

Summary
The past twenty years have seen many advances in our understanding of protein-protein interactions (PPI) and how to target them with small-molecule therapeutics. In 2004, we reviewed some early successes; since then, potent inhibitors have been developed for diverse protein complexes, and compounds are now in clinical trials for six targets. Surprisingly, many of these PPI clinical candidates have efficiency metrics typical of ‘lead-like’ or ‘drug-like’ molecules and are orally available. Successful discovery efforts have integrated multiple disciplines and make use of all the modern tools of target-based discovery - structure, computation, screening, and biomarkers. PPI become progressively more challenging as the interfaces become more complex, i.e., as binding epitopes are displayed on primary, secondary, or tertiary structures. Here, we review the last ten years of progress, focusing on the properties of PPI inhibitors that have advanced to clinical trials and prospects for the future of PPI drug discovery.
Introduction
Protein-protein interactions (PPI) represent a vast class of therapeutic targets both inside and outside the cell. PPI are central to all biological processes and are often dysregulated in disease. Despite the importance of PPI in biology, this target class has been extremely challenging to convert to therapeutics. Twenty years ago, PPI were deemed ‘intractable.’ High-resolution structures in the 1980–1990s showed PPI interfaces are generally flat and large (roughly 1000–2000 A2 per side)(Hwang et al., 2010), in stark contrast to the deep cavities that typically bind small molecules (ca. 300–500 A2)(Fuller et al., 2009). Unlike enzymes or GPCRs, nature did not offer simple small molecules that can start a chemical discovery process, and high-throughput screening (HTS) had not provided validated hits.
Between 1995–2005, hopeful signs were emerging. A clinically approved integrin antagonist (tirofiban), and natural products like taxanes, rapamycin, and cyclosporine inspired confidence that PPI could be modulated by small molecules. Mutational analysis of protein interfaces showed that not all residues at the PPI interface were critical but rather small “hot spots” conferred most of the binding energy (Arkin and Wells, 2004; Clackson and Wells, 1995). Hot spots tended to cluster at the center of the interface, to cover an area comparable to the size of a small molecule, to be hydrophobic, and to show conformational adaptivity. These features suggested that at least some PPI might have small-molecule-sized patches that could dynamically adjust to bind a drug-like molecule. By 2005, about a half-dozen small molecules had been reported to bind with the affinities one would expect for drug leads, at binding sites defined by high-resolution structures (Wells and McClendon, 2007). In parallel, computation and chemical technologies were being developed that might be well suited to PPI. For instance, fragment-based lead discovery (FBLD) has had a particularly strong impact. FBLD used biophysical methods, including crystallography, surface plasmon resonance, and NMR, or disulfide trapping (Tethering) to identify low-molecular weight, low-complexity molecules that bound weakly to subsites on the protein surface (Erlanson et al., 2004; Hajduk and Greer, 2007; Winter et al., 2012)].
The last decade has seen amazing progress in tackling challenging PPI targets with synthetic molecules. More than 40 PPIs have now been targeted (Basse et al., 2013; Higueruelo et al., 2009; Labbe et al., 2013), and several inhibitors have reached clinical trials. With this advance, it is important to reconsider the distinction between ligandability (‘druggability’) and our ability to convert PPI inhibitors into drugs. Historically, PPI inhibitors have been larger and more hydrophobic than typical orally available drugs (Wells and McClendon, 2007). Two commonly used metrics to assess the drug-like quality of a compound (or to compare a series of compounds) are ‘ligand efficiency’ (ΔG/HA) and ‘lipophilic ligand efficiency’ (pIC50 - logD or logP) (Hopkins et al., 2014). The LE for small molecule inhibitors of PPI have hovered around 0.24, whereas LE ~ 0.3 or higher is desired. Values of LLE > 5 are considered favorable for in vivo activity. Encouragingly, recent PPI inhibitors are approaching these ‘drug-like’ values for several targets (see below). Even inhibitors with properties outside average ranges for oral drugs have been made orally bioavailable. Clinically successful PPI inhibitors may therefore expand our understanding of the types of molecules that can be made into drugs.
Also during the past fifteen years, there has been very promising progress with designing peptides that target PPI and show promising cell based (and even in vivo) activities (Azzarito et al., 2013; Bernal et al., 2010; Boersma et al., 2012; Chang et al., 2013; DeLano et al., 2000; Gavenonis et al., 2014). While these approaches are outside the scope of the current review, they represent a parallel strategy that can also inform small-molecule design.
Although PPI come in many shapes and sizes, most of the clinical-stage inhibitors target PPI where the hot-spot residues are concentrated in small binding pockets (250 – 900 A2)(Basse et al., 2013; Smith and Gestwicki, 2012) and partner proteins are characterized by short primary sequences at the interface (London et al., 2013; Perkins et al., 2010). At one extreme, the peptide epitope is recapitulated by a linear peptide epitope 1–4 amino acids long; there are four clinical-stage examples of small-molecule mimetics of these epitopes. PPI that contain a single unit of secondary structure, such as an alpha helix, binding to a hydrophobic groove have also proven tractable to small-molecule inhibition. Globular interfaces, requiring tertiary structure on both sides of the PPI, have fewer published successes. In developing guidelines for PPI-inhibitor discovery, it is instructive to consider each clinical success story in the context of the type of interface (Figure 1).
Figure 1.
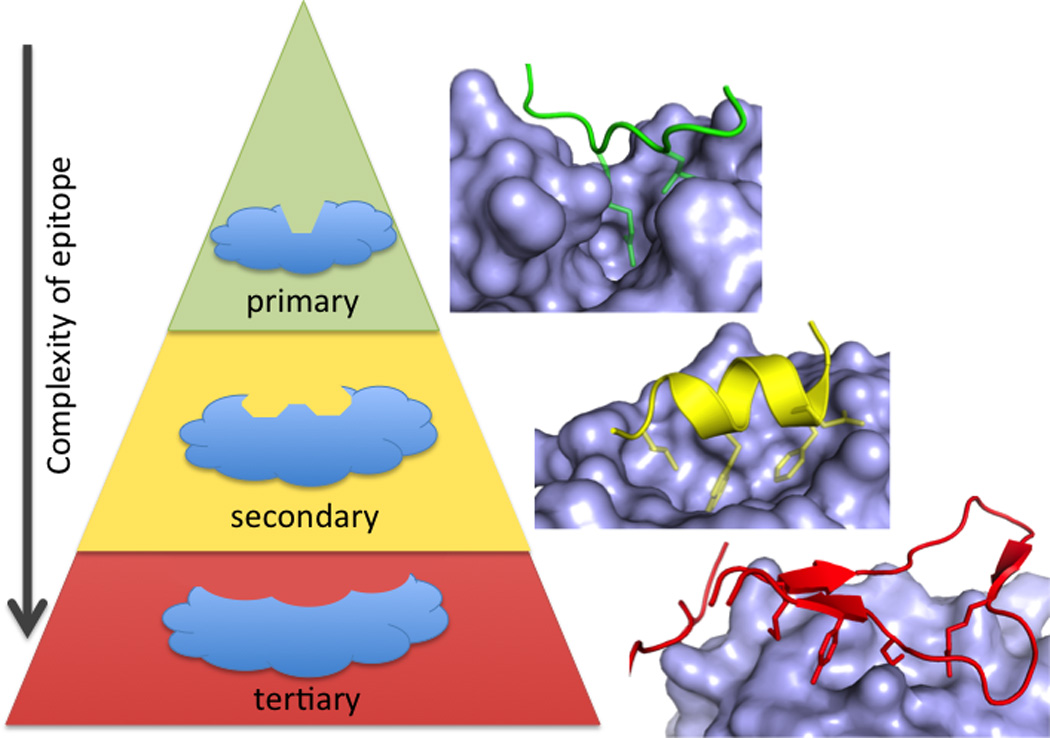
PPI can be classified by whether one side of the interface consists of a primary (linear) protein sequence (green), a single region of secondary structure (such as an alpha helix, yellow), or multiple sequences requiring tertiary structure (red). There are fewer examples of small-molecule inhibitors of PPI as the interface becomes more complex (from primary, to secondary, to tertiary epitopes).Structures shown are BRDt/histone (green, 2WP1), MDM2/p53 (yellow, 1YCR), and IL-2/IL-2Ra (red, 1Z92).
Primary peptide epitopes: Short, continuous, linear peptides
One of the first examples of a clinically successful PPI inhibitor is tirofiban, which binds to the integrin IIbIIIa. Tirofiban was designed to mimic the linear tripeptide Arg-Gly-Asp (RGD), the epitope of fibrinogen that binds to the “I - like” domain of IIbIIIa (Hartman et al., 1992). While this small, linear peptide epitope seems like a special case of PPI, this motif is observed in turns, at protein termini, and in unstructured regions (Perkins et al., 2010). Unsurprisingly, these types of interfaces have been particularly amenable to inhibition by small molecules, and four such targets have recently entered clinical trials, which we discuss below.
LFA1
Leukocyte function-associated antigen-1 (LFA1) has been the target of drug discovery efforts aimed at reducing inflammatory immune responses. LFA1 is a beta2 integrin involved in T-cell activation and adhesion (Ford and Larsen, 2009) through binding to its ligand ICAM1. LFA1 is a heterodimer consisting of an alpha chain (CD11a/aL) and a beta chain (CD18, β2). ICAM1 binds to an inserted (I-domain) located on CD11a. CD18 also has an I-like domain that is located near the I-domain. Three types of drugs have been in the clinic for LFA-1. The anti-LFA-1 antibody efalizumab was approved for psoriasis in 2003, but was withdrawn in 2009 due to rare but fatal viral infections associated with immunosuppression (Schwab et al., 2012). Small molecules that bound to an allosteric site on the I-domain were in clinical trials for psoriasis, but no development has been reported since 2010 (Guckian and Scott, 2013). Lifitigrast (SAR1118), however, recently completed its registrational phase III trial, and a new drug application will be filed in early 2015 (2014b).
Lifitegrast was discovered at Sunesis, building from a series originally published by Genentech (Gadek et al., 2002), and was developed clinically by SARcode/Shire (Zhong et al., 2012). There are no crystal structures of lifitegrast or its analogs bound to LFA1. The mechanism of action of the compound is still under debate; it either binds directly to the ICAM site on the I-domain (Keating et al., 2006) or to the related site on the I-like domain (Shimaoka et al., 2003) (Figure 2), analogous to the binding of tirofiban. SARcode developed lifitegrast for the inflammatory disorder Dry Eye Syndrome, the most common eye disease in humans (Schaumberg et al., 2003). SAR1118 has an optimal pharmacokinetic profile for ocular formulation, with low systemic exposure and high aqueous solubility. Since this indication does not require systemic exposure, lifitegrast could achieve the efficacy that eluded I-domain antagonists (Guckian and Scott, 2013) and avoid the toxicity that led to efalizumab’s withdrawal (Schwab et al., 2012).
Figure 2.
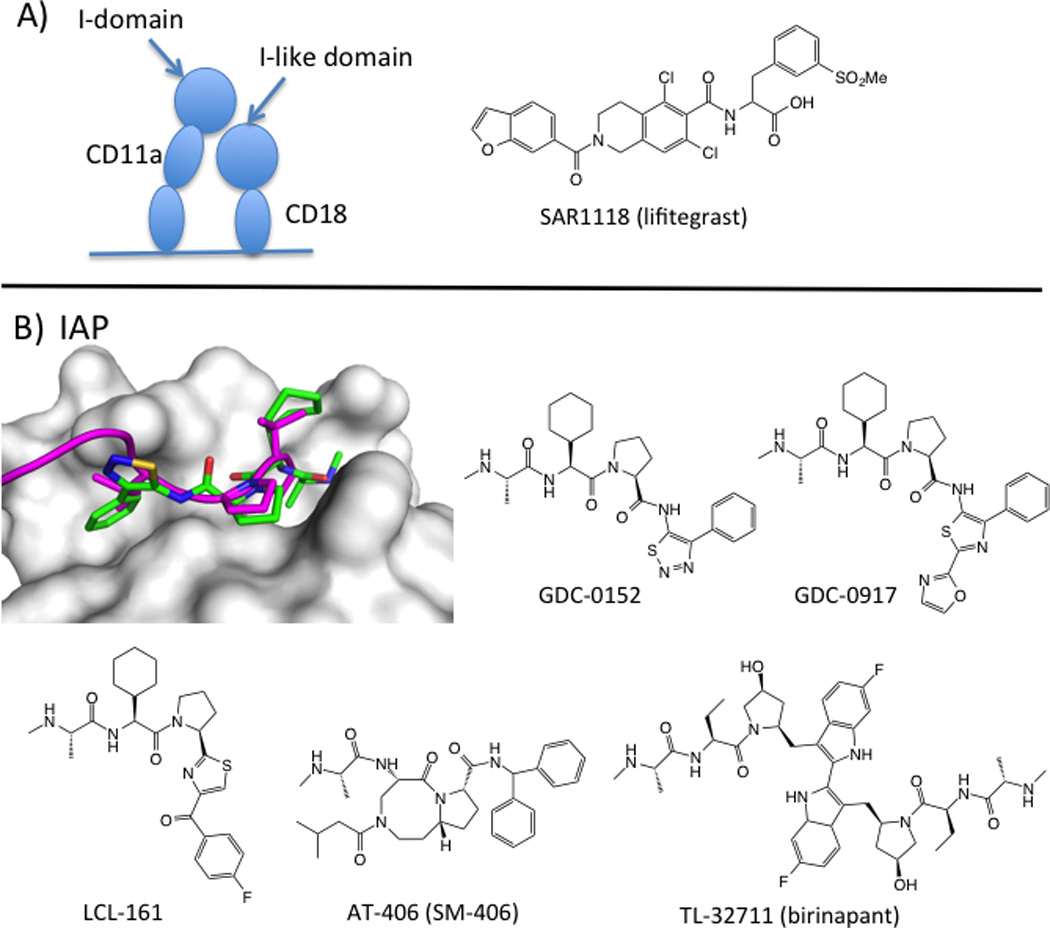
Peptide-like inhibitors of PPI with primary epitopes. A) SAR1 118 (lifitegrast) inhibits binding of LFA1 (CD11a/CD18) to its ligand ICAM. The mechanism either involves binding directly to the ICAM site in the I-domain of CD11a (Keating et al., 2006) or allosteric inhibition through binding to the I-like domain in CD18 (Shimaoka et al., 2003). B) X-ray structure of a cIAP1/XIAP chimera (white surface) bound to GDC-0152 (green sticks; PDB: 3UW4) and overlaid with SMAC peptide (magenta cartoon; PDB: 1G73). Chemical structures of SMAC mimetics in clinical trials.
IAPs
Inhibitor of apoptosis proteins (IAPs), including cIAP1, cIAP2, and XIAP, are important regulators of cell fate, including apoptotic cell death and immunity (Dubrez et al., 2013; Fulda and Vucic, 2012). The IAPs have PPI interaction domains called BIR domains and a RING domain E3 ubiquitin-ligase domain. The BIRs bind directly to an amino-terminal tetrapeptide sequence exposed during proteolytic activation of caspases-3, −7, and 9, thus inhibiting the activity of these pro-apoptotic proteases. IAPs ubiquitin-ligase activity also leads to the ubiquitylation and degradation of caspases (Dueber et al., 2011) and other proteins involved in apoptosis and NFkB signaling (reviewed in (Fulda and Vucic, 2012)). Current thinking holds that XIAP inhibition directly affects caspase activity, whereas cIAP inhibition leads to stronger ubiquitin-mediated effects. Developing homolog-selective inhibitors of the IAPs will allow evaluation of these pathways in cancer therapy and toxicity (Condon et al., 2014; Ndubaku et al., 2009; Sun et al., 2014).
An endogenous inhibitor of IAPs, called Smac (second mitochondrial activator of caspases), competes with caspase binding to BIR domains and thereby stimulates apoptosis (Liu et al., 2000). Small-molecule IAP inhibitors mimic the tetrapeptide-binding motif on Smac and are therefore called Smac mimetics (SMs). The Smac peptide motif, Ala-Val-Pro-Ile (AVPI), binds to two adjacent subpockets on BIR domains (Liu et al., 2000). Hot-spot residues on the XIAP BIR3 domain focus on the pocket that binds the N-terminal amine (E314) and alanine residue (L307, W310) and W323, which interacts with proline. Smac peptides are impressively tight binders; AVPI-amide binds cIAP1 with a Kd value of 2 nM (Condon et al., 2014). Since the first peptidomimetic inhibitor of the IAP/Smac (and IAP/caspase) interaction was disclosed (Oost et al., 2004), several groups have developed inhibitors with improved potency, PK, and in vivo activity. These compounds mimic the key interactions between AVPI and BIR domains (Figure 2). Seven SMs have reached clinical trials, and five compounds remain active in clinical development.
Clinical IAP inhibitors fall into two structural camps. The monovalent antagonists, LCL161 (Novartis) (Dubrez et al., 2013), GDC-0917/CUDC-427 (Genentech/Curis) (Flygare et al., 2012; Wong et al., 2013) and SM-406/AT-406 (Wang lab/Ascenta) (Cai et al., 2011) were designed from the AVPI peptide and have IC50 values in the 2–60 nM range and LE ~0.3 (LLEs are in the drug-like range of 6–7.5). The bivalent antagonists, birinapant (TetraLogic) (Benetatos et al., 2014; Condon et al., 2014) and SM-1387 (Bai et al., 2014) are dimerized SMs, designed based on the idea that the Smac homodimer simultaneously binds to XIAP’s BIR2 and BIR3 domains (Huang et al., 2003). These dimers demonstrate better cellular potencies than their monovalent analogs. Unlike the orally active, monovalent compounds, the bivalent drug candidates are dosed intravenously. SM-406 and birinapant bind ca. 50-fold more tightly to cIAP1 than XIAP, and may allow clinical evaluation of whether XIAP inhibition is required for efficacy.
Identifying patients who will respond to IAP inhibitors as single agents has been challenging. SMs induce apoptosis in ~15% of cancer cell lines (Oost et al., 2004), suggesting that single-agent activity of clinical compounds could be unpredictable. Transcription profiling and genetic data suggest that certain tumor types might be more sensitive, particularly MALT lymphomas with constitutively active IAP-protein fusions (Fulda and Vucic, 2012). Importantly, preclinical and clinical studies have identified serum cytokines and cIAP levels as potential biomarkers of response (Dubrez et al., 2013), which could be used to quickly assess whether a patient is resistant to SM treatment. Furthermore, preclinical data provides a strong rationale for several combinations with radiation and chemotherapeutics, including the apoptotic agonist TRAIL (Fulda and Vucic, 2012). The preclinical data also point to the potential for on-target toxicities (Erickson et al., 2013; Yang and Novack, 2013). To date, clinical trials are too early to assess anticancer activity, but the clinical compounds appear to behave well and are tolerated at doses that show strong pharmacodynamics effects (Jeffrey R. Infante, 2010; Tolcher et al., 2013).
Bromodomains
The cellular decision to transcribe a gene requires the complex integration of signal transduction, the action of DNA-binding proteins that repress or stimulate transcription, and epigenetic signals on the DNA and DNA-bound histones. Transcriptional complexes are highly dynamic and contain multiple enzymes, scaffolding proteins, and DNA-binding proteins (Lessard and Crabtree, 2010). These complexes represent multiple opportunities to activate or repress transcription using small molecule modulators of PPI.
Bromodomains are epigenetic ‘readers’ that recognize acetylated lysines (Kacs) on histone tails and direct transcription complexes to turn on genes (Filippakopoulos and Knapp, 2014). Bromodomains share a conserved hydrophobic pocket formed by a left-handed four-helical bundle and loop regions of various lengths and charges. An asparagine residue and five ordered water molecules recognize Kac-histone peptides with ~100 µM affinity (Vollmuth et al., 2009). This deep pocket has been exploited by synthetic Kac-mimetics that bind with much higher affinity than the native peptide. One of the first published examples, (+)−JQ1, stimulated strong interest in bromodomain-containing protein 4 (BRD4) as a cancer target and in the druggability of bromodomains as a class (Filippakopoulos et al., 2010; Vidler et al., 2012). (+)-JQ1 binds to BRD4 with an affinity from 49 nM. Crystal structures show that (+)-JQ1 completely occupies the acetyl-lysine binding groove, including hydrophobic interactions not seen in the histone H3 peptide/BRD4 complex (Figure 3). In the past five years, several potent inhibitors have been described and have been used to explore the biology of bromodomains in cancer and inflammation (reviewed in (Filippakopoulos and Knapp, 2014)).
Figure 3.

Small-molecule inhibitors of PPI with primary epitopes. A) Crystal structure of BRD4 bromodomain (white surface) bound to (+)-JQ1 (green sticks, PDB: 3MXF) overlaid with acetylated histone peptide (magenta cartoon, PDB: 2WP1). Clinical compounds or their closest published analogs are shown below. B) Crystal structure of the dimerization interface of HIV integrase (white and cyan surface) bound to compound 16 (green sticks, PDB: 4NYF) with overlaid epitope from LEDGF (magenta, PDB: 2B4J). Compound 16 is a precursor to the clinical compound BI 224436. C) Von-Hippel Lindau (VHL) protein (white surface) with bound inhibitor compound 51 (green sticks, PDB: 4B9K). The central hydroxyproline mimics the binding of a peptide derived from HIF1α (PDB: 4AJY, not shown).
Five compounds have already advanced to clinical trials. I-BET762 (Glaxo Smith-Klein) (Mirguet et al., 2013), CPI-0610 (Constellation) (Gehling et al., 2013), Ten-010 (Tensha)(Rohn, 2012), and OTX15 (OncEthix)(Herait et al., 2014) are based on the JQ1 scaffold (Figure 3) and are in clinical trials for cancer, including NUT midline carcinoma, a rare cancer caused by BRD4-NUT fusions (Filippakopoulos et al., 2010). RVX-208, is a quinazolinone that binds deeply into the Kac pocket, making a hydrogen bond to the Kac-recognizing asparagine residue; unlike the JQ1-like compounds, it binds preferentially to the second bromodomain of BRD3 (McLure et al., 2013). RVX-208 has been in phase II trials for atherosclerosis, with promising results. The clinical compounds were identified through a range of methods. For instance, I-BET762 and RVX-208 were found in cell-based HTS focused on ApoA1 upregulation (for atherosclerosis) and were later found to bind BET bromodomains (McLure et al., 2013; Mirguet et al., 2013); compound 3, a published analog of CPI-0610, was evolved from an isoxazole fragment merged with the core structure of JQ1 (Gehling et al., 2013). BET proteins may represent a best-case scenario for PPI inhibition, since the primary recognition element is a single amino acid. However, the ability to develop potent, ligand-efficient (average LE = 0.34, Table 1) inhibitors, even though histone peptides bind with low affinity, speaks to the potential to gain binding energy that is not used by the native PPI.
Table 1.
Properties of selected protein-protein interaction inhibitors
| PPI target/partnera | MW (Da) | IC50 (nM) | LEb | LLEc | oral bioavailability |
|---|---|---|---|---|---|
| PRIMARY EPITOPE | |||||
| LFA1/ICAM1 | |||||
| SAR1118 (lifitegrast) | 615 | 9 | 0.27 | 4.8 | − |
| cIAP/SMAC | |||||
| GDC-0152 | 499 | 14 | 0.31 | 6.3 | − |
| GDC-0917/CUDC-427 | 565 | 50d | 0.22 | 6.0 | + |
| AT-406/SM-406 | 562 | 2 | 0.30 | 7.5 | + |
| TL-32711 (birinapant) | 809 | 1 | 0.22 | 11.4 | − |
| bromodomain/histone | |||||
| I-BET762 (GSK 525762) | 424 | 630 | 0.29 | 2.9 | + |
| compound 3e | 400 | 26 | 0.39 | 3.5 | + |
| RVX-208 | 370 | 200 | 0.35 | 3.8 | nd |
| Integrase/LEDGF | |||||
| BI 224436 | 443 | 20 | 0.33 | 11.5 | + |
| VHL/HIF1alpha | |||||
| compound 51 | 450 | 900 | 0.26 | 4.4 | nd |
| SECONDARY EPITOPE | |||||
| BCL family/BH3 | |||||
| ABT263 (navitoclax) | 975 | 0.40 | 0.21 | −1.2 | + |
| ABT199 | 868 | 0.01 | 0.24 | 3.1 | + |
| compound 14 | 437 | 10 | 0.37 | 3.3 | + |
| MDM2/p53 | |||||
| RG7112 (Ro5045337) | 728 | 18 | 0.23 | 0.7 | + |
| RG7388 (Ro5503781) | 616 | 6 | 0.27 | 4.3 | + |
| MI-888f | 548 | 0.44 | 0.35 | 6.7 | + |
| PDK1/PIF-tide | |||||
| PS210 | 380 | 3000 | 0.32 | 4.5 | nd |
| Menin/MLL | |||||
| MIV-6R | 418 | 85 | 0.32 | 4.1 | nd |
| MIV-2-2 | 416 | 20 | 0.45 | 3.6 | nd |
| p300 CH1 domain/HIF1a | |||||
| OHM1 | 495 | 500 | 0.27 | 6.8 | nd |
| TERTIARY EPITOPE | |||||
| IL-2/IL-2R alpha | |||||
| SP4206 | 663 | 60 | 0.22 | 4.6 | nd |
| HPV11 E1/E2 | |||||
| BILH 434 | 608 | 40 | 0.25 | 2.3 | nd |
Compounds in green are in active clinical development. For PPI families (eg BCL family/BH3, bromodomains, IAPs), the highest affinity/lowest IC50 is reported. When available, Kd values are reported.
ligand efficiency, LE = ΔG/#heavy atoms;
lipophilic ligand efficiency, LLE = pIC50 - clogD.
Activity is “<60 nM” (Wong, et al, 2013).
compound 3 is an analog of the undisclosed clinical compound CPI-0610.
MI-888 is an analog of the undisclosed clinical compound MI-773.
HIV integrase (IN)
Viruses hijack host proteins to facilitate their replication and survival; these host/pathogen complexes represent important clinical targets. The homotetrameric protein HIV integrase (IN) integrates the viral genome into human DNA by catalyzing 3’-processing and strand transfer. The HIV drugs raltegravir and elvitegravir bind to the active site of IN and inhibit its enzymatic activity. Recently, a second class IN inhibitors targeting a host/pathogen PPI have reached the clinic (Engelman et al., 2013).
The human protein lens endothelial growth factor (LEDGF) acts as an IN cofactor, facilitating integration of the viral genome into the host chromosome, protecting IN from proteolytic degradation, and stimulating its catalytic activity (Engelman et al., 2013)]. The co-crystal structure of LEDGF binding domain with a dimer of IN catalytic domain shows a short turn of helix from LEDGF pointing into the IN dimer interface, with 400A2 buried surface area on IN (Cherepanov et al., 2005); 2P2I database (Basse et al., 2013). LEDGF-mimicking peptides provided proof-of-concept for inhibiting the LEDGF/IN PPI, and stimulated small-molecule discovery programs based on virtual screening, structure-aided design, and HTS (reviewed in (De Luca et al., 2011)). The most potent PPI inhibitors are tert-Butoxy-(4-phenyl-quinolin-3-yl)-acetic acid (tBPQA) derivatives (Figure 3), including the clinical compound BI-224436 (Boehringer-Ingelheim, Gilead), which has low nM antiviral potency, LE = 0.33, and LLE = 11.5 (Table 1) (Fader et al., 2014; Fenwick et al., 2014). tBPQAs were found in a HTS designed to identify inhibitors of IN-catalyzed 3’-processing of DNA; structural studies later revealed the compound bound to LEDGF binding pocket at the IN dimer interface. The quinolone core positions the two side-chain mimetics; as with BRD4 inhibitors, BI-224436 binds to pockets that the LEDGF turn does not (Figure 3).
tBPQAs are dual inhibitors. In addition to inhibiting the LEDGF/IN complex, they also allosterically inhibit IN catalytic activity by preventing the formation of functional IN tetramer. Recent studies suggest that the mechanism of these inhibitors is complex, and altering tetramerization may be more therapeutically important than inhibiting LEDGF per se (Balakrishnan et al., 2013; Sharma et al., 2014). BI-224436 could provide a new mechanism for HIV treatment that will likely have a different resistance profile than active site IN inhibitors. Furthermore, its story highlights the fact that enzyme/protein complexes have multiple effects on the enzyme; small molecule inhibitors might alter one or more of these functions.
Prospects for primary sequence epitopes
Inhibitors of PPI that consist of primary sequence epitopes have been discovered using multiple approaches. Fragment-based, biochemical, and cell-based screening have all yielded hits for this PPI class (Fader et al., 2014; Gehling et al., 2013; Mirguet et al., 2013). The success of screening for primary-epitope PPI is consistent with the theory of Hann, et al, which posits that less complex interfaces are more likely to yield to diversity-based screening (Hann et al., 2001). Design strategies based on the peptide epitope are also logical, since the native ligand is close to the size of a drug-sized inhibitor. Such epitope-mimetic design was recently demonstrated for the HIF1a/ the von Hippel-Lindau protein (VHL) interaction (Buckley et al., 2012a; Buckley et al., 2012b). Similar to BRD4, HIF1a recognition focuses on a single, modified amino acid. Here, Crews and coworkers virtually screened a library of compounds to mimic a tripeptide epitope on VHL that centers on hydroxyproline. They identified a oxazole-hydrozyproline scaffold, optimized the compound to arrive at a 1 µM inhibitor of the PPI, and showed by crystallography that these compounds bind at the hydroxyproline site. Finally, since inhibitors are either peptidic or compact, LE and LLE values tend to be high. Hence, both the variety of discovery strategies and the chemical properties of hits are favorable for PPI with linear epitopes.
Secondary structure epitopes: α-helix, β-sheet or extended peptides
It has been estimated that up to 40% of PPI consist of a single peptide from one protein that binds into a groove on the other (Petsalaki and Russell, 2008). 60% of PPI in the protein databank utilize an α-helix and 60% of these bind a single face of the helix (Raj et al., 2013). In general, hot spots on the target protein are centered on 2–3 sub-pockets that bind hydrophobic residues on the partner peptide. Such interfaces have been described as ‘hot sequences’ (London et al., 2010) or ‘secondary-structural epitopes,’ reflecting the fact that the key residues on the peptide are not next to each other in the primary sequence, but that an isolated peptide can often mimic the binding affinity and pose of the PPI.
BCL family
BCL-family proteins are central effectors of apoptosis. Pro-apoptotic family members, including BAD and BAK, are kept in an inhibited state through binding to the anti-apoptotic family members, including BCL2, BCL-xL, and MCL1. The anti-apoptotic members are helical proteins with an open groove that binds to a single helix (the BH3 domain) on the pro-apoptotic partner. Upregulation of the anti-apoptotic BCL family members is an important mechanism through which cancer cells avoid cell death in the face of pathway dysregulation, radiation, and chemotherapy. The structure of BCL-xL bound to BAK peptide (16-mer, Kd = 340 nM) suggested that this PPI might be particularly druggable (Sattler et al., 1997). The binding site on BCL-xL is comprised of two large (~200 A2) nearby pockets and three hydrophobic residues from the BH3 peptides define the PPI hot spot (Oltersdorf et al., 2005). Indeed, between 2000–2004, several laboratories published small-molecule inhibitors with potencies in the 0.1 – 10 µM range that bound in the BAK-binding site (reviewed in (Arkin and Wells, 2004)).
The major preclinical breakthrough was the 2005 disclosure of ABT-737, which was discovered through NMR-based fragment discovery, structure-based design, and medicinal chemistry (Oltersdorf et al., 2005). ABT-737 binds to BCL2, BCL-xL, and BCL-W with sub nanomolar affinity, but it is also a large compound (MW = 813 Da), with LE = 0.21. ABT-737 demonstrated impressive single-agent activity in cancer xenograft models, but was not orally bioavailable (Oltersdorf et al., 2005). In 2008, Abbott disclosed the orally available analog ABT-263 (Tse et al., 2008), which entered phase 1 and 2 clinical trials for solid tumors and chronic lymphocytic leukemia (CLL) (Roberts et al., 2012; Rudin et al., 2012). During these early clinical trials, thrombocytopenia (platelet loss) was an important dose-limiting toxicity linked to BCL-xL inhibition.
Current clinical BCL-family inhibitors are selective for BCL-2 or BCL-xL. ABT-199 was designed to be selective towards BCL2 to avoid thrombocytopenia (Souers et al., 2013). To engineer selectivity into ABT263, a stripped-down scaffold was co-crystalized with BCL2. A tryptophan residue from a neighboring BCL2 molecule sat very nearby to the inhibitor; linking an indole at that position led to ABT199, which binds BCL2 10-fold more tightly than ABT263, and shows 5000-fold selectivity for BCL2 compared to BCLxL. Like its precursors, ABT-199 accesses the BAK-binding pockets, but does not mimic the peptide backbone structure or side-chain orientation (Figure 4). ABT199 is currently in phase 1 trials for CLL, and preliminary results look very promising, with response rates of >80% (2014a). BCL-xL-selective inhibitors (e.g., compound 14, Figure 4) have also been developed, with the aim of treating solid tumors while avoiding the lymphocytic effects of BCL2 (Koehler et al., 2014). The L-shaped BCL-xL-selective series has a distinct binding mode from ABT-199 and appears to be buried more deeply in the peptide-binding groove. It is noteworthy that both ABT-199 and compound 14 are more ligand-efficient and have better LLE than ABT-737 and ABT-263 (Table 1). Interestingly, cell-based activity for BCL-family inhibitors is generally 100–1000-fold weaker than binding affinity. This may be due to permeability, protein binding, or the cellular context of BCL2/xL, and seems to hold for several members of this target class. Thus, very high-affinity compounds have been required (Souers et al., 2013). While it is still early days in the clinical life of BCL-family antagonists, the impressive story of their discovery highlights the realities of drug discovery for a novel and challenging target; often, several iterations of clinical candidates are needed to strike the ideal balance of efficacy, safety, and dosing regimen.
Figure 4.

BCL-2/BCL-xL and MDM2 interfaces contain secondary epitopes. A) Structure of BCL-2 (white surface) bound to ABT-199 (green sticks, PDB: 4MAN) with overlaid BAX BH3 peptide (magenta cartoon, PBD: 2XA0). Chemical structures of selected BCL-2 and BCL-xL inhibitors. B) Structure of MDM2 (white surface) bound to RG-7112 (green sticks, PDB: 4IPF) with overlaid p53 peptide (magenta cartoon, PDB: 1YCR). Chemical structures of compounds in clinical testing or their closest published analogs.
MDM2
The transcription factor p53 is a master tumor suppressor that regulates cell-fate decisions such as senescence, cell-cycle arrest, and apoptosis. P53 or its regulators are mutated or altered in most cancers, resulting in reduction or loss of p53 function. The ubiquitin E3 ligase responsible for p53 degradation is MDM2 (murine double minute 2) and the MDM2/MDMx heterodimer (Khoo et al., 2014). This system is overexpressed or amplified in several cancers with wild-type p53, and inhibiting p53 ubiquitylation by MDM2 should lead to increased p53 levels and activity. The MDM2-p53 complex can be minimized to an α-helix from the N-terminal transactivation domain of p53 that binds into a pocket on the surface of the N-terminal domain of MDM2 (Kussie et al., 1996). The MDM2/p53 interface is more compact than that found for BCL-xL/BH3 peptide: the helix is only two turns, and the three critical hotspot residues (Phe19, Trp23, Leu26) point towards a deep pocket in the center of the peptide-binding groove. The isolated peptide binds with ~ 500 nM affinity to the N-domain of MDM2.
The breakthrough for inhibiting p53/MDM2 came from the discovery of the Nutlins, identified in a large high-throughput screen (Vassilev et al., 2004). The optimized, ~100 nM inhibitor contains a central imidazole ring with four substitutions. Three hydrophobic substitutions mimic the binding of the three key residues from p53, and the fourth substituent binds on the protein surface. Soon after the discovery of the Nutlins, several labs described compounds with different cores but similar molecular-recognition features (Fry, 2012; Khoo et al., 2014). A general feature is a tripod shape that pushes at least two hydrophobic elements deep into the binding site; these are fairly rigid structures that use a central ring to enforce the side-chain geometries. Some compounds also bind further along the p53-binding groove. Compounds also differ in their ability to bind MDMX, which might be important for efficacy and overcoming resistance in some contexts (Khoo et al., 2014). The key observation from these diverse MDM2 ligands is that many different scaffolds (including peptides) can direct the hydrophobic groups into the p53-binding pocket. (Fry, 2012). Thus, it is not critical to mimic precisely the peptide backbone or the peptide’s side-chain orientation.
One of the puzzling aspects of MDM2 inhibitor design has been the role of the fourth substituent. Modifications to this group alter the compounds’ physicochemical characteristics, and have a modest effect on binding to the N-terminal domain of MDM2. However, changes often have a disproportionate effect on cell-based potency (Ding et al., 2013; Zhao et al., 2013). Recent data suggest that parts of MDM2 outside the p53-binding domain may play a role in the binding interaction. For instance, MDM2 mutations that render cells resistant to Nutlins often occur outside the p53-binding domain (Wei et al., 2013). The direct demonstration of a structural interaction has yet to be made, but the general point rings true: in minimizing the PPI for biochemical and structural studies, we are likely to miss long-range interactions that the compounds will face when they enter the cell.
Three scaffolds have been developed into experimental therapeutics. Roche initially took RG7112 (Figure 4) into phase 1 trials (Tovar et al., 2013), and is currently testing the more potent and ligand-efficient analog RG7388 (Ding et al., 2013). The spirooxindole class of compounds, exemplified by MI-888, was discovered by the Wang lab at University of Michigan and is being developed by Sanofi (MI-773, structure not disclosed) (Zhao et al., 2013). Roche recently disclosed Ro8994, a 5 nM (95% bioavailable) analog based on a spiroindolinone scaffold that combines features of RG7388 and MI 888 (Zhang et al., 2014). Finally, Daiichi Sankyo is developing an undisclosed MDM2 inhibitor based on a dihydroimidazothiazole scaffold (Miyazaki et al., 2013). These clinical-stage inhibitors show impressive tumor regressions in xenograft models and selective killing of cells containing wild-type p53 vs those with mutant p53 (Tovar et al., 2013). Their drug-like properties are also promising, with LE = 0.27 – 0.35, LLE in the range of 4.3 – 6.7, and very high oral bioavailabilities despite molecular weights in the range of 550–730.
MDM2 inhibitors are being developed for single-agent use and in combination with existing drugs. Early trials for RG7112 have shown pharmacodynamic activity, including 3–5-fold increases in levels of p53 and p53 target genes (Ray-Coquard et al., 2012), and potential therapeutic activity in acute leukemia (Michael Andreeff, 2012). The biological challenges facing clinical positioning of these compounds has been recently discussed (Khoo et al., 2014). Despite decades of work on p53, its precise roles in survival and death pathways remain to be elucidated; these clinical MDM2 antagonists are poised to address some of these critical issues.
Selected emerging examples of secondary structure epitope inhibitors
Diverse methods have been used to develop secondary structural mimetics and prospects for developing inhibitors of this class of PPI are good. One instructive example is the N-lobe of the kinase PDK1. This site binds to a 10 residue peptide called a PDK1-interacting fragment (PIF-tide) from substrate proteins. Binding serves to colocalize the proteins and to allosterically activate PDK1 kinase activity (Gold et al., 2006). Fragments that mimic the PIF-tide have been found through virtual screening (Engel et al., 2006), NMR (Stockman et al., 2009), and Tethering (Sadowsky et al., 2011).
For Tethering, six cysteine residues were introduced around the PIF-tide binding site and were screened for binding to a library of disulfide-containing fragments. Remarkably, fragments tethered at the same cysteine residues could be either activators or inhibitors of PDK-1 kinase activity. Co-crystal structures showed that activators pushed the C-helix toward the active site and better organized the catalytic apparatus, whereas inhibitors shifted the C-helix outward into a less competent state (Figure 5). Thus, small changes in the small-molecule ligand can have positive or negative effects on enzyme allostery. Virtual screening, followed by chemical optimization, provided cell active inhibitors of the PDK1/PIF-tide interaction. PS210 (LE= 0.35) mimics two hot-spot phenylalanine residues on the PIF-tide (Busschots et al., 2012). In biochemical assays, this compound activates the kinase towards peptide substrates, as does the PIF-tide. Paradoxically, in cells, a cell-permeable version of PS210 inhibits phosphorylation of the subset of PDK1 substrates that require binding at the PIF-tide site. We recently identified cell active compounds from an HTS that monitored displacement of an optimized PIF-tide (Rettenmaier, T.J. et al, unpublished results). These compounds serve as additional examples of peptide mimicry by small molecules and provide ligand efficient scaffolds that may be further optimized.
Figure 5.
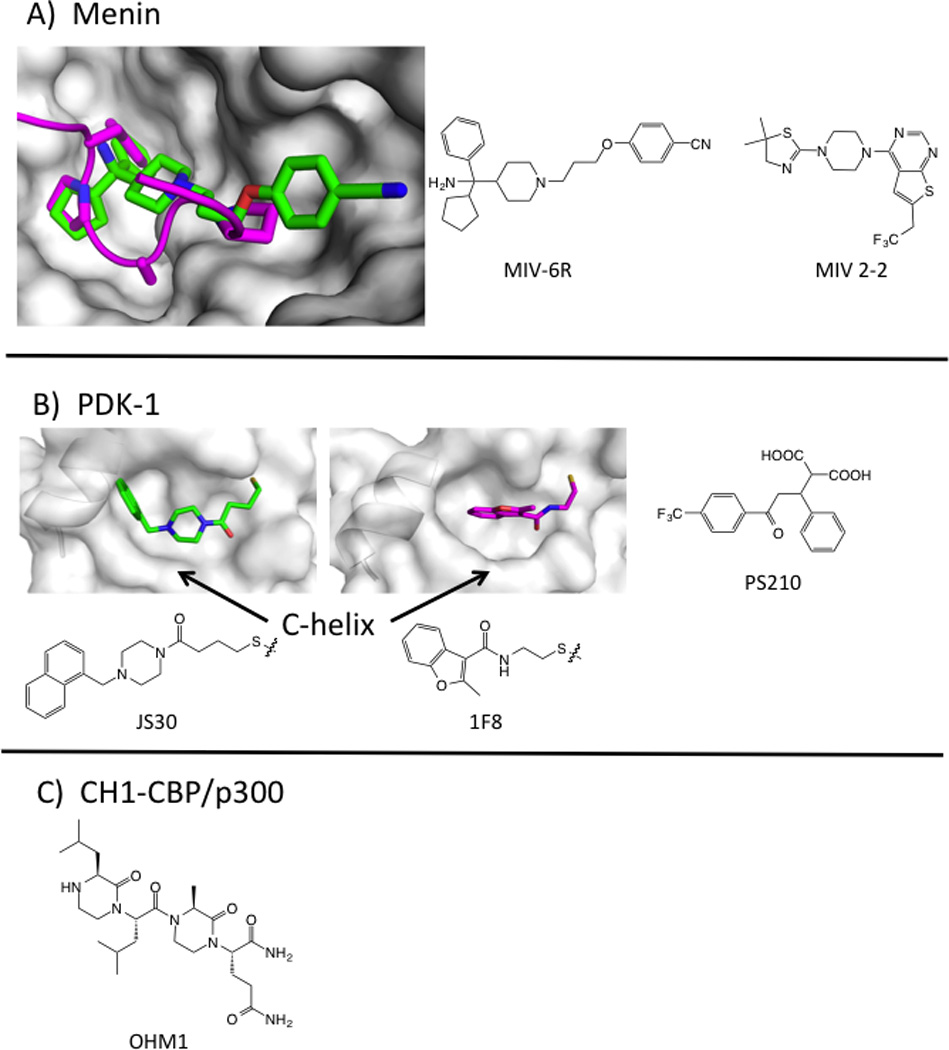
Preclinical examples of inhibitors of PPI with secondary epitopes. A) Crystal structure of menin (white surface) bound to MIV-6R (green sticks, PDB: 4OG8) with MLL peptide overlaid (magenta cartoon, PDB: 3U85). Structures of MLL inhibitors MIV-6R and MIV 2-2. B) Crystal structures of PDK-1, showing the PIF-tide binding site bound with Tethered activator (JS30, green sticks, PDB: 3OTU) or inhibitor (1F8, magenta sticks, 3ORX). JS30 pushes the C-helix into a catalytically competent conformation. Chemical structure of noncovalent PIF-tide inhibitor PS210. C) Structure of OHM1, which binds to CBP at the HIF1α peptide-binding site.
Multiple approaches have also led to inhibitors of the transcription factor mixed-lineage leukemia protein (MLL) at two different domains. MLL binds to the scaffolding protein menin, and this interface can be minimized to an 11-mer peptide from the N-terminal domain of MLL. HTS followed by structural biology and medicinal chemistry led to MIV2-2 and MIV-6, potent, ligand-efficient, and cell-active inhibitors that closely mimic the MLL peptide hotspots (Figure 5, Table 1) (He et al., 2014; Shi et al., 2012). MLL also binds to the KIX domain of the acetyltransferase CBP using an alpha helix in the C-terminal domain of MLL. Mapp and colleagues have designed synthetic molecules that bind to the KIX domain at the MLL site (Buhrlage et al., 2009). They have also used Tethering to identify disulfide-bound compounds that bind in the MLL site and allowed the first x-ray structure of the KIX domain (Wang et al., 2013b). Their ultimate goal is to develop bifunctional molecules that activate transcription by bringing together DNA-binding proteins and transcriptional coactivators.
Prospects for secondary structure epitope design
Design approaches have focused on both the binding pocket and the partner peptide. When focusing on the binding pocket, virtual methods should allow for side-chain flexibility, since hot spots at these interfaces often show small motions that create deeper binding pockets (Johnson and Karanicolas, 2013; Wells and McClendon, 2007). Pocket-based design is justified by the observation that inhibitors such as ABT199 and RG7112 mimic the function of hot-spot residues, but rarely mimic trajectories seen in the peptide structures. Others have focused on strategies to mimic the peptide partner directly. For example, Lao et al designed an α-helical mimetic of the HIF1a transactivation domain that binds to the cysteine-histidine rich 1 (CH1) domain on CBP/p300. The optimized compound OHM1 is based on a bis-oxopiperazine scaffold; OHM1 binds CH1 with a Kd = 500 nM (Table 1), down-regulates hypoxia inducible genes, and inhibits tumor growth in a xenograft model (Lao et al., 2014). Computational approaches to systematically replace peptide residues have also seen some promising success (Guo et al., 2014). This type of epitope is also highly amenable to stabilized peptide approaches (Azzarito et al., 2013; Bernal et al., 2010; Boersma et al., 2012; Chang et al., 2013). Historically, inhibitors of PPI with secondary-structural epitopes were large and had low LE; however, the second-generation clinical compounds, and the PDK1, menin, and HIF1α lead compounds all fall into more favorable ranges for LE and LLE.
Tertiary structural epitopes: discontinuous binding sites
IL-17
The larger, shallower interfaces that originally discouraged drug discovery remain challenging. Recently, Ensemble Therapeutics disclosed the development of a macrocyclic compound that inhibits IL-17’s binding to its receptor (Livingston et al., 2012), which represents a breakthrough in targeting hormone/receptor interfaces with synthetic macrocyles. The undisclosed structure has 3 nM affinity, MW~750Da and LE ~ 0.24. We eagerly await publication of the structural and preclinical data surrounding this program.
IL-2 and HPV-11
Two examples of PPI inhibitors highlight the dynamic nature of globular PPI. A series of compounds, exemplified by SP4206, bind to interleukin-2 (IL-2) at the receptor-alpha interface (reviewed in (Wilson and Arkin, 2011)). These compounds were identified serendipitously and optimized through fragment-based methods, including Tethering. IL-2 presents a relatively flat surface in the apo- and IL-2Ra bound states, but shows pockets upon binding SP4206. SP4206 binds at the receptor-binding hotspot and mimics the receptor’s hot-spot residues and charge distribution, even though the residues on are separated over multiple regions of the receptor’s structure. The conformational changes seen across the SP4206-binding surface include side-chain rotations and loop rearrangements, larger changes than those observed for the peptide epitopes described above. Molecular dynamics simulations suggest that these structural adaptations are better thought of as part of the protein conformational ensemble rather than induced fits.
BILH 434 (Figure 5) is another example of an inhibitor of a tertiary structural epitope (Wang et al., 2004). Human papilloma virus-11 (HPV11) requires the replication initiation factor E1 helicase to bind to the E2 transcription factor at specific DNA sites. BILH 434 was identified in a screen for inhibitors of the E1/E2/DNA complex, and binds to the transactivation domain (TAD) of E2 protein with a Kd of 40 nM (LE = 0.25). The x-ray structure of the compound/E2 complex shows the compound nestled into the elbow of the E2 TAD. Several side chains in this protein interface change conformations to create a deep pocket for BILH 434 to bind. Again, changes to the structure were unanticipated and were crucial for creating a binding site at an otherwise flat surface. Importantly, both the IL-2 and E2 TAD discoveries relied heavily on biophysical validation of the binding mode that was confirmed by x-ray crystallography.
Prospects for tertiary epitope binders
This class is probably the most challenging and there are only a handful of successful programs that have broken into the nM level of potency with modest LEs (typically 0.24 kcal/HA). These interfaces tend to be much more dynamic than the primary and secondary class epitopes, and thus less amenable to virtual screening. Small molecules can however find deeper pockets in these interfaces and thus bind with higher LE than their protein partner counterpart. The small molecules also bind hot spots predicted from alanine scanning. This class is probably the most prevalent class for extracellular PPI. Strong validation of the biology will certain make this class tempting – if high-hanging – fruit. For example, four groups have recently reported allosteric and orthosteric PPI inhibitors of Ras, a very challenging cancer target with no apparent deep pockets in the interface. Interestingly, three groups took fragment-based approaches, using NMR (Maurer et al., 2012; Sun et al., 2012) and Tethering (Ostrem et al., 2013), and one group identified inhibitors through virtual screening (Shima et al., 2013). Finally, PPI-binding peptides discovered from phage display could provide another avenue towards peptide-based therapies (DeLano et al., 2000).
Allosteric mechanisms of inhibition
Functional screening for PPI inhibitors has also serendipitously led to allosteric mechanisms of inhibition. For instance, BIO8898, an inhibitor of the trimeric ligand CD40L, alters the conformation of the trimer, thus inhibiting its binding to CD40 (Silvian et al., 2011). Similarly, compounds have been found that bind and disrupt trimer formation for TNF (He et al., 2005). For multicomponent complexes, one way to harness this serendipity is “grey box” screening, where the protein network is assembled in vitro. This approach has yielded interesting modulators of the HSP70 chaperone network. Depending on the biological context and how these HSP70 modulators bind, they have opposite effects on folding and thus utility in different disease states (Li et al., 2013; Wang et al., 2013a). Directed (as opposed to serendipitous) allosteric modulation of PPI provides potential way forward for the most difficult targets (Nussinov and Tsai, 2014).
How has our understanding of PPI evolved in the past ten years?
The discovery of small, drug-like molecules that inhibit PPI continues to be challenging, but the diverse success stories demonstrate that it is possible for some systems. General strategies are being sought, but it is likely that PPI is not properly thought of as a target class analogous to class I GPCRs or kinases. PPI are extremely diverse in the size and shapes of the interfaces and the binding affinities and dynamics of assembly. Given that there are (conservatively) 100,000 PPI (Venkatesan et al., 2009), our current database of PPI inhibitors is small and biased. Nevertheless, here is what we know so far.
Analyses of PPI where crystal structures are available for both the protein-protein and protein-small-molecule reveal some provocative themes (Johnson and Karanicolas, 2013; Smith and Gestwicki, 2012; Wells and McClendon, 2007). First, PPI target proteins tend to have extended binding grooves which may be divided into three or more (visible) subpockets (Fuller et al., 2009). Hot-spot residues are centered in or around these pockets, and are complementary on both sides of the interface. Second, small conformational changes in the binding site present a deeper pocket to small molecules than to the partner protein. These pockets are also more readily formed at the PPI interfaces than elsewhere on the protein surfaces (Johnson and Karanicolas, 2013). The consensus is that ligand-binding surfaces (including PPI) have a higher propensity for binding (to cognate ligands, solvent molecules, inhibitors) than the rest of the protein surface. Third, PPI with small-molecule inhibitors tend to have small, high-affinity interfaces, and include a hot segment that can recapitulate the binding of the partner protein. The observation that high affinity complexes have more small-molecule ligands (Smith and Gestwicki, 2012) could imply that they are inherently ‘sticky.’ Fortunately, however, there are many examples where peptides, mutated proteins, and small molecules – such as bromodomain inhibitors – bind to hot-spot sites with higher affinities than the native PPI. The preponderance of inhibitors for primary- and secondary-structural epitopes is probably due to their inherent druggability, the fact that there are many such interfaces (Petsalaki and Russell, 2008) and the availability of design approaches. In general, success seems to follow the simplicity of the epitopes to mimic: primary is easier than secondary is easier than tertiary.
PPI inhibitors bind at the hot spot and tend to mimic the types of binding interactions (e.g. hydrophobic) made by the partner. However, they generally use different functional groups and approach the binding site from different orientations than in the PPI. To reach each subpocket in an extended interface, molecules often have 3 distinct pharmacophores arrayed on a scaffold that serves to orient them. Hence, orthosteric inhibitors have tended to be larger than 500 Da and to be more three-dimensional than the average compound in an HTS library (Basse et al., 2013; Labbe et al., 2013). To address the potential lack of PPI-inhibitor-like compounds in HTS libraries, investigators have proposed design of libraries that mimic the shapes of current PPI inhibitors or protein secondary structures. To date, more traction has been gained through virtual screening and the use of non-traditional libraries, including larger, more natural-product like macrocycles on the one hand and fragment-based lead discovery on the other. Fragment screening has been a particularly successful strategy, perhaps because the pocket sizes at PPI are close to fragment size and the bias in chemical composition is lower. Experimental and virtual fragment screens have even be used to define the druggability of a PPI (Hajduk and Greer, 2007; Kozakov et al., 2011).
On the other hand, second-generation clinical candidates are generally smaller and more efficient than their precursors, and so perhaps the inherency of undrug-likeness of PPI inhibitors has been overstated (Table 1), particularly for small interfaces. Concerns for LE and LLE originate from the observation that larger molecules are more difficult to tune for in vivo properties. More important than molecular weight per se is finding the balance of hydrophobicity and electrostatics that provides tight, specific binding to the protein target and acceptable pharmacokinetics and pharmacodynamics. These issues are by no means specific to PPI drug discovery.
Screening methodologies have also become more sophisticated in the past ten years. Foremost, there is an expanded use of multiple drug discovery technologies, including an integrated use of biochemistry, biophysics, structural biology, and computational studies. Initial PPI inhibitors have been discovered using a wide variety of methods; we conclude that the screening format itself is less crucial than the compounds going into the screen and the post-screening evaluation of hits. True positives should be validated by demonstrating that the inhibitor binds to a single site on the PPI target using biophysical and structural methods. The importance of mechanism of action over potency at the screening stage cannot be overstated.
What we expect to see more of in the near future
Twenty years ago, PPIs were considered impossible fruit to reach. Today, there are dozens of targets for which early stage compounds have been discovered, and nearly a dozen compounds have been in the clinic. Clearly much progress has been made, and generated the courage to move forward, even with enthusiasm.
Ten years from now, how far will we have come? In terms of methodology, new computational methods will allow better understanding of allostery and conformational ensembles that are grist for improved virtual screening algorithms. There will be a continued emphasis on fragment-based design, and also a resurgence in natural products and natural product-like molecules, since biology has evolved these molecules for binding at protein interfaces. Popular target classes will include epigenetic and transcription-factor complexes, signal transduction networks, and protein quality control systems, reflecting the need for chemical tools to dissect these complex protein networks. Building on the function of natural products, there will also be greater emphasis on synthetic stabilizers of PPI (Milroy et al., 2014). Advances in cell biology will allow faster discovery of compounds’ mechanism of action and more translatable cell-based assays. Importantly, the PPI inhibitors in clinical trials today will be drugs benefitting patients. To us the writing is on the wall: PPIs are a challenging but tractable class for small molecule discovery if the biology is compelling and the will is strong.
Figure 6.
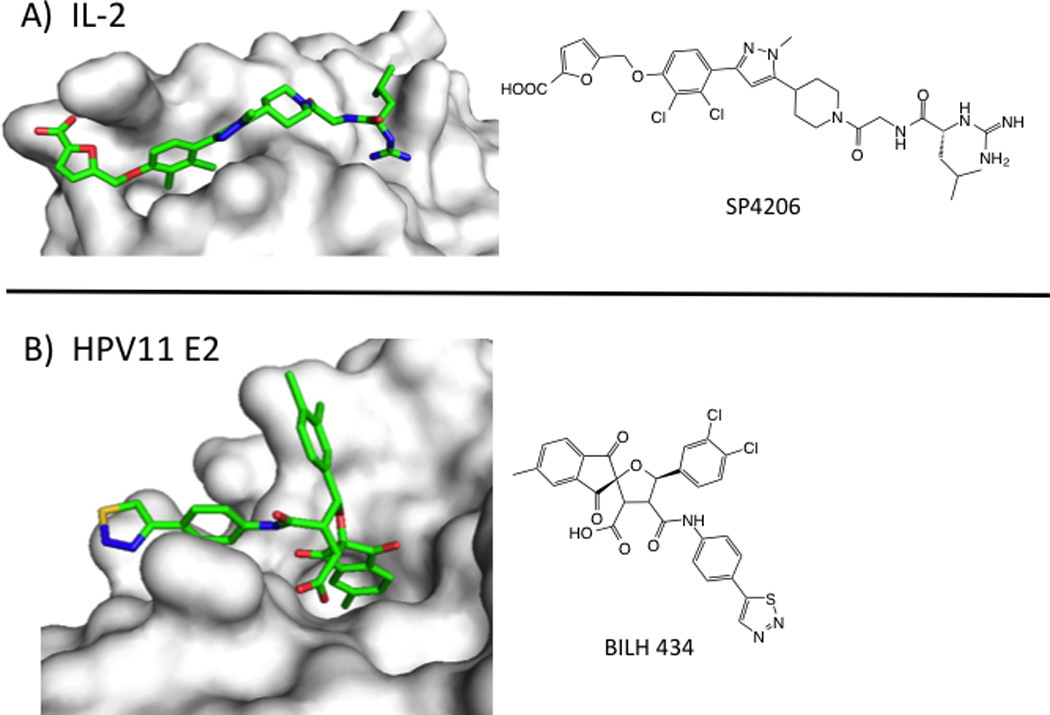
Small-molecule inhibitors of PPI with tertiary epitopes. A) Structure of IL-2 (white surface) bound to SP4206 (green sticks, PDB: 1PY2). B) Structure of transactivation domain of E2 protein from HPV11 (white surface) bound to BILH 434 (green sticks, PDB: 1R6N).
Acknowledgments
We thank our colleagues in the PPI-inhibitor field for their beautiful work and apologize to those whose work we could not describe due to space considerations. We also thank Jason Gestwicki for his thoughtful edits.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- BCL-2 inhibitor yields high response in CLL and SLL. Cancer discovery. 2014a;4:OF5. doi: 10.1158/2159-8290.CD-NB2013-178. [DOI] [PubMed] [Google Scholar]
- Shire Plans to Submit a New Drug Application to FDA for Lifitegrast for Dry Eye Disease in Adults. Lexington, Mass: 2014b. [Google Scholar]
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nature chemistry. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Bai L, Smith DC, Wang S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacology & therapeutics. 2014 doi: 10.1016/j.pharmthera.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan M, Yant SR, Tsai L, O’Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PloS one. 2013;8:e74163. doi: 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse MJ, Betzi S, Bourgeas R, Bouzidi S, Chetrit B, Hamon V, Morelli X, Roche P. 2P2Idb: a structural database dedicated to orthosteric modulation of protein-protein interactions. Nucleic acids research. 2013;41:D824–D827. doi: 10.1093/nar/gks1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G, Seipel ME, Kapoor GS, Laporte MG, Rippin SR, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kappaB activation, and is active in patient-derived xenograft models. Molecular cancer therapeutics. 2014;13:867–879. doi: 10.1158/1535-7163.MCT-13-0798. [DOI] [PubMed] [Google Scholar]
- Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer cell. 2010;18:411–422. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, Gellman SH. Evaluation of diverse alpha/beta-backbone patterns for functional alpha-helix mimicry: analogues of the Bim BH3 domain. J Am Chem Soc. 2012;134:315–323. doi: 10.1021/ja207148m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angewandte Chemie. 2012a;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc. 2012b;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. Amphipathic small molecules mimic the binding mode and function of endogenous transcription factors. ACS chemical biology. 2009;4:335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busschots K, Lopez-Garcia LA, Lammi C, Stroba A, Zeuzem S, Piiper A, Alzari PM, Neimanis S, Arencibia JM, Engel M, et al. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chemistry & biology. 2012;19:1152–1163. doi: 10.1016/j.chembiol.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R, et al. A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem. 2011;54:2714–2726. doi: 10.1021/jm101505d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110:E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A. 2005;102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, Alexander MD, Kumar PT, Hendi MS, Lee YH, et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem. 2014;57:3666–3677. doi: 10.1021/jm500176w. [DOI] [PubMed] [Google Scholar]
- De Luca L, Ferro S, Morreale F, De Grazia S, Chimirri A. Inhibitors of the interactions between HIV-1 IN and the cofactor LEDGF/p75. ChemMedChem. 2011;6:1184–1191. doi: 10.1002/cmdc.201100071. [DOI] [PubMed] [Google Scholar]
- DeLano WL, Ultsch MH, de Vos AM, Wells JA. Convergent solutions to binding at a protein-protein interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- Dubrez L, Berthelet J, Glorian V. IAP proteins as targets for drug development in oncology. Onco Targets and therapy. 2013;9:1285–1304. doi: 10.2147/OTT.S33375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, Zobel K, Maurer B, Varfolomeev E, Wu P, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334:376–380. doi: 10.1126/science.1207862. [DOI] [PubMed] [Google Scholar]
- Engel M, Hindie V, Lopez-Garcia LA, Stroba A, Schaeffer F, Adrian I, Imig J, Idrissova L, Nastainczyk W, Zeuzem S, et al. Allosteric activation of the protein kinase PDK1 with low molecular weight compounds. EMBO J. 2006;25:5469–5480. doi: 10.1038/sj.emboj.7601416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Kessl JJ, Kvaratskhelia M. Allosteric inhibition of HIV-1 integrase activity. Curr Opin Chem Biol. 2013;17:339–345. doi: 10.1016/j.cbpa.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RI, Tarrant J, Cain G, Lewin-Koh SC, Dybdal N, Wong H, Blackwood E, West K, Steigerwalt R, Mamounas M, et al. Toxicity profile of small-molecule IAP antagonist GDC-0152 is linked to TNF-alpha pharmacology. Toxicological sciences : an official journal of the Society of Toxicology. 2013;131:247–258. doi: 10.1093/toxsci/kfs265. [DOI] [PubMed] [Google Scholar]
- Erlanson DA, Wells JA, Braisted AC. Tethering: fragment-based drug discovery. Annual review of biophysics and biomolecular structure. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med Chem Lett. 2014;5:422–427. doi: 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick C, Amad M, Bailey MD, Bethell R, Bos M, Bonneau P, Cordingley M, Coulombe R, Duan J, Edwards P, et al. Preclinical Profile of BI 224436, a Novel HIV-1 Non-Catalytic-Site Integrase Inhibitor. Antimicrobial agents and chemotherapy. 2014;58:3233–3244. doi: 10.1128/AAC.02719-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S, Cohen F, Deshayes K, Doerner K, Eckhardt SG, et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152) J Med Chem. 2012;55:4101–4113. doi: 10.1021/jm300060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ML, Larsen CP. Translating costimulation blockade to the clinic: lessons learned from three pathways. Immunological reviews. 2009;229:294–306. doi: 10.1111/j.1600-065X.2009.00776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DC. Small-molecule inhibitors of protein-protein interactions: how to mimic a protein partner. Current pharmaceutical design. 2012;18:4679–4684. doi: 10.2174/138161212802651634. [DOI] [PubMed] [Google Scholar]
- Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- Fuller JC, Burgoyne NJ, Jackson RM. Predicting druggable binding sites at the protein-protein interface. Drug Discov Today. 2009;14:155–161. doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Gadek TR, Burdick DJ, McDowell RS, Stanley MS, Marsters JC, Jr., Paris KJ, Oare DA, Reynolds ME, Ladner C, Zioncheck KA, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295:1086–1089. doi: 10.1126/science.295.5557.1086. [DOI] [PubMed] [Google Scholar]
- Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, Kritzer JA. Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nature chemical biology. 2014;10:716–722. doi: 10.1038/nchembio.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehling VS, Hewitt MC, Vaswani RG, Leblanc Y, Cote A, Nasveschuk CG, Taylor AM, Harmange JC, Audia JE, Pardo E, et al. Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors. ACS Med Chem Lett. 2013;4:835–840. doi: 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Barford D, Komander D. Lining the pockets of kinases and phosphatases. Curr Opin Struct Biol. 2006;16:693–701. doi: 10.1016/j.sbi.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Guckian KM, Scott DM. 8. Inhibition of LFA-1/ICAM Interaction for the Treatment of Autoimmune Diseases. In: Domling A, editor. In Protein-Protein Interactions in Drug Discovery. Weinheim, Germany: Wiley-VCH Verlag GbmH and Co KGaA; 2013. [Google Scholar]
- Guo W, Wisniewski JA, Ji H. Hot spot-based design of small-molecule inhibitors for protein-protein interactions. Bioorganic & medicinal chemistry letters. 2014;24:2546–2554. doi: 10.1016/j.bmcl.2014.03.095. [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- Hann MM, Leach AR, Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. Journal of chemical information and computer sciences. 2001;41:856–864. doi: 10.1021/ci000403i. [DOI] [PubMed] [Google Scholar]
- Hartman GD, Egbertson MS, Halczenko W, Laswell WL, Duggan ME, Smith RL, Naylor AM, Manno PD, Lynch RJ, Zhang G, et al. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J Med Chem. 1992;35:4640–4642. doi: 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
- He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, Cancilla MT, Wang J, Lugovskoy AA, Yoburn JC, et al. Small-molecule inhibition of TNF-alpha. Science. 2005;310:1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- He S, Senter TJ, Pollock J, Han C, Upadhyay SK, Purohit T, Gogliotti RD, Lindsley CW, Cierpicki T, Stauffer SR, et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem. 2014;57:1543–1556. doi: 10.1021/jm401868d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herait P, Berthon C, Theiblemont C, Raffoux E, Magarotto V, Stathis A, Thomas X, Leleu X, CGomez-Roca C, Odore E, et al. BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at non-toxic doses: Interim results of an ongoing phase I trial in hematologic malignancies. AACR Annual Meeting. 2014 Apr;:5–9. 2014 http://www.oncoethix.com/news.
- Higueruelo AP, Schreyer A, Bickerton GR, Pitt WR, Groom CR, Blundell TL. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: the TIMBAL database. Chemical biology & drug design. 2009;74:457–467. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Keseru GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13:105–121. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Huang Y, Rich RL, Myszka DG, Wu H. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. The Journal of biological chemistry. 2003;278:49517–49522. doi: 10.1074/jbc.M310061200. [DOI] [PubMed] [Google Scholar]
- Hwang H, Vreven T, Janin J, Weng Z. Protein-protein docking benchmark version 4.0. Proteins. 2010;78:3111–3114. doi: 10.1002/prot.22830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey R, Infante ECD, Burris Howard A, Leigh Zawel, Sager Jason A, Claudina Stevenson, Kathryn Clarke, Shyeilla Dhuria, Dale Porter, Sen Suman K, Erika Zannou, Sushil Sharma, Cohen Roger B. In AACR 101st Annual Meeting. Washington, DC: Cancer Research; 2010. A phase I study of LCL161, an oral IAP inhibitor, in patients with advanced cancer. Abstract 2775. [Google Scholar]
- Johnson DK, Karanicolas J. Druggable protein interaction sites are more predisposed to surface pocket formation than the rest of the protein surface. PLoS computational biology. 2013;9:e1002951. doi: 10.1371/journal.pcbi.1002951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating SM, Clark KR, Stefanich LD, Arellano F, Edwards CP, Bodary SC, Spencer SA, Gadek TR, Marsters JC, Jr., Beresini MH. Competition between intercellular adhesion molecule-1 and a small-molecule antagonist for a common binding site on the alphal subunit of lymphocyte function-associated antigen-1. Protein science : a publication of the Protein Society. 2006;15:290–303. doi: 10.1110/ps.051583406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–236. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- Koehler MF, Bergeron P, Choo EF, Lau K, Ndubaku C, Dudley D, Gibbons P, Sleebs BE, Rye CS, Nikolakopoulos G, et al. Structure-Guided Rescaffolding of Selective Antagonists of BCL-XL. ACS Med Chem Lett. 2014;5:662–667. doi: 10.1021/ml500030p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, Beglov D, Pelletier J, Whitty A, Vajda S. Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci U S A. 2011;108:13528–13533. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Labbe CM, Laconde G, Kuenemann MA, Villoutreix BO, Sperandio O. iPPI-DB: a manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov Today. 2013;18:958–968. doi: 10.1016/j.drudis.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Lao BB, Grishagin I, Mesallati H, Brewer TF, Olenyuk BZ, Arora PS. In vivo modulation of hypoxia-inducible signaling by topographical helix mimetics. Proc Natl Acad Sci U S A. 2014;111:7531–7536. doi: 10.1073/pnas.1402393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard JA, Crabtree GR. Chromatin regulatory mechanisms in pluripotency. Annual review of cell and developmental biology. 2010;26:503–532. doi: 10.1146/annurev-cellbio-051809-102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, Zuiderweg ER, Sun D, Gestwicki JE. Analogs of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, as Anti-Cancer Agents. ACS Med Chem Lett. 2013;4 doi: 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- Livingston DJ, Alexander S, Bond J, Briggs T, Fraley A, Hale S, Landsman T, Martinelli R, Shortsleeves K, Terrett N, et al. Therapeutics E, editor. Identification and Characterization of Synthetic Small Molecule Macrocycle Antagonists of Human IL17A. American College of Rheumatology 2012 Annual Meeting. 2012 In http://wwwensemblediscoverycom/news/pdfs/ACR%20Poster%20DJL-v1-1%20(2)pdf.
- London N, Raveh B, Movshovitz-Attias D, Schueler-Furman O. Can self-inhibitory peptides be derived from the interfaces of globular protein-protein interactions? Proteins. 2010;78:3140–3149. doi: 10.1002/prot.22785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Raveh B, Schueler-Furman O. Druggable protein-protein interactions--from hot spots to hot segments. Curr Opin Chem Biol. 2013;17:952–959. doi: 10.1016/j.cbpa.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLure KG, Gesner EM, Tsujikawa L, Kharenko OA, Attwell S, Campeau E, Wasiak S, Stein A, White A, Fontano E, et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PloS one. 2013;8:e83190. doi: 10.1371/journal.pone.0083190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael Andreeff KRK, Karen Yee, Sarit Assouline, Roger Strair, Leslie Popplewell, David Bowen, Giovanni Martinelli, Drummond Mark W, Paresh Vyas, Mark kirschbaum, Swaminathan Padmanabhan Iyer, Kensuke Kojima, David Geho, Steven Blotner, Suzanne Cheng, Lyubomir Vassilev, Meichun Ding, Jianguo Zhi, Steven Middleton, Gwen Nichols. Results of the Phase 1 Trial of RG7112, a Small-Molecule MDM2 Antagonist, in Acute Leukemia. In Blood (ASH Annual Meeting Abstracts) 2012 Abstract 675. [Google Scholar]
- Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C. Modulators of protein-protein interactions. Chemical reviews. 2014;114:4695–4748. doi: 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Naito H, Sugimoto Y, Kawato H, Okayama T, Shimizu H, Miyazaki M, Kitagawa M, Seki T, Fukutake S, et al. Lead optimization of novel p53-MDM2 interaction inhibitors possessing dihydroimidazothiazole scaffold. Bioorganic & medicinal chemistry letters. 2013;23:728–732. doi: 10.1016/j.bmcl.2012.11.091. [DOI] [PubMed] [Google Scholar]
- Ndubaku C, Varfolomeev E, Wang L, Zobel K, Lau K, Elliott LO, Maurer B, Fedorova AV, Dynek JN, Koehler M, et al. Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS chemical biology. 2009;4:557–566. doi: 10.1021/cb900083m. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ. Unraveling structural mechanisms of allosteric drug action. Trends in pharmacological sciences. 2014;35:256–264. doi: 10.1016/j.tips.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem. 2004;47:4417–4426. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Petsalaki E, Russell RB. Peptide-mediated interactions in biological systems: new discoveries and applications. Current opinion in biotechnology. 2008;19:344–350. doi: 10.1016/j.copbio.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Raj M, Bullock BN, Arora PS. Plucking the high hanging fruit: a systematic approach for targeting protein-protein interactions. Bioorganic & medicinal chemistry. 2013;21:4051–4057. doi: 10.1016/j.bmc.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The lancet oncology. 2012;13:1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn J. Tensha therapeutics. Nature biotechnology. 2012;30:305. doi: 10.1038/nbt0412-305. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. American journal of ophthalmology. 2003;136:318–326. doi: 10.1016/s0002-9394(03)00218-6. [DOI] [PubMed] [Google Scholar]
- Schwab N, Ulzheimer JC, Fox RJ, Schneider-Hohendorf T, Kieseier BC, Monoranu CM, Staugaitis SM, Welch W, Jilek S, Du Pasquier RA, et al. Fatal PML associated with efalizumab therapy: insights into integrin alphaLbeta2 in JC virus control. Neurology. 2012;78:458–467. doi: 10.1212/WNL.0b013e3182478d4b. discussion 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, et al. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS pathogens. 2014;10:e1004171. doi: 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J, Cierpicki T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A. 2013;110:8182–8187. doi: 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small molecule integrin antagonists that bind to the beta2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity. 2003;19:391–402. doi: 10.1016/s1074-7613(03)00238-3. [DOI] [PubMed] [Google Scholar]
- Silvian LF, Friedman JE, Strauch K, Cachero TG, Day ES, Qian F, Cunningham B, Fung A, Sun L, Shipps GW, et al. Small molecule inhibition of the TNF family cytokine CD40 ligand through a subunit fracture mechanism. ACS chemical biology. 2011;6:636–647. doi: 10.1021/cb2000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert reviews in molecular medicine. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Stockman BJ, Kothe M, Kohls D, Weibley L, Connolly BJ, Sheils AL, Cao Q, Cheng AC, Yang L, Kamath AV, et al. Identification of allosteric PIF-pocket ligands for PDK1 using NMR-based fragment screening and 1H-15N TROSY experiments. Chemical biology & drug design. 2009;73:179–188. doi: 10.1111/j.1747-0285.2008.00768.x. [DOI] [PubMed] [Google Scholar]
- Sun H, Lu J, Liu L, Yang CY, Wang S. Potent and selective small-molecule inhibitors of cIAP1/2 proteins reveal that the binding of Smac mimetics to XIAP BIR3 is not required for their effective induction of cell death in tumor cells. ACS chemical biology. 2014;9:994–1002. doi: 10.1021/cb400889a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angewandte Chemie. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher AW, Papadopoulos KP, Patnaik A, Fairbrother WJ, Wong H, Budha NR, Darbonne WC, Peale FV, Mamounas MJ, Royer-Joo S, et al. Phase I study of safety and pharmacokinetics (PK) of GDC-0917, an antagonist of inhibitor of apoptosis (IAP) proteins in patients (Pts) with refractory solid tumors or lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31 [Google Scholar]
- Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer research. 2013;73:2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, et al. An empirical framework for binary interactome mapping. Nature methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem. 2012;55:7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmuth F, Blankenfeldt W, Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. The Journal of biological chemistry. 2009;284:36547–36556. doi: 10.1074/jbc.M109.033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nature chemical biology. 2013a;9:112–118. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Majmudar CY, Pomerantz WC, Gagnon JK, Sadowsky JD, Meagher JL, Johnson TK, Stuckey JA, Brooks CL, 3rd, Wells JA, et al. Ordering a dynamic protein via a small-molecule stabilizer. J Am Chem Soc. 2013b;135:3363–3366. doi: 10.1021/ja3122334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Coulombe R, Cameron DR, Thauvette L, Massariol MJ, Amon LM, Fink D, Titolo S, Welchner E, Yoakim C, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. The Journal of biological chemistry. 2004;279:6976–6985. doi: 10.1074/jbc.M311376200. [DOI] [PubMed] [Google Scholar]
- Wei SJ, Joseph T, Sim AY, Yurlova L, Zolghadr K, Lane D, Verma C, Ghadessy F. In vitro selection of mutant HDM2 resistant to Nutlin inhibition. PloS one. 2013;8:e62564. doi: 10.1371/journal.pone.0062564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Wilson CG, Arkin MR. Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Curr Top Microbiol Immunol. 2011;348:25–59. doi: 10.1007/82_2010_93. [DOI] [PubMed] [Google Scholar]
- Winter A, Higueruelo AP, Marsh M, Sigurdardottir A, Pitt WR, Blundell TL. Biophysical and computational fragment-based approaches to targeting protein-protein interactions: applications in structure-guided drug discovery. Quarterly reviews of biophysics. 2012;45:383–426. doi: 10.1017/S0033583512000108. [DOI] [PubMed] [Google Scholar]
- Wong H, Gould SE, Budha N, Darbonne WC, Kadel EE, 3rd, La H, Alicke B, Halladay JS, Erickson R, Portera C, et al. Learning and confirming with preclinical studies: modeling and simulation in the discovery of GDC-0917, an inhibitor of apoptosis proteins antagonist. Drug metabolism and disposition: the biological fate of chemicals. 2013;41:2104–2113. doi: 10.1124/dmd.113.053926. [DOI] [PubMed] [Google Scholar]
- Yang C, Novack DV. Anti-cancer IAP antagonists promote bone metastasis: a cautionary tale. Journal of bone and mineral metabolism. 2013;31:496–506. doi: 10.1007/s00774-013-0479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ding Q, Liu JJ, Zhang J, Jiang N, Chu XJ, Bartkovitz D, Luk KC, Janson C, Tovar C, et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorganic & medicinal chemistry. 2014 doi: 10.1016/j.bmc.2014.05.072. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yu S, Sun W, Liu L, Lu J, McEachern D, Shargary S, Bernard D, Li X, Zhao T, et al. A potent small-molecule inhibitor of the MDM2-p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J Med Chem. 2013;56:5553–5561. doi: 10.1021/jm4005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Gadek TR, Bui M, Shen W, Burnier J, Barr KJ, Hanan EJ, Oslob JD, Yu CH, Zhu J, et al. Discovery and Development of Poten LFA-1 /ICAM-1 Anatagonist SAR1118 as an Ophthalmic Solution for Treating Dry Eye. ACS medicinal chemistry letters. 2012;3:203–206. doi: 10.1021/ml2002482. [DOI] [PMC free article] [PubMed] [Google Scholar]