular and viral factors to membranes.
Proteins from the cellular secretory pathway are known to be recruited to the poliovirus replication complexes and to facilitate RNA replication. GBF1, a guanine nucleotide exchange factor for a small cellular GTPase, Arf1, is recruited to the replication complexes of poliovirus and CVB3 through interactions with viral protein 3A; inhibition of GBF1 with brefeldin A strongly suppresses viral replication (35–38). Another cellular factor, phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ), was shown to be important for replication of different positive-strand RNA viruses and was proposed to be recruited to the replication complexes in a GBF1-dependent manner (15). To determine if the new small-molecule inhibitors of viral replication interfere with the recruitment of these important host factors to the replication complexes, we translated poliovirus RNA in a HeLa cell extract in the presence of the inhibitors, collected the membranes, and analyzed the associated proteins by Western blotting. The presence of A4(1) in the translation reaction mixture resulted in somewhat dose-dependent increases of both GBF1 and PI4KIIIβ recruitment to membranes (Fig. 6A). E5(1) did not have any significant effect on the poliovirus-specific association of these proteins with membranes (Fig. 6B). Interestingly, samples incubated with E7(2) showed a dose-dependent decrease of recruitment of GBF1 to the membranes but had no effect on PI4KIIIβ, supporting a recent report that recruitment of these proteins may be independent of each other (39). Similarly, we did not see any significant effect on the association with membranes of another cellular protein, acyl coenzyme A (acyl-CoA) binding protein 3 (ACBD3), which was shown previously to be able to modulate replication of diverse picornaviruses, including poliovirus (40–42) (not shown). Since GBF1 recruitment to membranes was diminished in the presence of E7(2), we tested whether its inhibitory effect on viral replication can be rescued by overexpression of GBF1. We monitored poliovirus replicon replication in cells transfected with a GBF1-expressing vector in the presence of 10 and 25 μM E7(2). No differences were observed compared to the control samples transfected with an empty vector (data not shown), arguing that the reduced amount of GBF1 recruited to the replication complexes in the presence of this inhibitor is not a limiting factor for viral replication. We similarly investigated the effect of these compounds on the recruitment of the viral proteins to membranes. A4(1) and E5(1) did not significantly affect binding to membranes of viral proteins 2C, 3A, and 3D and some uncleaved precursors containing these sequences (Fig. 6A and B). E7(2), on the other hand, induced a significant increase in accumulation on the membranes of polyprotein fragment P3 and 3CD but not of any other viral proteins assessed (Fig. 6C). Thus, the inhibition of poliovirus replication in the presence of E7(2) is likely related to the aberrant composition of the cellular and viral factors in the replication complexes.
FIG 6.

Recruitment of host and viral proteins to replication complexes in the presence of the inhibitors. Poliovirus RNA was translated in HeLa cell extracts in the presence of the inhibitors. The membranes were collected by centrifugation, and the membrane-associated proteins were analyzed by Western blotting. Calnexin is shown as a loading control. (A) Recruitment of the cellular proteins GBF1 and PI4KIIIβ and poliovirus proteins P3, 3CD, 3D, 2C, and 3A/3AB to membranes in the presence of A4(1). (B) Recruitment of cellular proteins GBF1 and PI4KIIIβ and poliovirus proteins P3, 3CD, 3D, 2C, and 3A/3AB to membranes in the presence of E5(1). (C) Recruitment of cellular proteins GBF1 and PI4KIIIβ and poliovirus proteins P3, 3CD, 3D, 2C, and 3A/3AB to membranes in the presence of E7(2). KI, kinase inhibitor.
A4(1) and E5(1) are refractory to emergence of resistant mutants, while resistance to E7(2) is conferred by a mutation previously shown to rescue replication in the presence of enviroxime-like compounds.
To obtain deeper insight into the mechanism of inhibition of the viral life cycle and to evaluate the potential of the inhibitors as antiviral therapeutics, we attempted to isolate resistant mutants by serially passaging poliovirus in the presence of the compounds. For the first passage, HeLa cells were infected with poliovirus at a multiplicity of infection (MOI) of 10 PFU/cell, and cells and extracellular medium were harvested at 6 h p.i. and subjected to three cycles of freeze-thawing to release the intracellular virus. An aliquot of this material was used for the next round of infection, and the rest was stored for later analysis. Initially, we passaged the virus in the presence of 50 μM the inhibitors. Under these conditions, no resistant mutants emerged for any of the inhibitors.
When we passaged poliovirus in the presence of 10 μM A4(1), we saw constant low levels of viral replication with no apparent increase between passages, indicative of an emergence of resistant mutants. At passages 5 and 10, the titer was ∼10E7 PFU/ml, about 2.5 logs lower than that for control replication (Fig. 7A). We sequenced viral RNA from passage 10 and found no mutations leading to amino acid changes.
FIG 7.
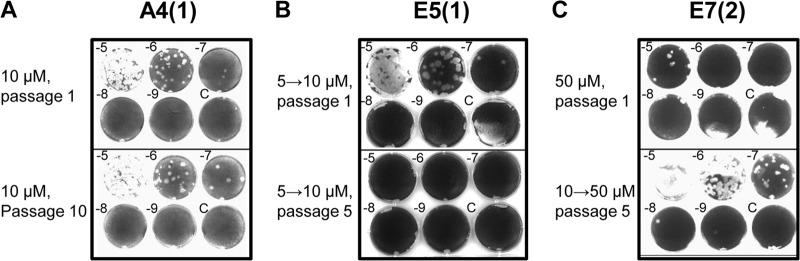
Selection of poliovirus mutants resistant to the inhibitors. Poliovirus yield was determined by a plaque assay on HeLa cell monolayers. Log dilution factors are indicated; C denotes the control uninfected wells. (A) Poliovirus yield after passage 1 (top) and passage 10 (bottom) in the presence of 10 μM A4(1). (B) Poliovirus yield after passage 1 with 10 μM E5(1) of the population previously passaged 10 times with 5 μM the inhibitor (top) and disappearance of the virus after four subsequent passages with 10 μM E5(1) (bottom). (C) Poliovirus yield after passage 1 with 50 μM E7(2) (top) and propagation of the resistant population selected after 5 passages with 50 μM the inhibitor after 10 preliminary passages with 10 μM the inhibitor (bottom).
Propagation of poliovirus in the presence of 10 μM E5(1) resulted in the complete disappearance of the virus at passage 5. We then decreased the concentration of the inhibitor to 5 μM and propagated the virus for several passages to enrich the population for possible mutations that may facilitate selection of resistant mutants at a higher concentration. At 5 μM E5(1), the virus replicated to an ∼0.5- to 1-log-lower level than that of control replication (not shown). We took the material collected at passage 10 with 5 μM the inhibitor and continued passaging with 10 μM the inhibitor. Again, after passage 5, we saw a complete disappearance of the virus, showing that no resistant mutants were selected under these conditions (Fig. 7B).
To further extend the chances of selecting resistant mutants, we performed 10 additional passages of poliovirus in the presence of 10 μM A4(1) and 5 μM E5(1). Viral populations selected after 20 passages showed the same absence of increased resistance as the populations selected after 10 passages. The population propagated for 20 passages with 5 μM E5(1) again disappeared after 5 passages when the concentration of the compound was increased to 10 μM (not shown).
After the initial failure to isolate resistant mutants with 50 μM E7(2), we propagated the virus for 10 passages with 10 μM the inhibitor and then used this material for five subsequent passages with 50 μM the inhibitor. This scheme resulted in the selection of a resistant population (Fig. 7C). We isolated two individual plaques from passage 5 with 50 μM E7(2) and sequenced the entire coding region of the viral RNA. We found that the viral RNAs from both plaques contained one nucleotide substitution, G5318A, resulting in an amino acid change from alanine to threonine in the 3A sequence. The same mutation was reported previously to confer resistance to a diverse class of so-called enviroxime-like compounds, believed to target a PI4KIIIβ-dependent step in the viral replication cycle (10, 43–46). We confirmed that this mutation was responsible for the resistance to E7(2) by genetically engineering the corresponding change into the poliovirus genome (not shown).
Thus, A4(1) and E5(1) are novel strong inhibitors of poliovirus replication that cannot be overcome by resistance mutations, at least in HeLa cells, while E7(2) is another enviroxime-like compound targeting a 3A-dependent process in the viral life cycle.
Another enviroxime-like compound affects poliovirus replication and polyprotein processing similarly to E7(2).
The emergence of the same mutation upon selection of poliovirus in the presence of E7(2) as the one previously described for other enviroxime-like compounds prompted an investigation of the effect of another enviroxime-like compound, GW5074 (10), on formation of poliovirus replication complexes and polyprotein processing. We performed an in vitro translation-replication experiment where the compounds were added during either the translation step (formation of the replication complexes) or the replication step. The inhibitory effect of E7(2) added during the replication step was more pronounced in this experiment, likely reflecting variations among HeLa extract preparations. However, both enviroxime-like compounds were much more effective if they were present during the formation of the replication complexes (Fig. 8A). Moreover, in the presence of GW5074, polyprotein processing defects similar to those induced by E7(2) were observed (Fig. 8B). When we assessed the replication of the resistant G5318A mutant in the in vitro system, we observed that this mutation partially rescues polyprotein processing defects in the presence of E7(2) (Fig. 8B). Interestingly, the level of RNA replication of this mutant was practically the same in the presence and in the absence of the inhibitor, but it was significantly lower than the control replication level of wild-type RNA (Fig. 8B). These results show that inhibition of formation of poliovirus replication complexes by at least some enviroxime-like compounds may be mediated at least in part by polyprotein processing defects.
FIG 8.

Enviroxime-like compounds E7(2) and GW5074 inhibit formation of the replication complexes and interfere with polyprotein processing. (A) Both E7(2) and GW5074 inhibit formation of the functional replication complexes and affect polyprotein processing. The inhibitors were added during either the translation step (formation of the replication complexes) or the replication step (functioning of the replication complexes). DMSO (solvent) was added as a control where indicated. An aliquot of the in vitro translation mixture was labeled with [35S]methionine to measure viral polyprotein synthesis and processing. Replication was performed in the presence of [α-32P]CTP to label the newly synthesized RNA. (B) The G5318A mutation partially rescues the polyprotein processing defect and replication in the presence of E7(2) but compromises replication in the absence of the inhibitor. The inhibitor (50 μM) was present during the translation step (formation of the replication complexes). DMSO (solvent) was added as a control where indicated. An aliquot of the in vitro translation mixture was labeled with [35S]methionine to measure viral polyprotein synthesis and processing. Replication was performed in the presence of [α-32P]CTP to label the newly synthesized RNA. WT, wild type.
A4(1) delays the onset of paralysis in a murine model of poliomyelitis.
The novel antipicornaviral compounds A4(1), E5(1), and E7(2) are practically insoluble in water, which significantly limits their usefulness as antiviral drugs for in vivo studies. To enhance solubility, we utilized two novel acyclic cucurbit[n]uril (CB[n]) molecular containers known as M1 and M2 (21) (Fig. 8A). These containers were previously shown to increase the solubility of at least 10 pharmacologically relevant drugs and to be nontoxic to several cell lines and tolerated by mice at doses as high as 1,230 mg/kg (21). Based on the large aromatic surfaces of inhibitors A4(1), E5(1), and E7(2), we decided to first test solubilization by using M2, which features the larger aromatic naphthalene rings as side walls. Experimentally, we separately mixed each insoluble inhibitor (1 mg) with M2 (14 mM; 0.7 ml) in 20 mM sodium phosphate buffer (pH 7.4 at room temperature [RT]). The mixture was then filtered, and the concentration of each inhibitor was determined by 1H nuclear magnetic resonance (NMR) spectroscopy by comparing the integrals of the inhibitor with those of 1,3,5-benzene tricarboxylic acid added as an internal standard with a known concentration. Container M2 (14 mM) increased the solubility of A4(1) up to 2.01 mM (1.24 mg/ml) and increased the solubility of E7(2) up to 0.90 mM (0.33 mg/ml) but did not solubilize E5(1). Accordingly, we tested inhibitor E5(1) with container M1, but once again, we were unable to enhance its solubility.
We evaluated whether container M2-solubilized A4(1) and E7(2) retained their antiviral activity in the poliovirus replicon replication assay. Empty container M2 did not have any inhibitory effect on poliovirus RNA replication (Fig. 9B and C). Container M2-dissolved A4(1) reduced poliovirus replicon replication but not as effectively as the DMSO-dissolved compound (Fig. 9B). Container M2-dissolved E7(2) did not inhibit replicon replication at both 10 μM and 25 μM (Fig. 9C), suggesting a strong molecular interaction between the inhibitor and container M2. Container M2-dissolved compounds also did not show increased cellular toxicity compared to the DMSO-dissolved ones (Fig. 9B and C). Since container M2-dissolved A4(1) retained its antiviral properties in cell culture, we further evaluated it as an antiviral drug in a murine model of poliomyelitis (22).
FIG 9.

A4(1) retains antiviral activity in cell culture and delays poliovirus-induced paralysis in vivo after solubilization enhancement with molecular container M2. (A) Chemical structures of acyclic CB[n]-type molecular containers M1 and M2. (B and C) HeLa cells grown on 96-well plates were transfected with poliovirus replicon RNA with a Renilla luciferase gene. DMSO-dissolved or container M2-dissolved A4(1) (B) or E7(2) (C) was added after transfection, the cells were incubated for 16 h in the presence of a cell-permeable Renilla luciferase substrate, and the signal was measured every hour for 16 h posttransfection. Cell toxicity was measured in the same plate after the replication experiments were finished. (D) A4(1) delays the onset of paralysis in a murine model of poliomyelitis. Container M2-dissolved A4(1) in PBS [14 mM M2–2.01 mM A4(1)] was administered to TgPVR21 mice at a dose of 10 mg/kg 3 h before challenge of mice with 10 PD50s of type 1 wild-type poliovirus (Mahoney strain) (PV), and a second dose was administered 24 h after challenge. Control groups were treated with PBS or container M2 in PBS only.
A group of 10 transgenic mice expressing human poliovirus receptor (5 males and 5 females) were inoculated intraperitoneally with 10 mg/kg of A4(1) in complex with container M2 in PBS on day 1, 3 h before intramuscular challenge with 10 PD50s of poliovirus type 1 strain Mahoney. Control groups were treated with either empty container M2 in PBS or PBS alone. Mice received the second dose of the compound or the control solution at 24 h postinfection. The treatment was stopped after the second dose due to signs of toxicity in animals treated with A4(1). Hind limb paralysis developed in both control groups treated with container M2 only (5 out of 10 mice) and PBS (6 out of 10 mice) on day 3, while mice treated with A4(1) did not show signs of disease. On day 4, mice in all groups were paralyzed (Fig. 9D). Thus, A4(1), in spite of the toxic effect, demonstrated protective antipoliovirus potential in vivo.
DISCUSSION
In this study, we identified and characterized three novel molecules with antipicornavirus activity by screening commercially available libraries of cell-permeable kinase inhibitors. The selected inhibitors effectively suppressed replication of poliovirus replicon RNA with IC50s ranging from 3.2 μM for A4(1) to ∼12 μM for E5(1) and ∼16 μM for E7(2). These compounds similarly blocked infection by poliovirus; another related enterovirus, CVB3; as well as the much more distantly related EMCV of the Cardiovirus genus. Enteroviruses and cardioviruses are known to be markedly different in their responses to at least some other inhibitors targeting either viral or cellular proteins. Low concentrations of guanidine-HCl strongly interfere with enterovirus but not cardiovirus replication. The effect of this compound is believed to be mediated by inhibition of the ATPase activity of enterovirus protein 2C (47–50). Similarly, brefeldin A, an inhibitor of a GTPase-activating function of a cellular protein, GBF1, strongly inhibits replication of poliovirus and some other picornaviruses but is completely ineffective against cardioviruses (24, 25, 35, 36, 51). The identification of compounds that can block replication of cardioviruses and enteroviruses equally suggests that at least some components of the replication complexes of these viruses are conserved. The inhibitors were selected for their activity in the replicon assay. Since the introduction of replicon RNA into cells by transfection skips the viral attachment/penetration process, the inhibitors should be acting on viral RNA expression and/or replication. The experiments with the in vitro translation-replication system revealed that all inhibitors interfered with RNA replication, while having a minimal effect on the uridylylation of the viral protein VPg, which serves as a primer for RNA synthesis. A4(1) was the strongest inhibitor and interfered with both the formation and functioning of the replication complexes. E5(1) and E7(2), on the other hand, blocked only formation of the replication complexes and could not stop replication if they were added to already formed complexes. Neither A4(1) nor E5(1) showed any effect on accumulation of the viral proteins, while E7(2) interfered with polyprotein processing. It is likely that the aberrant processing of the viral polyprotein contributed to the defect of the replication complexes formed in the presence of E7(2). Since the inhibitors were effective in the in vitro system, which is based on postnuclear cell lysates, they likely directly inhibit the formation and/or functioning of viral replication complexes rather than induce an alteration of cellular metabolism making cells generally nonpermissive for viral infection. The inhibitory effect in the in vitro system required higher concentrations of the compounds than those in in vivo assays such as replicon replication or virus propagation. A similar phenomenon was also previously observed for another inhibitor of poliovirus replication, brefeldin A, which acts by blocking the activity of the cellular protein GBF1 (18, 52). This may reflect the fact that in live cells, translation and replication of the viral RNAs are not separated, and inhibition of any process is amplified in a feedback loop, while in the two-step in vitro system, the amount of available RNA templates is the same, so higher concentrations of the compounds may be required to achieve a similar inhibitory effect on the replication readout signal. Since the compounds effectively blocked infection by such diverse picornaviruses as enteroviruses and cardioviruses, it is likely that they are targeting cellular rather than viral proteins; however, the identity of their targets remains to be established. Originally, we anticipated that screening of libraries of characterized kinase inhibitors may reveal the cellular metabolism regulation pathways that may be important for viral infection. Our results show, however, that the antiviral action of the inhibitors described in this paper is unlikely to be mediated by their annotated kinase targets, since other inhibitors of the same kinases often showed markedly different effects on viral replication. Supporting the off-target effect mediating the antiviral activity of A4(1), E5(1), and E7(2) is the fact that their IC50s for the annotated cellular targets are in the nanomolar range (see Tables S1 and S2 in the supplemental material), while the inhibitors were practically ineffective against viral propagation at concentrations of <10 μM. Interestingly, A4(1) was also reported previously to block replication of negative-strand RNA viruses, such as respiratory syncytial virus and vesicular stomatitis virus, by an unknown mechanism unrelated to the Akt pathway targeted by this molecule (53). Those and our results underscore the importance of the off-target effects of the small-molecule compounds used to decipher cellular pathways hijacked during viral infection. This inhibitor also showed a negative effect on replication of vaccinia virus (53). It is an intriguing possibility that all these diverse viruses may share a requirement for the same cellular factor(s).
A major problem in antiviral drug development is the emergence of resistant mutants. Picornaviruses are known to easily overcome compounds targeting either cellular or viral proteins (8–11). Both A4(1) and E5(1) were refractory to the emergence of viral resistance, at least in cell culture, making these inhibitors promising candidates for the development of effective broad-spectrum antivirals. After multiple attempts, we were able to select viruses that were resistant only to E7(2). The resistance mutation was localized in the hydrophobic domain of the 3A protein. The mutation was the same as the previously described one conferring resistance to so-called enviroxime-like compounds; thus, E7(2) can be added to this broad class of seemingly unrelated molecules. They are believed to target either PI4KIIIβ (major enviroxime-like compounds) (10, 45) or oxysterol binding protein (OSBP) (minor enviroxime-like compounds) (14). The amount of PI4KIIIβ associated with the replication membranes in the presence of E7(2) was not affected; however, this inhibitor induced a decrease in recruitment of GBF1 to membranes. These data support a recent report that the GBF1 and PI4KIIIβ proteins are recruited to the replication complex independently (39). It was suggested that the activity of PI4KIIIβ on the replication membranes, resulting in their enrichment in phosphatidylinositol 4 phosphate (PI4P), may facilitate binding of viral protein 3D (15). Interestingly, we found that E7(2) actually did not have a significant effect on the recruitment of 3D but significantly increased the binding of P3 and 3CD to membranes in apparently nonfunctional configurations, since the complexes formed in the presence of this compound were defective in RNA replication. Both E7(2) and another enviroxime-like compound, GW5074, induced defects in polyprotein processing, affecting 3C- and 2A-dependent cleavages, while the resistance mutation G5318A partially rescued these defects. While this mutation rescued replication in the presence of the inhibitor to a certain level, it was detrimental for replication in the absence of the drug. Collectively, these data suggest that at least some enviroxime-like compounds likely interfere with the formation of a proper membrane microenvironment in a way that prevents the correct presentation of the proteolytic sites of the viral polyprotein and results in aberrant recruitment of some viral proteins from the P3 genomic region (but not the polymerase 3D) to the replication membranes and that developing resistance to this changed environment requires a tradeoff leading to lower levels of replication.
We were able to partially evaluate the protective properties of A4(1) in vivo in a murine model of poliomyelitis. To render the compound water soluble, we used the novel molecular container M2 (21). Unfortunately, A4(1) was noticeably toxic to mice, and we had to stop its administration after only two doses; however, even this limited treatment delayed the onset of paralysis. It is likely that a slight modification of the chemical structure of the inhibitors identified in this study may render these structures more water soluble and increase their antiviral potency in vivo.
These molecules will provide important tools for understanding the fundamental aspects of the formation and functioning of picornavirus replication complexes as well as for the development of broad-spectrum antivirals refractory to the emergence of resistant mutants.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christopher Moore for his insightful suggestions.
This work was supported by University of Maryland start-up funds (G.A.B.) and the National Institutes of Health (CA-168365) (L.I.).
Footnotes
Published ahead of print 9 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01877-14.
REFERENCES
- 1.Fendrick AM, Monto AS, Nightengale B, Sarnes M. 2003. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Arch. Intern. Med. 163:487–494. 10.1001/archinte.163.4.487 [DOI] [PubMed] [Google Scholar]
- 2.Bertino JS. 2002. Cost burden of viral respiratory infections: issues for formulary decision makers. Am. J. Med. 112(Suppl 6A):42S–49S. 10.1016/S0002-9343(01)01063-4 [DOI] [PubMed] [Google Scholar]
- 3.Arden KE, Mackay IM. 2009. Human rhinoviruses: coming in from the cold. Genome Med. 1:44. 10.1186/gm44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fujinami RS, von Herrath MG, Christen U, Whitton JL. 2006. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin. Microbiol. Rev. 19:80–94. 10.1128/CMR.19.1.80-94.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller W, Kandolf R, Schultheiss HP. 2005. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 111:887–893. 10.1161/01.CIR.0000155616.07901.35 [DOI] [PubMed] [Google Scholar]
- 6.Yeung WC, Rawlinson WD, Craig ME. 2011. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ 342:d35. 10.1136/bmj.d35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collett MS, Neyts J, Modlin JF. 2008. A case for developing antiviral drugs against polio. Antiviral Res. 79:179–187. 10.1016/j.antiviral.2008.04.002 [DOI] [PubMed] [Google Scholar]
- 8.Pfeiffer JK, Kirkegaard K. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. U. S. A. 100:7289–7294. 10.1073/pnas.1232294100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crotty S, Saleh MC, Gitlin L, Beske O, Andino R. 2004. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J. Virol. 78:3378–3386. 10.1128/JVI.78.7.3378-3386.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2011. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 85:2364–2372. 10.1128/JVI.02249-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thibaut HJ, De Palma AM, Neyts J. 2012. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem. Pharmacol. 83:185–192. 10.1016/j.bcp.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 12.Geller R, Vignuzzi M, Andino R, Frydman J. 2007. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 21:195–205. 10.1101/gad.1505307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 86:302–312. 10.1128/JVI.05937-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2013. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J. Virol. 87:4252–4260. 10.1128/JVI.03546-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. 10.1016/j.cell.2010.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belov GA, Altan-Bonnet N, Kovtunovych G, Jackson CL, Lippincott-Schwartz J, Ehrenfeld E. 2007. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 81:558–567. 10.1128/JVI.01820-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herold J, Andino R. 2000. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 74:6394–6400. 10.1128/JVI.74.14.6394-6400.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belov GA, Fogg MH, Ehrenfeld E. 2005. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. J. Virol. 79:7207–7216. 10.1128/JVI.79.11.7207-7216.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fogg MH, Teterina NL, Ehrenfeld E. 2003. Membrane requirements for uridylylation of the poliovirus VPg protein and viral RNA synthesis in vitro. J. Virol. 77:11408–11416. 10.1128/JVI.77.21.11408-11416.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teterina NL, Rinaudo MS, Ehrenfeld E. 2003. Strand-specific RNA synthesis defects in a poliovirus with a mutation in protein 3A. J. Virol. 77:12679–12691. 10.1128/JVI.77.23.12679-12691.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma D, Hettiarachchi G, Nguyen D, Zhang B, Wittenberg JB, Zavalij PY, Briken V, Isaacs L. 2012. Acyclic cucurbit[n]uril molecular containers enhance the solubility and bioactivity of poorly soluble pharmaceuticals. Nat. Chem. 4:503–510. 10.1038/nchem.1326 [DOI] [PubMed] [Google Scholar]
- 22.Koike S, Taya C, Kurata T, Abe S, Ise I, Yonekawa H, Nomoto A. 1991. Transgenic mice susceptible to poliovirus. Proc. Natl. Acad. Sci. U. S. A. 88:951–955. 10.1073/pnas.88.3.951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koike S, Taya C, Aoki J, Matsuda Y, Ise I, Takeda H, Matsuzaki T, Amanuma H, Yonekawa H, Nomoto A. 1994. Characterization of three different transgenic mouse lines that carry human poliovirus receptor gene—influence of the transgene expression on pathogenesis. Arch. Virol. 139:351–363. 10.1007/BF01310797 [DOI] [PubMed] [Google Scholar]
- 24.Irurzun A, Perez L, Carrasco L. 1992. Involvement of membrane traffic in the replication of poliovirus genomes: effects of brefeldin A. Virology 191:166–175. 10.1016/0042-6822(92)90178-R [DOI] [PubMed] [Google Scholar]
- 25.Maynell LA, Kirkegaard K, Klymkowsky MW. 1992. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 66:1985–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belov GA, Romanova LI, Tolskaya EA, Kolesnikova MS, Lazebnik YA, Agol VI. 2003. The major apoptotic pathway activated and suppressed by poliovirus. J. Virol. 77:45–56. 10.1128/JVI.77.1.45-56.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arita M, Wakita T, Shimizu H. 2009. Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J. Gen. Virol. 90:1869–1879. 10.1099/vir.0.012096-0 [DOI] [PubMed] [Google Scholar]
- 28.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. 2003. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 4:463–476. 10.1016/S1535-6108(03)00303-9 [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM, Cheng JQ. 2004. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 64:4394–4399. 10.1158/0008-5472.CAN-04-0343 [DOI] [PubMed] [Google Scholar]
- 30.Feldman ME, Shokat KM. 2011. New inhibitors of the PI3K-Akt-mTOR pathway: insights into mTOR signaling from a new generation of Tor kinase domain inhibitors (TORKinibs). Curr. Top. Microbiol. 347:241–262. 10.1007/82_2010_64 [DOI] [PubMed] [Google Scholar]
- 31.Matsuno K, Ushiki J, Seishi T, Ichimura M, Giese NA, Yu JC, Takahashi S, Oda S, Nomoto Y. 2003. Potent and selective inhibitors of platelet-derived growth factor receptor phosphorylation. 3. Replacement of quinazoline moiety and improvement of metabolic polymorphism of 4-[4-(N-substituted (thio)carbamoyl)-1-piperazinyl]-6,7-dimethoxyquinazoline derivatives. J. Med. Chem. 46:4910–4925. 10.1021/jm020505v [DOI] [PubMed] [Google Scholar]
- 32.Nam S, Buettner R, Turkson J, Kim D, Cheng JQ, Muehlbeyer S, Hippe F, Vatter S, Merz KH, Eisenbrand G, Jove R. 2005. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. U. S. A. 102:5998–6003. 10.1073/pnas.0409467102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barton DJ, Black EP, Flanegan JB. 1995. Complete replication of poliovirus in vitro: preinitiation RNA replication complexes require soluble cellular factors for the synthesis of VPg-linked RNA. J. Virol. 69:5516–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barton DJ, Morasco BJ, Flanegan JB. 1999. Translating ribosomes inhibit poliovirus negative-strand RNA synthesis. J. Virol. 73:10104–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanke KH, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJ. 2009. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 83:11940–11949. 10.1128/JVI.01244-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E. 2008. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 4:e1000216. 10.1371/journal.ppat.1000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wessels E, Duijsings D, Lanke KH, Melchers WJ, Jackson CL, van Kuppeveld FJ. 2007. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J. Virol. 81:5238–5245. 10.1128/JVI.02680-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wessels E, Duijsings D, Niu TK, Neumann S, Oorschot VM, de Lange F, Lanke KH, Klumperman J, Henke A, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell 11:191–201. 10.1016/j.devcel.2006.06.005 [DOI] [PubMed] [Google Scholar]
- 39.Dorobantu CM, van der Schaar HM, Ford LA, Strating JR, Ulferts R, Fang Y, Belov G, van Kuppeveld FJ. 2014. Recruitment of PI4KIIIbeta to coxsackievirus B3 replication organelles is independent of ACBD3, GBF1, and Arf1. J. Virol. 88:2725–2736. 10.1128/JVI.03650-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasaki J, Ishikawa K, Arita M, Taniguchi K. 2012. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 31:754–766. 10.1038/emboj.2011.429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J. Virol. 86:3605–3616. 10.1128/JVI.06778-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teoule F, Brisac C, Pelletier I, Vidalain PO, Jegouic S, Mirabelli C, Bessaud M, Combelas N, Autret A, Tangy F, Delpeyroux F, Blondel B. 2013. The Golgi protein ACBD3, an interactor for poliovirus protein 3A, modulates poliovirus replication. J. Virol. 87:11031–11046. 10.1128/JVI.00304-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arita M, Takebe Y, Wakita T, Shimizu H. 2010. A bifunctional anti-enterovirus compound that inhibits replication and the early stage of enterovirus 71 infection. J. Gen. Virol. 91:2734–2744. 10.1099/vir.0.023374-0 [DOI] [PubMed] [Google Scholar]
- 44.Heinz BA, Vance LM. 1996. Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses. J. Virol. 70:4854–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Schaar HM, Leyssen P, Thibaut HJ, de Palma A, van der Linden L, Lanke KH, Lacroix C, Verbeken E, Conrath K, Macleod AM, Mitchell DR, Palmer NJ, van de Poel H, Andrews M, Neyts J, van Kuppeveld FJ. 2013. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIbeta. Antimicrob. Agents Chemother. 57:4971–4981. 10.1128/AAC.01175-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Schaar HM, van der Linden L, Lanke KH, Strating JR, Purstinger G, de Vries E, de Haan CA, Neyts J, van Kuppeveld FJ. 2012. Coxsackievirus mutants that can bypass host factor PI4KIIIbeta and the need for high levels of PI4P lipids for replication. Cell Res. 22:1576–1592. 10.1038/cr.2012.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barton DJ, Flanegan JB. 1997. Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J. Virol. 71:8482–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfister T, Wimmer E. 1999. Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J. Biol. Chem. 274:6992–7001. 10.1074/jbc.274.11.6992 [DOI] [PubMed] [Google Scholar]
- 49.McCormick W, Penman S. 1968. Replication of mengovirus in HeLa cells preinfected with nonreplicating poliovirus. J. Virol. 2:859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prather SO, Taylor MW. 1975. Host-dependent restriction of mengovirus replication. IV. Effect of some quaternary ammonium ions on the restricted replication of mengovirus in Madin-Darby bovine kidney cells. J. Virol. 15:1033–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gazina EV, Mackenzie JM, Gorrell RJ, Anderson DA. 2002. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J. Virol. 76:11113–11122. 10.1128/JVI.76.21.11113-11122.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuconati A, Molla A, Wimmer E. 1998. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 72:6456–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunn EF, Fearns R, Connor JH. 2009. Akt inhibitor Akt-IV blocks virus replication through an Akt-independent mechanism. J. Virol. 83:11665–11672. 10.1128/JVI.01092-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.