

ABSTRACT
In an attempt to explore infectious agents associated with nasopharyngeal carcinomas (NPCs), we employed our high-throughput RNA sequencing (RNA-seq) analysis pipeline, RNA CoMPASS, to investigate the presence of ectopic organisms within a number of NPC cell lines commonly used by NPC and Epstein-Barr virus (EBV) researchers. Sequencing data sets from both CNE1 and HONE1 were found to contain reads for human papillomavirus 18 (HPV-18). Subsequent real-time reverse transcription-PCR (RT-PCR) analysis on a panel of NPC cell lines identified HPV-18 in CNE1 and HONE1 as well as three additional NPC cell lines (CNE2, AdAH, and NPC-KT). Further analysis of the chromosomal integration arrangement of HPV-18 in NPCs revealed patterns identical to those observed in HeLa cells. Clustering based on human single nucleotide variation (SNV) analysis of two separate HeLa cell lines and several NPC cell lines demonstrated two distinct clusters with CNE1, as well as HONE1 clustering with the two HeLa cell lines. In addition, duplex-PCR-based genotyping showed that CNE1, CNE2, and HONE1 do not have a HeLa cell-specific L1 retrotransposon insertion, suggesting that these three HPV-18+ NPC lines are likely products of a somatic hybridization with HeLa cells, which is also consistent with our RNA-seq-based gene level SNV analysis. Taking all of these findings together, we conclude that a widespread HeLa contamination may exist in many NPC cell lines, and authentication of these cell lines is recommended. Finally, we provide a proof of concept for the utility of an RNA-seq-based approach for cell authentication.
IMPORTANCE Nasopharyngeal carcinoma (NPC) cell lines are important model systems for analyzing the complex life cycle and pathogenesis of Epstein-Barr virus (EBV). Using an RNA-seq-based approach, we found HeLa cell contamination in several NPC cell lines that are commonly used in the EBV and related fields. Our data support the notion that contamination resulted from somatic hybridization with HeLa cells, likely occurring at the point of cell line establishment. Given the rarity of NPCs, the long history of NPC cell lines, and the lack of rigorous cell line authentication, it is likely that the actual prevalence and impact of HeLa cell contamination on the EBV field might be greater. We therefore recommend cell line authentication prior to performing experiments using NPC cell lines to avoid inaccurate conclusions. The novel RNA-seq-based cell authentication approach reported here can serve as a comprehensive method for validating cell lines.
INTRODUCTION
Nasopharyngeal carcinoma (NPC) is an epithelial malignancy arising within the posterior nasopharynx with a high incidence among Southeast Asian, Alaskan Eskimo, Greenland, and Central and North African populations (1). The World Health Organization (WHO) categorizes NPCs into three histological subtypes: well-differentiated squamous cell carcinoma (WHO type I), nonkeratinizing carcinoma (WHO type II), and undifferentiated carcinoma (WHO type III) (2). Although the etiology of NPC is still unclear, both genetic and environmental factors have been linked to the development of NPC. Among the environmental factors, infection with Epstein-Barr virus (EBV) has been extensively studied and shown to play a critical etiological role in NPCs, particularly the undifferentiated nasopharyngeal carcinoma subtype (WHO type III). However, EBV is less commonly found in other types of NPC, such as WHO type I.
Due to the rarity of this disease and the limited availability of pathological specimens, NPC cell lines have been important model systems to study its pathophysiology. The unique tropism of EBV to NPC tissues also makes NPC cell lines valuable systems to study EBV's biology and pathogenesis. The majority of NPC cell lines were established around 10 to 30 years ago, with very few NPC cell lines stably harboring natural EBV infection (e.g., c666-1). Although most of NPC cell lines used today are EBV negative, it is believed that they were once EBV positive and that the EBV genome was lost due to long-term culture (3). Whether other exogenous agents are present in these NPC cell lines has not yet been documented.
The use of next-generation sequencing (NGS) technology has successfully been applied to the discovery and investigation of pathogens associated with cancer. This approach utilizes an unbiased method for the global assessment of all exogenous agents within a cancer sample with high sensitivity and specificity. Several laboratories have successfully utilized NGS and specifically high-throughput RNA sequencing (RNA-seq) for the discovery and investigation of exogenous agents associated with various cancers (4–11).
In this study, we utilized RNA-seq technology along with our computational analysis pipeline RNA CoMPASS (12) to explore the exogenous agents associated with nasopharyngeal carcinomas. To our surprise, most of the NPC cell lines analyzed were positive for human papillomavirus 18 (HPV-18). Further transcriptome and comparative analyses revealed that these HPV-18-positive NPC cell lines, CNE1, CNE2, HONE1, AdAH, and NPC-KT, are likely HeLa derivatives.
MATERIALS AND METHODS
Cell culture.
CNE1 is a well-differentiated nasopharyngeal carcinoma cell line established by the Laboratory of Tumor Viruses of Cancer Institute in China (13) and was obtained from three independent sources: Jack Strominger (Harvard) for CNE1, Yixin Zeng (China) for CNE1-CN, and Asuka Nanbo (Japan) for CNE1-JPN. CNE2 is a poorly differentiated nasopharyngeal carcinoma cell line established by Gu and colleagues (14) and was obtained from Jack Strominger (Harvard). AdAH is a human nasopharyngeal adenoid epithelial cell line established by Toru Takimoto's group in Japan (15) and was obtained from Erik Flemington (Tulane). NPC-KT is an EBV-positive hybrid cell line which was generated by the fusion of EBV genome-positive NPC epithelial explant culture cells with AdAH cells in Takimoto's laboratory in Japan (16) and was obtained from Erik Flemington (Tulane). HONE1 is a poorly differentiated nasopharyngeal carcinoma cell line established by Ronald Glaser and colleagues (17) and was obtained from Asuka Nanbo (Japan). The cell line c666-1 is an EBV-infected undifferentiated NPC cell line established by Dolly Huang's group in China (18) and was obtained from Bryan Cullen (Duke). HeLa is a well-known and characterized human cervical adenocarcinoma cell line (19) and was obtained from the ATCC and from Erik Flemington (Tulane).
All cells (except c666-1, which was cultured in RPMI 1640) were grown in Dulbecco modified Eagle medium (DMEM; Thermo Scientific, Waltham, MA) supplemented with10% fetal bovine serum (FBS; Invitrogen-Gibco, Grand Island, NY) and 0.5% penicillin-streptomycin (Invitrogen-Gibco). Cells were grown at 37°C in a humidified, 5% CO2 incubator. In our laboratory, we have never used or generated any HPV plasmid or HPV recombinant plasmid.
Sequencing data set acquisition.
RNA-seq data sets from c666-1, HK1, NP460, and x666-1 were downloaded from the Gene Expression Omnibus under accession number GSE54174. In addition, paired-end HeLa cells RNA-seq data sets were downloaded from the European Nucleotide Archive and the Sequence Read Archive under run accession numbers ERR049804 (20) and SRR309265 (21), respectively.
Sample preparation and next-generation sequencing.
All cell lines were freshly thawed, grown to 90% confluence on a 10-cm plate, and harvested for RNA using 0.25% trypsin with 1 mM EDTA. Total RNA was extracted from each cell line culture using the miRNeasy minikit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Three cell lines were selected for RNA-sequencing, including CNE1 (CNE1 cells from Harvard), CNE1-JPN (CNE1 cells from Japan), and HONE1. All cDNA libraries were prepared from ribosomal RNA-depleted (ribodepleted) RNAs using the Illumina Truseq Stranded Total RNA Sample Prep kit (RS-122-2101). The ribodepleted RNA cDNA library was subjected to 1 × 100-base single-end strand-specific sequencing on an Illumina HiSeq 2000 instrument.
RNA CoMPASS analysis.
RNA CoMPASS is an automated computational pipeline that seamlessly analyzes RNA-seq data sets (12). Briefly, RNA-seq data sets were first aligned to the human reference genome, hg19 (Genome Reference Consortium GRCH37), plus a splice junction database (which was generated using the make transcriptome application from Useq [22]; splice junction radius set to the read length minus 4) using Novoalign version 3.00.05 (Novocraft) (-o SAM, default options). Nonmapped reads were isolated and subjected to consecutive BLAST version 2.2.27 (23) searches against the Human RefSeq RNA database and then to the NCBI NT database to identify reads corresponding to known exogenous organisms (24). Results from the NT BLAST searches were filtered to eliminate matches with an E value of greater than 10e−6. The results were then fed into the taxonomic classifier MEGAN 4 version 4.70.4 (25) for visualization of taxonomic classifications within the specimens being analyzed. RNA CoMPASS was run in parallel on three 2x2.66 GHz 12 core Intel Xeon Mac Pro computers with 64 to 96 GB of memory each.
Real-time RT-PCR analysis.
Total RNA was reverse transcribed using the SuperScript III First-Strand Synthesis System for reverse transcription-PCR (RT-PCR; Invitrogen, Carlsbad, CA). Random hexamers were used along with 1 μg of RNA in a 20-μl reaction volume according to the manufacturer's instructions. For the incubation steps (25°C for 10 min followed by 50°C for 50 min), a Mastercycler ep (Eppendorf, Hamburg, Germany) was used. The resulting cDNA was subjected to quantitative (real-time) PCR using sequence-specific forward and reverse primers (Integrated DNA Technologies) (see Table S1 in the supplemental material). For real-time PCR, 1 μl of the resulting cDNA was used in a 10-μl reaction volume that included 5 μl of SybrGreen and a 500 nM concentration each of forward and reverse primers. Amplification was carried out using the following conditions: 95°C for 3 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. Melt curve analysis was performed at the end of every quantitative RT-PCR (qRT-PCR) run. Samples were tested in triplicates. No-template controls were included in each PCR run. PCRs were performed on a Bio-Rad CFX96 Real Time System, and data analysis was performed using CFX Manager 3.0 software. Relative detection levels were calculated by normalizing with the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as a reference gene.
Viral transcriptome analysis.
Raw sequence data were aligned to a reference genome containing a human genome (hg19; Genome Reference Consortium GRCH37) plus a library of virus sequences (including sequences from all known human viruses documented by NCBI and PAvE (Papillomavirus Episteme Database [http://pave.niaid.nih.gov/]) (see Table S2 in the supplemental material). The alignments were performed using Spliced Transcripts Alignment to a Reference (STAR) aligner version 2.3.0 (--clip5pNbases 6, default options) (26) and were subjected to visual inspection using the Integrative Genomics Viewer (IGV) genome browser (27). Transcript data from STAR were subsequently analyzed using RSEM version 1.2.09 (28) for quantification of human and HPV-18 gene expression. Signal maps (i.e., the total number of reads covering each nucleotide position) were generated using IGV tools, and read coverage maps were visualized using the IGV genome browser (27).
Identification of HPV-18 integration sites and breakpoints.
The ribodepleted single-end strand-specific reads aligned previously were analyzed further to determine HPV-18 integration sites and breakpoints in NPCs. For each 100-base single-end read, we extracted the first 25-mers from both ends, and these paired 25-mer reads were then aligned independently of each other to a reference genome containing both human hg19 and HPV-18 genomes. If one 25-mer end was aligned to human and the other corresponding 25-mer end mapped to the viral genome, their corresponding single-end read represents a fusion read. The corresponding 25-mer read pairs from the identified fusion reads were collected and visualized in the IGV browser to pinpoint the viral integration sites in the host genome and to identify the breakpoints in the viral genome.
SNV analysis.
The coverage of reads aligning to human and HPV-18 from the STAR aligner was visualized using IGV. Mutations in the HPV-18 genome were color-coded and manually inspected. Human single nucleotide variants (SNVs) were identified using a detection pipeline consisting of samtools version 0.1.19 mpileup (-q 10, default parameters) (29) and VarScan version 2.3.6 (mpileup2snp --p-value 0.01 -output -vcf 1, default parameters) (30) to identify candidate SNVs and output in variant call format (VCF) format. In dealing with SNVs from RNA-seq data, the potential candidate SNVs not called may be due to lack of expression or not meeting the default parameter thresholds. To overcome this obstacle, coverage maps for each chromosome were generated from expression data for each RNA-seq data set with a minimum of 10 reads using IGVtools (27). The coverage maps were compared to the candidate SNV output in order to determine SNVs that were from uniform gene expression for all RNA-seq data sets analyzed. This was a necessary step prior to cluster analysis using PLINK version 1.9 (https://www.cog-genomics.org/plink2) (31) to eliminate cluster bias based on SNVs originating from genes expressed only in certain cell types. The human SNVs in VCF format from commonly expressed genes for each cell line were merged using vcftools version 0.1.11 (vcf-merge, default parameters) (32). The merged VCF file was converted to binary PED files using PLINK 1.9 (--vcf, --make-bed). The resulting binary PED files were subjected to clustering (-cluster) and a subsequent cluster dendrogram was generated in R using plot(hclust(dist(x))) (33).
Genomic DNA isolation and genotyping of the HeLa-specific L1 retrotransposon marker.
Genomic DNA was isolated from freshly thawed NPC and HeLa cell lines using the DNeasy Blood & Tissue kit (Qiagen catalog no. 69504) by following the manufacturer's protocol. DNA samples were quantified using a NanoDrop 2000 spectrophotometer (Thermo Scientific).
To detect the L1 retotransposon marker, 500 ng of the genomic DNA was used in a 50-μl reaction volume that included 1× standard Taq reaction buffer (NEB catalog no. B9014), 500 nM each primer (VM164A, VM164B, and RB164K2), 400 μM deoxynucleotide solution mix (NEB catalog no. N0447), and 2.5 U of Taq DNA polymerase (NEB catalog no. M0273). A list of primers can be found in Table S1 in the supplemental material. Amplification was carried out on a Mastercycler ep (Eppendorf, Hamburg, Germany), using the following conditions: 96°C for 30 s, followed by 30 cycles of 96°C for 30 s, 54°C for 30 s, and 72°C for 1 min, and then 1 cycle of 72°C for 5 min.
Thirty microliters of PCR product was run on a 1% agarose gel and visualized by ethidium bromide staining on a Bio-Rad gel documentation system. The product size was estimated by comparison to a Quick-Load 2-log DNA ladder (NEB N0469).
Nucleotide sequence accession numbers.
Sequence data (RNA-seq) for CNE1, CNE1-JPN, and HONE1 can be retrieved from the National Center for Biotechnology Information Sequence Read Archive (accession no. SRP043686). Fasta files, bed format annotation files (for visualization of annotation data on a genome browser), and .gtf format annotation files (for quantification of RNA-seq data) are available on request.
RESULTS
Detection of HPV-18 in nasopharyngeal carcinoma cells.
To elucidate the metatranscriptome of NPC cells that are commonly used in NPC and EBV research, RNA-seq data sets generated from our lab (CNE1 and HONE1) as well as from publicly available sequencing data sets (HK1, c666-1, x666-1, and NP460) were analyzed using RNA CoMPASS. Analysis of the virome component of RNA CoMPASS showed that these samples were free of most known DNA viruses (see Fig. S1 to S6 in the supplemental material). As expected, Epstein-Barr virus was detected in both c666-1 (Fig. 1A; see also Fig. S1) and x666-1 (see Fig. S2) but not in the other data sets analyzed. In addition to the EBV findings, we were surprised to find high read numbers for the high-risk human papillomavirus 18 (HPV-18) in the CNE1 (Fig. 1A; see also Fig. S3) and HONE1 (see Fig. S4) data sets.
FIG 1.

Detection of human papillomavirus 18 in nasopharyngeal carcinomas. (A) RNA-seq data from each NPC sample was analyzed using RNA CoMPASS. A representative virome branch of the taxonomy tree (CNE1) for each HPV+ sample was generated using the metagenome analyzer MEGAN 4. c666-1 was positive for both EBV (human herpesvirus 4) and MuLV. (B) In-depth analysis of the virome. Raw reads from each sample were aligned to the hg19 genome with the custom human virus genome using the genome aligner STAR. Reads are displayed as viral reads per million human mapped reads. Blue, HPV-16 reads; green, HPV-18 reads; and red, EBV reads. (C) Using quantitative real-time PCR, six NPC cell lines were surveyed for the presence of HPV-16 (blue columns), HPV-18 (green columns), and EBV (red columns). Five out of the six NPC cell lines demonstrated evidence of HPV-18, but none of the NPC cell lines was infected with HPV-16. Both NPC-KT and c666-1 were positive for EBV EBER. Samples were normalized to GAPDH and compared to c666-1 for HPV-16 or -18 expression and compared to NPC-KT for EBV expression.
The murine leukemia virus (MuLV), an RNA oncogenic virus, was detected in both the c666-1 and x666-1 cell lines (see Fig. S1, S2, and S7 in the supplemental material) and represented active infection (M. J. Strong and Z. Lin, unpublished data). To our knowledge no study has previously reported the detection of MuLV within these 2 NPC cell lines, and we reason that the infection most likely occurred before 1999 at the source institution, since both cell lines were established through passage in laboratory mice. MuLV was also detected in NP460 (see Fig. S7). Detailed MuLV transcriptome analysis in this cell line revealed that the MuLV reads may be derived from MuLV-based retroviral expression vectors since coverage was restricted to the LTR regions (Strong and Lin, unpublished).
To confirm the virus findings from RNA CoMPASS, the NPC RNA-seq data sets were aligned to a reference genome containing a human genome (hg19; Genome Reference Consortium GRCH37) plus a library of virus sequences (including sequences from all known human viruses documented by NCBI and PAvE) (see Table S2 in the supplemental material) using the aligner Spliced Transcripts Alignment to a Reference (STAR) version 2.3.0 with default options. Consistent with our RNA CoMPASS analysis, CNE1 and HONE1 data sets were positive for HPV-18 but not HPV-16, with viral reads per million human mapped reads of 170 and 503 for CNE1 and HONE1, respectively (Fig. 1B). Furthermore, c666-1 and x666-1 samples were negative for HPV-16 and HPV-18 but positive for EBV, with 457 and 387 viral reads per million human mapped reads, respectively. Finally, HK1 was negative for all known viruses (see Fig. S5 in the supplemental material).
The detection of HPV-18 in these NPC cell lines was unexpected because HPV-18 typically infects the genital epithelium and is generally not associated with the head and neck region (34). To verify that the presence of HPV-18 in these cells was not due to contamination from a particular lab, CNE1 cells were obtained from two additional independent laboratories in China (CNE1-CN) and Japan (CNE1-JPN) and analyzed by real-time PCR using primers specific for HPV-18 and HPV-16. The results confirmed the presence of HPV-18 but not HPV-16 in these CNE1 cell lines (see Fig. S8 in the supplemental material). Furthermore, total RNA from CNE1 cells obtained from Japan was subjected to RNA sequencing and subsequent virome analysis (see Fig. S9). The results were consistent with our first CNE1 RNA-seq data set, with more than 500 HPV-18 reads per million human mapped reads (Fig. 1B).
An additional three NPC cell lines (CNE2, AdAH, and NPC-KT) were surveyed for the prevalence of HPV as well as EBV using real-time PCR. Cell lines (CNE1, HONE1, and c666-1) were also included in the assay to serve as positive controls and to further validate the RNA-seq results. As shown in Fig. 1C, CNE1, CNE2, AdAH, NPC-KT, and HONE1 cells were all positive for HPV-18 but not HPV-16, with NPC-KT also positive for EBV. In contrast, c666-1 was negative for both HPV-18 and HPV-16 but positive for EBV.
HPV transcriptome analysis in NPC cells.
The depth of coverage in our NPC RNA-seq data sets were sufficient for HPV transcriptome analysis, which indicated robust expression of HPV-18 (see Fig. S10 in the supplemental material). Further transcriptome analysis revealed that the viral early genes E6 and E7 were actively expressed at relatively high levels (see Table S3). As a comparison, two publicly available HeLa RNA-seq data sets from Nagaraj et al. (20) and Cabili et al. (21) were obtained and subjected to HPV transcriptome analysis. The average transcripts per million (TPM) for HPV-18 E6 in HeLa and HPV+ NPC cell lines were 461.6 (20% of total transcripts) and 132.4 (23% of total transcripts), respectively. The average TPM for HPV-18 E7 in HeLa cells and HPV+ NPC cell lines were 1,228.4 (52% of total transcripts) and 297.7 (50% of total transcripts), respectively. Although there was abundant expression of the E1 gene (average TPM, 622.3 [26% of total transcripts] and 122.3 [21% of total transcripts] for HeLa cells and HPV+ NPC cell lines, respectively), the expression of the E1 open reading frame (ORF) is disrupted, leading to a premature mRNA product (see Fig. S10).
Since the NPC cell lines CNE1, CNE1-JPN, and HONE1 were subjected to strand-specific RNA sequencing, we were able to distinguish transcripts initiated from either the sense or antisense direction (strand-specific viral transcripts). In addition to the transcripts observed from the E6/E7 region, large numbers of reads were observed in the E1/E2 and L1/URR regions (see Fig. S10 in the supplemental material). The middle of the HPV-18 genome is void of coverage due to the viral genome rearrangement during integration into human chromosome 8 (see details below). Strand-specific transcriptome analysis further revealed that nearly all reads were derived from the sense strand of HPV-18. There were very low read levels (<15) from the antisense strand, possibly resulting from inefficiencies in the strand-specific sequencing approach (see Fig. S10).
HPV+ NPCs have a common pattern of viral and cellular genomic rearrangement as HeLa cells.
In HPV-associated cancers, the HPV genome is typically found to be integrated into the host genomes. We therefore examined the transcriptionally active integration sites in NPCs. Our single-end ribodepleted strand-specific RNA-seq data set was analyzed by extracting the first 25-mer reads from each end of the 100-base read and aligning them independently of each other to a reference genome containing both the human hg19 and HPV-18 genomes. Next, fusion reads were identified as those in which one 25-mer end mapped to one genome and the other corresponding 25-mer end mapped to the other genome. These fusion reads were then utilized to pinpoint the integration sites/breakpoints through visualizing the read coverage peaks in the IGV genomic browser. This analysis revealed that all examined HPV+ NPCs demonstrated similar viral and cellular genomic rearrangements within a 12-kb fragile region of human chromosome 8 (Fig. 2). Further, the HPV-18 integrated in a reverse manner and exhibited five genome breakpoints (numbered 1 to 5 with corresponding read coverage peaks) resulting in the four potential HPV-18 integration patterns (colored and boxed). For each HPV-18 integration pattern, there is a corresponding read coverage peak for chromosome 8 (labeled a to e). All HPV-18 integration patterns started with the L1 breakpoint and shared the same upstream insertion point on chromosome 8, as demonstrated by the high coverage peaks, respectively. Analysis of reads spanning the rearranged HPV-18 integrated genome demonstrated consistent coverage across all segments, suggesting active transcription in this rearranged configuration (Fig. 2).
FIG 2.

HPV+ NPCs demonstrate same pattern of viral and cellular genomic rearrangement as HeLa cells. Strand-specific sequencing data from ribodepleted CNE1 RNA were analyzed for integration sites and breakpoints. First, 25-mer reads were extracted from both ends of each read and aligned to a reference genome containing both the human hg19 and HPV-CNE1 genomes. Next, fusion reads that were aligned to both human and HPV genomes were identified and visualized in the IGV genomic browser. Potential HPV integration patterns are represented as colored boxes. Read coverage peaks represent chromosome 8 integration sites (a to e) and HPV genome breakpoints (1 to 5). The rearranged HPV integrated genome is shown with breakpoints highlighted by numbers and corresponding read coverage.
Since HPV-18 has been shown to integrate in the same region of chromosome 8 in HeLa cells (35), and since HeLa cells have been plaguing in vitro cell cultures since the 1950s, we compared the precise integration sites and viral breakpoints between NPCs and HeLa cells. Reference genomes that span the five HPV-18/human chromosome 8 junctions in NPCs were generated, and reads from HPV+ NPCs and the two HeLa data sets were aligned using the STAR aligner with default parameters. The results showed that all HPV-18/human chromosome 8 junctions in HPV+ NPCs were identical (down to the nucleotide) to those observed in the two HeLa cells (see Fig. S11 to S15 in the supplemental material). Furthermore, nucleotide insertions within a junction sequence specifically identified in HeLa cells (35) showing no homology to human or HPV-18 were also identified in analyzed HPV+ NPCs (see Fig. S15).
SNV analysis of NPCs further substantiates HeLa cell contamination in NPC cell lines.
To further elucidate the cell origins of these HPV+ NPCs, we investigated both viral and human single nucleotide variations (SNVs) in the three NPCs and the two HeLa data sets. Analysis of the viral SNV showed that all five data sets have the same integrated HPV-18 genome, exhibiting 29 identical SNVs relative to a reference HPV-18 consensus genome (Fig. 3A; see also Fig. S16 in the supplemental material). Additional SNVs observed in the HeLa-1 sample are likely from sequencing errors, since those were not preserved in the HeLa-2 sample.
FIG 3.

Single nucleotide variant analysis of NPCs further substantiates HeLa cell contamination in NPC cell lines. (A) HPV-18 SNVs were analyzed in all HPV-18-positive NPCs and visualized using IGV. The nucleotide mutation coordinates and corresponding amino acid changes are represented in Fig. S16 in the supplemental material. The y axis represents the number of reads at each nucleotide position of the genome. The maximum scale for the coverage graphs is 4,950 reads for HeLa-1 and 1,650 reads for the other graphs. Coverage graph insets represent blowup regions of the genome with maximum scales of 60 reads for HeLa-1 and 200 reads for HeLa-2 and HONE1. (B) Universally expressed human-specific SNVs in each cell line were also identified and clustered using PLINK. CNE1 and HONE1 were clustered with HeLa cells in one major branch, whereas c666-1, x666-1, HK1, and NP460 were clustered in another branch.
Analysis of the human SNVs for all RNA-seq data sets was also performed. A total of 13,551 human SNVs were identified within genes commonly expressed in all nine data sets. These SNVs were subjected to clustering using PLINK (31). As shown in Fig. 3B, two distinct clusters were identified. The two HeLa cell samples cluster tightly with CNE1, CNE1-JPN, and HONE1, forming one major cluster. In contrast, cell lines c666-1, x666-1, HK1, and NP460 formed a distinct major cluster. Notably, CNE1 and HONE1 are within the HeLa cell cluster but not tightly clustering with the other two HeLa cell samples, suggesting that these NPC cell lines were not entirely of HeLa cell origin.
In addition to cluster analysis, in-depth investigation of the SNV patterns at the gene level was performed. By comparing individual SNVs, we found that HPV+ NPC cells exhibited distinct SNV patterns in certain genomic regions compared to HeLa cells. For example, isoenzyme analysis of glucose-6-phosphate dehydrogenase (G6PD) revealed G6PD type A for both HeLa cell samples, a signature variant that has been previously used to identify HeLa cells (36). Interestingly, CNE1, CNE1-JPN, and HONE1 were all heterozygous for this G6PD type A variant (see Fig. S17A in the supplemental material), while HK1, c666-1, x666-1, and NP460 were homozygous for the wild-type G6PD type B variant. Further, a unique SNV within the p53 gene was found in CNE1, CNE1-JPN, and HONE1 but not HeLa or the other NPC cell lines (see Fig. S17B). Finally, SNV analysis of phosphoglucomutase 1 (PGM1) showed a heterozygous variant in the 3′ untranslated region in HeLa, CNE1, CNE1-JPN, and HONE1 but not in HK1, c666-1, x666-1, or NP460 (see Fig. S17C). Taken together, these data suggest that HPV+ NPCs, CNE1 and HONE1, are likely derivatives of HeLa and another cell of unknown origin.
HeLa-specific L1 retrotransposon marker is present in some NPC cell lines.
To further confirm our findings, we screened the six NPC cell lines with a single duplex detection PCR assay, which targets a HeLa-specific L1 retrotransposon insertion (37). As shown in Fig. 4, a 325-bp insertion-specific amplicon was readily detected in HeLa cells. This marker was also detected in the AdAH and NPC-KT cells. In contrast, other HPV+ NPCs such as CNE1, CNE1-JPN, CNE1-CN, and HONE1 were negative for this HeLa marker, indicating that these cells bear only part of the HeLa genomic sequences (partial HeLa genome) and lowering the possibility that these cell lines are simply cross-contaminants of HeLa cells.
FIG 4.
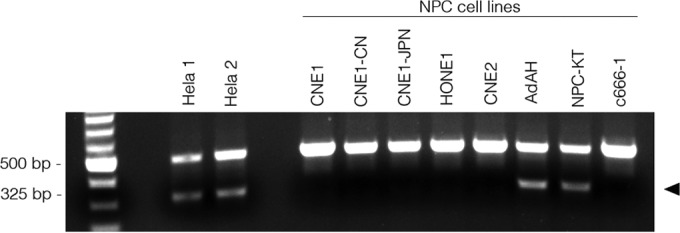
HeLa cell-specific L1 retrotransposon marker is present in a group of NPC cell lines. A single duplex detection PCR assay was conducted to detect a HeLa-specific L1 retrotransposon marker. Genomic DNAs from six NPC cell lines and two independent HeLa cell lines were subjected to PCR analysis. PCR products were fractionated on a 1% agarose gel. Two amplicons/bands were observed, and the HeLa-specific amplicon/band presents as a band of 325 bp.
DISCUSSION
CNE1 and other NPC cell lines serve as an essential widely used tool in both the NPC and the EBV research communities. Although the majority of NPC cell lines used today are currently EBV negative, it is believed that they were initially positive for EBV and that their EBV genomes were lost due to long culture (3). To date, whether other exogenous agents are present in NPC cell lines has not been determined. During our effort to fully assess the viromes of NPC cell lines, we identified active HPV-18 infection in several cell lines analyzed. Even though it is possible that these NPCs might have naturally acquired HPV infection (38), it is HPV-16, but not HPV-18, that is typically associated with tumors in the head and neck region (38). Considering the HPV-18 integration pattern and the host and viral SNV analyses, along with the widespread contamination of HeLa in several tissue culture systems, we reasoned that this HPV-18-positive cervical adenocarcinoma cell line is a likely source of contamination in these NPC cell lines.
Using our in-house RNA-seq based computational pipeline, we were able to identify the HPV/human genome rearrangements, which provided critical evidence to support the notion that CNE1 and HONE1 cells were of HeLa origin. Four complex integration patterns were detected in CNE1, HONE1, and HeLa data sets, which was consistent with a recent study showing similar integration patterns of HPV-18 in HeLa cells (35). Despite the similarity in HPV-18 integration patterns observed in our data and those of Adey et al., we observed an additional HPV-18 breakpoint and insertion. As shown in Fig. 2, there was a peak at the beginning of HPV-18 E1 (peak 3) indicating a pileup of fusion reads with the corresponding pileup shown as peak d on chromosome 8. This new observed breakpoint and integration pattern (see Fig. S13 in the supplemental material) may be due to differences in HeLa cell clones or may be allele specific, since we analyzed the average of both alleles, while Adrey et al. analyzed only a single allele from chromosome 8.
Our results indicated that several NPC cell lines analyzed (CNE1, CNE2, AdAH, NPC-KT, and HONE1) are contaminated with HeLa cells either during the initial establishment of these lines (e.g., CNE1) or during subsequent cell passage. Further, an earlier study identified the presence of HPV-18 E6 protein in AdAH and its variant (39), suggesting contamination with HeLa. In addition, since NPC-KT is also HPV-18 positive and was established in the same laboratory as AdAH, it is likely that NPC-KT is also contaminated with HeLa, with contamination possibly occurring during the initial establishment of this cell line. It is likely that other NPC cell lines not analyzed in this study that are commonly used in the field are also contaminated. For example, we detected HPV-18 in another NPC RNA-seq data set (SRA020971) (40) generated from 5-8F cells (a SUNE-1-derived cell line) (data not shown). Unfortunately, due to the nature of the experimental design (microRNA-seq study, which misses the majority of mRNA sequences), we do not have definitive evidence to call HeLa contamination in this cell line. Therefore, considering the rarity of NPCs, the long history of NPC cell lines, and the lack of rigorous authentication (since general cell line vendors such as the ATCC do not carry NPC cell lines), we strongly recommend cell line authentication prior to performing experiments. Otherwise, usage of HeLa-contaminated NPC cell lines in both NPC- and EBV-related studies might lead researchers to draw misleading or even false conclusions.
Our findings are consistent with a study conducted in Hong Kong by Chan and colleagues, in which they tested the authenticity of several NPC cell lines, including CNE1, CNE2, HK1, x666-1, and c666-1 (41). Chan et al. conducted a short tandem repeat (STR) profiling assay on these NPC cell lines and determined that CNE1 and CNE2 contain at least one allele identical to HeLa based on the STR patterns (41). Furthermore, Chan et al. identified the same CNE1-specific p53 SNV (Thr 280) that we observed in our CNE1 and HONE1 data sets (41).
The HeLa cell line was established in 1951 and is the first human cell line established from an aggressive cervical carcinoma. There has been widespread use of this cell line for a variety of experiments conducted in the United States and abroad. Since the establishment of this cell line more than 60 years ago, many cell lines have been confirmed to be contaminated with HeLa (42, 43). Despite massive collaborative efforts to ameliorate HeLa contamination and cell line misidentification within our cell line repertoire, these issues still persist today, as evidenced by this study. Recently, there has been a call to action to address cell line contamination and misidentification in the hope of avoiding the generation of misleading data due to cross-contaminated cell lines (44, 45).
To the best of our knowledge, an RNA-seq-based cell authentication method has not yet been reported. The existing methods all focus on analyzing the genetic DNA sequences of the cell lines. For instance, the most popular STR profiling measures the number of specific, short repetitive DNA sequences at specific loci in the genome (42). In this study, we explored the possibility of using transcriptome data analysis as an additional tool for cell authentication. Using this approach, we were able to obtain (from the RNA-seq data sets) critical information regarding the viral transcriptome, the viral and cellular genomic rearrangement, and the viral and cellular SNVs that were used for successful cell authentication. We believe that with increasing awareness of cell line contamination, the use of an RNA-seq-based approach may be another viable option in the pursuit of validating cell lines for experimental use.
Given our data along with data from Chan et al., we conclude that CNE1 is not of true nasopharyngeal origin and is therefore an inappropriate cell line to study NPC biology. The other NPC cell lines that tested positive for HPV-18 (HONE1, CNE2, AdAH, and NPC-KT) and/or HeLa markers (AdAH and NPC-KT) are strongly suspicious for HeLa contamination; however, additional testing from multiple sources is needed. For instance, our data strongly indicate that HONE1 is a HeLa-related somatic cell hybrid; however, without testing from multiple sources, a definitive conclusion cannot be drawn.
The three NPC cell lines analyzed in this study that are HPV-18 negative and are not contaminated with HeLa cells are c666-1, x666-1, and HK1. The normal nasopharyngeal cell line NP460 is also HPV-18 and HeLa negative. Even though c666-1 and x666-1 are free of HeLa contamination, the presence of murine leukemia virus (MuLV) in these cells might influence the results of EBV-related studies in unforeseen ways that may cause invalid or skewed conclusions. Caution must be exercised in interpreting experimental results from these two cell lines.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Louisiana Clinical & Translational Science Center Pilot Grant (1 U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health, which funds the Louisiana Clinical and Translational Science Center) to Z.L., Ruth L. Kirschstein National Research Service Award F30CA177267 from the NCI to M.J.S., and National Institutes of Health grant P20GM103518 to Prescott Deininger, which supports the Tulane Cancer Center next-generation sequencing bioinformatics core used in this study.
Footnotes
Published ahead of print 2 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01457-14.
REFERENCES
- 1.Chang ET, Adami H-O. 2006. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol. Biomarkers Prev. 15:1765–1777. 10.1158/1055-9965.EPI-06-0353 [DOI] [PubMed] [Google Scholar]
- 2.Shanmugaratnam K, Sobin L. 1993. The World Health Organization histological classification of tumours of the upper respiratory tract and ear. A commentary on the second edition. Cancer 71:2689–2697 [DOI] [PubMed] [Google Scholar]
- 3.Dittmer DP, Hilscher CJ, Gulley ML, Yang EV, Chen M, Glaser R. 2008. Multiple pathways for Epstein-Barr virus episome loss from nasopharyngeal carcinoma. Int. J. Cancer 123:2105–2112. 10.1002/ijc.23685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng H, Shuda M, Chang Y, Moore PS. 2008. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319:1096–1100. 10.1126/science.1152586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA. 2012. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22:299–306. 10.1101/gr.126516.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, Ogino S, Birren BW, Huttenhower C, Garrett WS, Meyerson M. 2012. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22:292–298. 10.1101/gr.126573.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Z, Puetter A, Coco J, Xu G, Strong MJ, Wang X, Fewell C, Baddoo M, Taylor C, Flemington EK. 2012. Detection of murine leukemia virus in the Epstein-Barr virus-positive human B-cell line JY, using a computational RNA-Seq-based exogenous agent detection pipeline, PARSES. J. Virol. 86:2970–2977. 10.1128/JVI.06717-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strong MJ, O'Grady T, Lin Z, Xu G, Baddoo M, Parsons C, Zhang K, Taylor CM, Flemington EK. 2013. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin's diffuse large B-cell lymphoma cohort by using RNA sequencing. J. Virol. 87:13059–13062. 10.1128/JVI.02380-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strong MJ, Xu G, Coco J, Baribault C, Vinay DS, Lacey MR, Strong AL, Lehman TA, Seddon MB, Lin Z, Concha M, Baddoo M, Ferris M, Swan KF, Sullivan DE, Burow ME, Taylor CM, Flemington EK. 2013. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog. 9:e1003341. 10.1371/journal.ppat.1003341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Z, Wang X, Strong MJ, Concha M, Baddoo M, Xu G, Baribault C, Fewell C, Hulme W, Hedges D, Taylor CM, Flemington EK. 2013. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J. Virol. 87:1172–1182. 10.1128/JVI.02517-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin Z, Xu G, Deng N, Taylor C, Zhu D, Flemington EK. 2010. Quantitative and qualitative RNA-Seq-based evaluation of Epstein-Barr virus transcription in type I latency Burkitt's lymphoma cells. J. Virol. 84:13053–13058. 10.1128/JVI.01521-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu G, Strong MJ, Lacey MR, Baribault C, Flemington EK, Taylor CM. 2014. RNA CoMPASS: a dual approach for pathogen and host transcriptome analysis of RNA-Seq datasets. PLoS One 9:e89445. 10.1371/journal.pone.0089445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LTVCI. 1978. Establishment of an epithelioid cell line and a fusiform cell line from a patient with nasopharyngeal carcinoma. Sci. Sin. 21:127–134 [PubMed] [Google Scholar]
- 14.Gu SU, Tann BF, Zeng Y, Zhou WP, Li K, Zhao MC. 1983. Establishment of an epithelial cell line (CNE-2) from an NPC patient with poorly differentiated squamous cell carcinoma. Chin. J. Cancer 2:70–72 [Google Scholar]
- 15.Takimoto T, Furukawa M, Hatano M, Umeda R. 1984. Epstein-Barr virus nuclear antigen-positive nasopharyngeal hybrid cells. Ann. Otol. Rhinol. Laryngol. 93:166–169 [DOI] [PubMed] [Google Scholar]
- 16.Takimoto T, Kamide M, Umeda R. 1984. Establishment of Epstein-Barr virus (EBV)-associated nuclear antigen (EBNA)-positive nasopharyngeal carcinoma hybrid cell line (NPC-KT). Arch. Otorhinolaryngol. 239:87–92. 10.1007/BF00454266 [DOI] [PubMed] [Google Scholar]
- 17.Glaser R, Zhang HY, Yao KT, Zhu HC, Wang FX, Li GY, Wen DS, Li YP. 1989. Two epithelial tumor cell lines (HNE-1 and HONE-1) latently infected with Epstein-Barr virus that were derived from nasopharyngeal carcinomas. Proc. Natl. Acad. Sci. U. S. A. 86:9524–9528. 10.1073/pnas.86.23.9524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung ST, Huang DP, Hui ABY, Lo KW, Ko CW, Tsang YS, Wong N, Whitney BM, Lee JCK. 1999. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int. J. Cancer 83:121–126. [DOI] [PubMed] [Google Scholar]
- 19.Gey G, Coffman W, Kubicek M. 1952. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 12:264–265 [Google Scholar]
- 20.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, Mann M. 2011. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7:548. 10.1038/msb.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25:1915–1927. 10.1101/gad.17446611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nix D, Courdy S, Boucher K. 2008. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 9:523. 10.1186/1471-2105-9-523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 24.Pruitt KD, Tatusova T, Brown GR, Maglott DR. 2012. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40:D130–D135. 10.1093/nar/gkr1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC. 2011. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21:1552–1560. 10.1101/gr.120618.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. 2012. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22:568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81:559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team. 2014. R: a language and environment for statistical computing. R. Foundation for Statistical Computing, Vienna, Austria: URL http://www.R-project.org/ [Google Scholar]
- 34.Kreimer AR, Clifford GM, Boyle P, Franceschi S. 2005. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol. Biomarkers Prev. 14:467–475. 10.1158/1055-9965.EPI-04-0551 [DOI] [PubMed] [Google Scholar]
- 35.Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500:207–211. 10.1038/nature12064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshida A, Watanabe S, Gartler SM. 1971. Identification of HeLa cell glucose 6-phosphate dehydrogenase. Biochem. Genet. 5:533–539. 10.1007/BF00485671 [DOI] [PubMed] [Google Scholar]
- 37.Rahbari R, Sheahan T, Modes V, Collier P, Macfarlane C, Badge RM. 2009. A novel L1 retrotransposon marker for HeLa cell line identification. Biotechniques 46:277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singhi AD, Califano J, Westra WH. 2012. High-risk human papillomavirus in nasopharyngeal carcinoma. Head Neck 34:213–218. 10.1002/hed.21714 [DOI] [PubMed] [Google Scholar]
- 39.Cayrol C, Flemington EK. 1996. The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO J. 15:2748–2759 [PMC free article] [PubMed] [Google Scholar]
- 40.Liao JY, Ma LM, Guo YH, Zhang YC, Zhou H, Shao P, Chen YQ, Qu LH. 2010. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3′ trailers. PLoS One 5:e10563. 10.1371/journal.pone.0010563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan SY-Y, Choy K-W, Tsao S-W, Tao Q, Tang T, Chung GT-Y, Lo K-W. 2008. Authentication of nasopharyngeal carcinoma tumor lines. Int. J. Cancer 122:2169–2171. 10.1002/ijc.23374 [DOI] [PubMed] [Google Scholar]
- 42.American Type Culture Collection Standards Development Organization Workgroup ASN-0002. 2010. Cell line misidentification: the beginning of the end. Nat. Rev. Cancer 10:441–448. 10.1038/nrc2852 [DOI] [PubMed] [Google Scholar]
- 43.Masters JR. 2002. HeLa cells 50 years on: the good, the bad and the ugly. Nat. Rev. Cancer 2:315–319. 10.1038/nrc775 [DOI] [PubMed] [Google Scholar]
- 44.Rojas A, Gonzalez I, Figueroa H. 2008. Cell line cross-contamination in biomedical research: a call to prevent unawareness. Acta Pharmacol. Sin. 29:877–880. 10.1111/j.1745-7254.2008.00809.x [DOI] [PubMed] [Google Scholar]
- 45.Lacroix M. 2008. Persistent use of “false” cell lines. Int. J. Cancer 122:1–4. 10.1002/ijc.23233 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.