

Abstract
Background
Plasma triglyceride levels are heritable and are correlated with the risk of coronary heart disease. Sequencing of the protein-coding regions of the human genome (the exome) has the potential to identify rare mutations that have a large effect on phenotype.
Methods
We sequenced the protein-coding regions of 18,666 genes in each of 3734 participants of European or African ancestry in the Exome Sequencing Project. We conducted tests to determine whether rare mutations in coding sequence, individually or in aggregate within a gene, were associated with plasma triglyceride levels. For mutations associated with triglyceride levels, we subsequently evaluated their association with the risk of coronary heart disease in 110,970 persons.
Results
An aggregate of rare mutations in the gene encoding apolipoprotein C3 (APOC3) was associated with lower plasma triglyceride levels. Among the four mutations that drove this result, three were loss-of-function mutations: a nonsense mutation (R19X) and two splice-site mutations (IVS2+1G→A and IVS3+1G→T). The fourth was a missense mutation (A43T). Approximately 1 in 150 persons in the study was a heterozygous carrier of at least one of these four mutations. Triglyceride levels in the carriers were 39% lower than levels in noncarriers (P<1×10−20), and circulating levels of APOC3 in carriers were 46% lower than levels in noncarriers (P = 8×10−10). The risk of coronary heart disease among 498 carriers of any rare APOC3 mutation was 40% lower than the risk among 110,472 noncarriers (odds ratio, 0.60; 95% confidence interval, 0.47 to 0.75; P = 4×10−6).
Conclusions
Rare mutations that disrupt APOC3 function were associated with lower levels of plasma triglycerides and APOC3. Carriers of these mutations were found to have a reduced risk of coronary heart disease. (Funded by the National Heart, Lung, and Blood Institute and others.)
In observational studies, plasma triglyceride levels are associated with the risk of coronary heart disease.1,2 Heritability accounts for more than 50% of the individual variation in triglyceride levels.3 Genomewide association studies have identified common DNA sequence variants at more than 150 genetic loci that are related to plasma lipids4,5 and have suggested that plasma triglyceride-rich lipoproteins directly influence the risk of coronary heart disease.6 These findings lead to two unanswered questions: first, to what extent do rare DNA sequence variants, particularly those in protein-coding sequences, contribute to individual variation in plasma triglyceride levels and the risk of coronary heart disease at the population level, and second, are there specific genetic variants that might lower triglyceride levels and reduce the risk of coronary heart disease?
Recent advances in DNA sequencing technology allow comprehensive detection of rare DNA sequence variants. When sequencing is performed in large populations, a sufficient number of mutation carriers can be identified to evaluate the correlation of genotype with phenotype. In particular, it is advantageous to focus sequencing on exons, the elements of the genome that code for proteins (collectively called the exome),7,8 since mutations in protein-coding sequences (e.g., missense, nonsense, or splice-site mutations) are most readily interpreted.
To address the two questions posed above, we sequenced the exomes of 3734 persons in the United States, identified mutations, and tested the mutations for association with plasma triglyceride levels. We subsequently investigated whether the same mutations were related to the risk of clinical coronary heart disease.
Methods
STUDY DESIGN AND OVERSIGHT
The study was conducted as part of the Exome Sequencing Project of the National Heart, Lung, and Blood Institute (NHLBI) (https://esp.gs.washington.edu/drupal).9,10 The study was designed by the Triglycerides and High-Density Lipoprotein (TG and HDL) Working Group of the Exome Sequencing Project (see the Appendix). The institutional review boards at the Broad Institute and all participating sites approved the NHLBI Exome Sequencing Project study protocols. The last two authors vouch for the accuracy and completeness of the data and all analyses.
DISCOVERY COHORTS
The Exome Sequencing Project study population includes enrollees in seven population-based cohorts (Atherosclerosis Risk in Communities,11 Coronary Artery Risk Development in Young Adults,12 the Cardiovascular Health Study,13 the Framingham Heart Study,14 the Jackson Heart Study,15 the Multiethnic Study of Atherosclerosis,16 and the Women’s Health Initiative17) and participants in a study of early-onset myocardial infarction (Myocardial Infarction Genetics Consortium18) (see Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org).
All participants provided consent for participation in genetic studies. Total cholesterol, HDL cholesterol, and triglyceride levels were measured in fasting venous blood samples, according to standard procedures. Low-density lipoprotein (LDL) cholesterol values were calculated according to the Friedewald formula, with the assignment of a missing value to persons with triglyceride levels higher than 400 mg per deciliter (4.5 mmol per liter).
EXOME SEQUENCING
Exome sequencing was performed at either the University of Washington or the Broad Institute. DNA sequence variants identified at either institution were then submitted to the University of Michigan, where they were subjected to quality control and filtering to exclude variants that were likely to be false positive findings. Protocols for exome sequencing and variant analysis are described in the Supplementary Appendix.
PRIMARY ANALYSES
Sequence Variants and Triglycerides
We used linear regression to test the association of plasma triglyceride levels with identified DNA sequence variants in race-specific models and in models including multiple races. The outcome variable was the natural logarithm of the triglyceride level. The primary independent variable was the number of copies of the variant allele (coded as 0, 1, or 2). Covariates included age, sex, two principal components of ancestry, an indicator variable for race (in the model in which races were combined), and indicator variables for the sequencing ascertainment scheme (see page 45 of the Supplementary Appendix for further in formation on the covariate of age). The single-variant association analysis was limited to variants with a frequency above 0.1%, corresponding to variants with approximately seven copies of the minor allele among the 3734 participants.
Since many of the identified variants occurred too infrequently to be analyzed individually, we aggregated rare variants within each gene; here, the gene (rather than an individual variant) became the unit of analysis. Within each gene, we included all missense, nonsense, and splice-site variants below a frequency threshold of 1% and considered whether plasma triglyceride levels differed between carriers of these mutations and noncarriers.19 Owing to considerations regarding statistical power, we limited the analysis to genes for which five or more persons carried minor alleles.
Replication of Association between APOC3 Genotypes and Plasma Lipids
The gene most strongly associated with plasma triglyceride levels in the discovery sample was the gene encoding apolipoprotein C3 (APOC3). To replicate this finding, we genotyped four APOC3 mutations (R19X, IVS2+1G→A, A43T, and IVS3+1G→T), using the HumanExome BeadChip (Illumina) in 41,671 participants of African or European ancestry in seven replication studies (Table S2 in the Supplementary Appendix). This cohort was separate from the discovery cohort. We tested the association of these four mutations with plasma lipid levels. The methods used for genotyping and association testing are described in the Supplementary Appendix.
APOC3 Genotypes, APOC3 Levels, and Coronary Heart Disease
We next tested the association of APOC3 mutations with coronary heart disease in participants of European, African, and Hispanic ancestry in 15 studies (for details, see Table S3 in the Supplementary Appendix). To investigate the association between APOC3 level and the risk of coronary heart disease, we used blood samples drawn from 3238 fasting participants in the Framingham Heart Study offspring cohort (examination cycle 5, 1991 to 1995).20 Plasma APOC3 levels were measured with the use of a commercially available immunochemical assay (Wako Diagnostics). All participants underwent continual surveillance for incident coronary heart disease events until December 31, 2010. As described in the Supplementary Appendix, we used proportional-hazards regression models to examine the relationship of plasma APOC3 levels (logarithmically transformed on a natural log scale) to the risk of incident coronary heart disease.
SECONDARY ANALYSES
In patients with angiographic coronary heart disease, we evaluated the association of plasma APOC3 levels with total and cardiovascular mortality. We also tested the association of APOC3 mutations with the presence of hepatic fat on computed tomography (CT). Details of the methods are presented in the Supplementary Appendix.
STATISTICAL ANALYSIS
For the single-variant analysis, we considered a P value of less than 5×10−7 to be significant because this threshold accounts for the 95,342 variants tested. For the gene-level analysis, we considered a P value of less than 3×10−6 to be significant to account for the testing of 18,666 genes. In the analyses of associations with coronary heart disease, we tested four variants and thus considered a P value of less than 0.0125 to be significant. Genetic association analyses were performed with use of PLINK (http://pngu.mgh.harvard.edu/~purcell/plink),21 SCORE-Seq (http://dlin.web.unc.edu/software/SCORE-Seq),22 SAS software, version 9.1, or R software, version R = 2.14 (www.r-project.org).
Results
BASELINE CHARACTERISTICS OF THE STUDY PARTICIPANTS
Among the 6823 persons for whom sequencing was performed through the NHLBI Exome Sequencing Project, 2861 were excluded because data on fasting triglyceride levels were not available, 149 were excluded because of relatedness, and 79 were excluded because of sample rejection during the quality-control process. These exclusions left 3734 persons eligible for the present investigation (the baseline characteristics of these participants are shown in Table S1 in the Supplementary Appendix). There were 2082 participants of European ancestry and 1652 of African ancestry.
EXOME SEQUENCING
We obtained sequence data for 256,143 exons from 18,666 genes. On average, targeted bases were read 89 times and 94% of targeted bases were covered with at least 20 sequencing reads. Each participant had an average of 46 mutations causing the gain or loss of a stop codon, 5712 had missense mutations, and 19 had mutations that altered canonical splice sites.
ASSOCIATION OF SINGLE VARIANTS WITH TRIGLYCERIDES
We tested the association of plasma triglyceride levels with 95,342 nonsense, missense, or splice-site mutations with an allele frequency greater than 0.1% (Fig. S1 and Tables S4, S5, and S6 in the Supplementary Appendix). In analyses of the full sample, the variant most strongly associated with triglyceride levels was the previously described S19W variant in APOA5,23 which had an allele frequency of 6% among participants of European ancestry (P = 3×10−7).
GENE-LEVEL ASSOCIATIONS WITH TRIGLYCERIDES AND REPLICATION
We next assessed whether rare mutations (i.e., missense, nonsense, or splice-site variants with a frequency of <1%) tested collectively within each gene were associated with plasma triglyceride levels. APOC3 was the gene most strongly associated with plasma triglyceride levels when analyses included only participants of European ancestry (P = 7×10−6) and when analyses included participants of European ancestry and participants of African ancestry (P = 1×10−5) (Table S7 in the Supplementary Appendix). An aggregate of rare alleles at APOC3 was associated with lower plasma triglyceride levels (Table 1 and Fig. 1). As compared with noncarriers, carriers of any rare APOC3 mutation had plasma triglyceride levels that were 39% lower (P = 6×10−9), HDL cholesterol levels that were 22% higher (P = 3×10−6), and LDL cholesterol levels that were 16% lower (P = 0.05) (Table 1). These rare mutations collectively explained 0.9% of the variance in triglyceride levels.
Table 1.
Rare Mutations in APOC3 and Plasma Levels of Triglycerides.*
| Chromosome Position | Mutation | Mutation Type | No. of Carriers | Mean Triglyceride Level | Mean HDL Cholesterol Level | Mean LDL Cholesterol Level |
|---|---|---|---|---|---|---|
| mg/dl | ||||||
| 11:116701284 | IVS1–2G→A | Splice site | 1 | 66.0 | 65.0 | 140.0 |
| 11:116701326 | A10T | Missense | 2 | 71.5 | 64.0 | 138.5 |
| 11:116701353 | R19X | Nonsense | 2 | 45.5 | 56.8 | 152.5 |
| 11:116701354 | IVS2+1G→A | Splice site | 10 | 71.0 | 59.6 | 111.7 |
| 11:116701560 | A43T | Missense | 12 | 79.0 | 64.5 | 110.4 |
| 11:116701613 | IVS3+1G→T | Splice site | 5 | 82.8 | 67.0 | 154.2 |
| 11:116703493 | D65 N | Missense | 1 | 415.0 | 32.0 | NA |
| Total APOC3 carriers | 33 | 84.5±86.0 | 61.9±16.6 | 122.8±54.8 | ||
| Total APOC3 noncarriers | 3701 | 137.5±90.1 | 50.7±15.6 | 146.2±61.8 | ||
| Carriers vs. noncarriers (%) | −38.5 | 22.1 | −16.0 | |||
| P value† | 6×10−9‡ | 4×10−6 | 0.05 |
Plus–minus values are means ± SD. The APOC3 mutations were identified by means of exome sequencing. To convert the values for cholesterol to millimoles per liter, multiply by 0.02586. To convert the values for triglycerides to millimoles per liter, multiply by 0.01129. HDL denotes high-density lipoprotein, LDL low-density lipoprotein, and NA not available.
P values were derived from a linear regression model, with adjustments for age, sex, ancestry, and principal components of ancestry.
The P value for the triglyceride phenotype is based on triglyceride levels that were logarithmically transformed on a natural log scale.
Figure 1. Rare Mutations in APOC3 and Reductions in Plasma Triglyceride Levels.
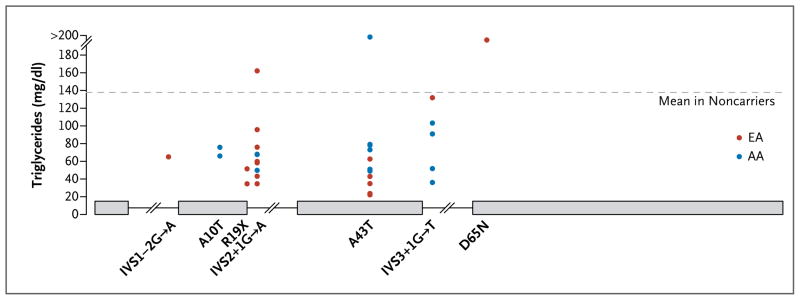
A schematic diagram of the gene encoding apolipoprotein C3 (APOC3) shows four exons and three introns, or intervening sequences (IVS). In the graph, each dot represents a single study participant who carried a specific mutation. Red dots denote persons of European ancestry (EA), and blue dots persons of African ancestry (AA). The mean level of triglycerides among all persons who did not carry any mutation in the coding sequence of APOC3 was 137 mg per deciliter (dashed line). To convert the values for triglycerides to millimoles per liter, multiply by 0.01129.
The association between plasma lipid levels and APOC3 was primarily attributable to four different sites within the gene. Three of the four mutations are predicted to severely disrupt the function of APOC3 — that is, lead to loss of function.24 Loss-of-function variants included a nonsense substitution (R19X) and two DNA sequence variants that disrupt a splice site (IVS2+1G→A and IVS3+1G→T). The missense A43T mutation has been shown to decrease APOC3 function in vitro.25,26 When the participants of European ancestry were combined with the participants of African ancestry, approximately 1 in 150 persons carried any one of these four loss-of-function mutations (Table S8 in the Supplementary Appendix).27
To replicate the results derived from exome sequencing, we genotyped one or more of these four mutations in 34,432 participants of European ancestry and 7239 participants of African ancestry, independently of the discovery sequencing study. When compared with participants who did not carry any of the four mutations, those who were heterozygous for any of the four mutations had lower plasma triglyceride levels among participants of European ancestry (42% lower, P<1×10−20), among participants of African ancestry (32% lower, P = 1×10−9), and among participants in the two groups combined (39% lower, P<1×10−20) (Table 2, and Table S9 in the Supplementary Appendix). APOC3 loss-of-function carrier status was associated with reduced levels of plasma triglycerides whether or not the S19W variant of APOA5 was present (Table S10 in the Supplementary Appendix). APOC3 loss-of-function mutation carriers also had significantly higher HDL cholesterol levels, with a trend toward lower LDL cholesterol levels (Table 2, and Table S9 in the Supplementary Appendix).
Table 2.
Association of Four APOC3 Mutations with Plasma Lipid Levels in Replication Studies.*
| Variable Affected by Mutation | Carriers of Any of Four APOC3 Mutations | ||
|---|---|---|---|
| European Ancestry (N = 34,432) | African Ancestry (N = 7239) | European and African Ancestries Combined (N = 41,671) | |
| Triglycerides (% difference) | −42 | −32 | −39 |
| P value | <1×10−20 | 1×10−9 | <1×10−20 |
| LDL cholesterol (mg/dl) | −9.3±3.4 | 10.7±5.4 | −3.8±2.9 |
| P value | 0.006 | 0.05 | 0.19 |
| HDL cholesterol (mg/dl) | 11.5±1.3 | 9.1±2.0 | 10.8±1.1 |
| P value | <1×10−20 | 8×10−6 | <1×10−20 |
Plus–minus values indicate effect size ±SE. Effect size represents the difference in mean lipid levels between carriers and noncarriers after adjustment for covariates. The four APOC3 mutations tested were R19X, splice-site mutation IVS2+1G→A, A43T, and splice-site mutation IVS3+1G→T. P values are for the comparison with noncarriers. P values were derived from a linear regression model, with adjustments for age, sex, ancestry, and principal components of ancestry. To convert the values for cholesterol to millimoles per liter, multiply by 0.02586. To convert the values for triglycerides to millimoles per liter, multiply by 0.01129.
APOC3 LOSS-OF-FUNCTION MUTATIONS, CORONARY HEART DISEASE, AND HEPATIC FAT
Among 110,970 study participants (34,002 patients with coronary heart disease and 76,968 controls) from 14 studies, 498 were heterozygous for at least one of the four APOC3 loss-of-function mutations. The risk of coronary heart disease for carriers of an APOC3 loss-of-function mutation was 40% lower than the risk for noncarriers (odds ratio, 0.60; 95% confidence interval [CI], 0.47 to 0.75; P = 4×10−6) (Fig. 2, and Tables S3 and S11 in the Supplementary Appendix). There was no evidence of heterogeneity across the 15 studies (Breslow–Day test for heterogeneity, P = 0.16).
Figure 2. Association of APOC3 Loss-of-Function Mutations with Risk of Coronary Heart Disease among 110,970 Participants in 15 Studies.

In each study, we tested the association of loss-of-function carrier status (heterozygous for any of four mutations: APOC3 R19X, IVS2+1G→A, IVS3+1G→T, or A43T) with the risk of coronary heart disease. We calculated P values for the association tests and confidence intervals for the odds ratios with the use of exact methods. We performed a meta-analysis with the use of the Cochran–Mantel–Haenszel statistics for stratified 2-by-2 tables. The Cochran–Mantel–Haenszel method combines score statistics rather than Wald statistics and is particularly useful when some observed odds ratios are zero. For each study, squares indicate the estimated odds ratios and the corresponding lines indicate the 95% confidence intervals. The diamond indicates the combined estimate of the odds ratio and the corresponding 95% confidence interval. HA denotes Hispanic ancestry. The full study names are as follows: ARIC Atherosclerosis Risk in Communities. ATVB Italian Atherosclerosis, Thrombosis, and Vascular Biology Study, EPIC European Prospective Study into Cancer and Nutrition, FHS Framingham Heart Study, FIA3 First Myocardial Infarction, in AC County 3, GoDARTS Genetics of Diabetes Audit and Research Tayside Study, HUNT Nord–Trøndelag Health Study, IPM Mt. Sinai Institute for Personalized Medicine Biobank, MDC–CVA Malmö Diet and Cancer Study Cardiovascular Cohort, OHS Ottawa Heart Study, PROCARDIS, Precocious Coronary Artery Disease Study, VHS Verona Heart Study, WHI Women’s Health Initiative, and WTCCC Wellcome Trust Case Control Consortium. All 15 studies are described in Table S3 in the Supplementary Appendix.
We did not observe homozygosity or compound heterozygosity for APOC3 loss-of-function variants in any participant. However, with a combined allele frequency of 1:300 and genotyping of 110,970 participants, the expected mean (±SD) number of persons with homozygosity or compound heterozygosity for APOC3 loss-of-function variants was only 1±1 (Table S12 in the Supplementary Appendix).
A total of 3051 participants had both an APOC3 loss-of-function allele and a phenotype of hepatic steatosis on CT. The presence of hepatic fat on CT did not differ significantly between carriers of an APOC3 loss-of-function allele (27 participants) and noncarriers (3034 participants) (P = 0.82) (Table S13 in the Supplementary Appendix).
APOC3 MUTATIONS AND PLASMA APOC3
In 3237 participants from the Framingham Heart Study, the mean plasma level of APOC3 was 16.6 mg per deciliter (Fig. S2 in the Supplementary Appendix). The APOC3 plasma level was associated with a number of cardiovascular risk factors, plasma lipid levels, lipoprotein subfractions, levels of other apolipoproteins, and levels of blood biomarkers (Table S14 in the Supplementary Appendix).
Genotypes had been obtained for 2707 of these Framingham Heart Study participants. A total of 13 persons carried one mutant allele (with heterozygosity for either R19X or splice-site IVS2+1G→A). As compared with noncarriers, these 13 persons had a plasma level of APOC3 that was 46% lower (P = 8×10−10) (Fig. S3 in the Supplementary Appendix).
PLASMA APOC3 LEVELS AND INCIDENT CORONARY HEART DISEASE
During a mean follow-up of 14.4 years, 303 participants in the Framingham Heart Study had a first coronary heart disease event. In models adjusted for age and sex, each decrease of 1 mg per deciliter in plasma levels of APOC3 was associated with a 4% decrease in the risk of incident coronary heart disease (hazard ratio, 0.96; 95% CI, 0.94 to 0.98; P<0.001) (Table S15 in the Supplementary Appendix). Participants with values in the lowest third of the distribution of plasma APOC3 levels had a reduced risk of incident coronary heart disease, as compared with participants with values in the highest third (odds ratio, 0.65; 95% CI, 0.48 to 0.87; P = 0.003) (Fig. 3, and Table S16 in the Supplementary Appendix). However, this association was not significant after adjustment for additional cardiovascular risk factors (Tables S15 and S16 in the Supplementary Appendix). In the Verona Heart Study cohort,18 which consisted of 794 patients with angiographic evidence of coronary artery disease, the lowest third of the distribution of APOC3 levels was associated with a reduced risk of death from cardiovascular disease, as compared with the highest third, in models that were minimally adjusted and fully adjusted for risk factors (Tables S17 through S20 in the Supplementary Appendix).
Figure 3. Cumulative Probability of Freedom from Coronary Heart Disease (CHD) According to Plasma Level of APOC3 at Baseline in the Framingham Heart Study.

Plasma APOC3 levels were 14.2 mg per deciliter or less in the lowest third of the population, 14.3 to 17.9 mg per deciliter in the middle third, and 18.0 mg per deciliter or more in the highest third. Median follow-up was 14.4 years. The numbers in parentheses are the numbers of study participants who were undergoing follow-up at the specified time points.
Discussion
By sequencing the protein-coding regions of the genome in 3734 persons, we identified several rare coding-sequence variants of APOC3 that were associated with a large effect on plasma triglyceride levels. Approximately 1 in 150 persons carried any one of four protein-altering or splice-site variants. As compared with noncarriers, carriers of any of these four APOC3 mutations had plasma triglyceride levels that were 39% lower, HDL cholesterol levels that were 22% higher, LDL cholesterol levels that were 16% lower, and circulating APOC3 levels that were 46% lower, and they had a risk of coronary heart disease that was reduced by 40%.
These results show that loss of APOC3 function confers protection against clinical coronary heart disease. In a previous study, Pollin and colleagues28 reported that approximately 5% of Amish persons in Lancaster County, Pennsylvania, carried the APOC3 R19X null allele and that these carriers had a favorable lipid profile and a reduced amount of coronary artery calcium, a surrogate marker for atherosclerosis. The A43T missense mutation in APOC3 (referred to as A23T in the earlier nomenclature) was initially identified in two Mayan Indians with low plasma levels of triglycerides and APOC3.25 Functional characterization in vitro showed deficient lipid binding25 and attenuated secretion of very-low-density lipoprotein from hepatocytes.26 Recently, in three families, 12 carriers of the A43T mutation or the two APOC3 splice-site mutations were identified, and as compared with family members who were not carriers, the carriers had lower levels of plasma triglycerides and higher levels of HDL cholesterol.29 In our study, which included 110,970 participants, carriers of APOC3 loss-of-function mutations had a reduced risk of clinical coronary heart disease.
Several mechanisms may link APOC3 to atherogenesis. 30 Resident on the surface of triglyceride-rich lipoproteins, APOC3 inhibits the lipolytic activity of lipoprotein lipase, thereby increasing the plasma level of atherogenic triglyceride-rich lipoproteins, including very-low-density lipoproteins and chylomicrons. APOC3 also delays the clearance of atherogenic, cholesterol-rich remnants of the postprandial metabolism of triglyceride-rich lipoproteins.31,32
The question of whether triglyceride-rich lipoproteins in plasma have a causal effect on coronary heart disease remains unanswered. Several treatments that lower plasma levels of triglycerides, including fibrates and fish oils, have failed to consistently reduce the risk of coronary heart disease in randomized, controlled trials.33,34 Our findings with respect to the human APOC3 loss-of-function alleles suggest that a lifelong decrease in APOC3 function is one means of reducing plasma levels of triglyceride- rich lipoproteins that may be atheroprotective. Recently, an antisense oligonucleotide therapeutic agent that decreased the production of APOC3 was shown to lower plasma levels of triglycerides and APOC3 in mice, nonhuman primates, and healthy human volunteers.35 Our data provide confidence that such a therapeutic strategy might be expected to reduce the risk of clinical coronary heart disease.
In more general terms, the results of our study highlight the potential usefulness of naturally occurring loss-of-function mutations in guiding the selection of therapeutic targets.24 For example, loss-of-function mutations in the gene encoding proprotein convertase subtilisin/kexin type 9 (PCSK9) were shown to reduce LDL cholesterol levels and lower the risk of coronary heart disease, a finding that prompted the development of monoclonal antibodies directed against PCSK9.36–38
Several limitations of our study deserve mention. First, we are unable to pinpoint the specific mechanism by which loss of APOC3 function might be atheroprotective. Carriers of APOC3 loss-of-function mutations have a range of associated biomarkers, including lower levels of triglycerides, higher levels of HDL cholesterol, lower levels of LDL cholesterol, and lower levels of APOC3. Additional work will be required to understand the primary mechanism linking APOC3 to coronary heart disease. Second, earlier reports are conflicting as to whether common DNA sequence variants at APOC3 affect hepatic steatosis.39,40 We found no significant difference in CT findings with respect to hepatic fat between study participants who carried APOC3 loss-of-function mutations and those who did not. However, larger samples may be required to confidently answer this question.
In conclusion, we identified rare DNA sequence variants in APOC3 that were associated with lifelong reductions in plasma levels of triglycerides and APOC3. These variants conferred protection against coronary heart disease.
Supplementary Material
Acknowledgments
Supported by the Grand Opportunity (GO) Exome Sequencing Project (ESP), through which grants were provided by the NHLBI (RC2 HL-103010 [HeartGO], RC2 HL-102923 [LungGO], RC2 HL-102924 [Women’s Health Initiative ESP], RC2 HL-102925 [BroadGO], and RC2 HL-102926 [SeattleGO]), the Russell and Diana Hawkins Family Foundation Discovery Fellowship, an award from the NHLBI (T32HL007208, to Dr. Peloso), an award from the British Heart Foundation (BHF) (to Dr. Samani, who holds a chair supported by the BHF), a Banting Fellowship from the Canadian Institutes of Health Research (to Dr. Do), a career-development award from the NIH and NHLBI (K08-HL114642, to Dr. Stitziel), a Research Scholar Award from the Massachusetts General Hospital (MGH) (to Dr. Kathiresan), the Howard M. Goodman Fellowship from MGH, the Donovan Family Foundation, a grant from the NIH (R01HL107816, to Dr. Kathiresan), an investigator-initiated research grant from Merck (to Dr. Kathiresan), a grant from Fondation Leducq (to Dr. Kathiresan), a grant from the Swedish Heart–Lung Foundation (2020389, to the Swedish First Myocardial Infarction in AC County 3 [FIA3] Study), and a grant from Regione Emilia-Romagna (RFPS-2007-3-644382, to the Italian Atherosclerosis, Thrombosis, and Vascular Biology Study).
We thank the site investigators, field staff, and study participants for their contributions to this study. A complete list of acknowledgements is provided in the Supplementary Appendix.
APPENDIX
The authors are as follows: Jacy Crosby, M.S., Gina M. Peloso, Ph.D., Paul L. Auer, Ph.D., David R. Crosslin, Ph.D., Nathan O. Stitziel, M.D., Ph.D., Leslie A. Lange, Ph.D., Yingchang Lu, M.D., Ph.D., Zheng-zheng Tang, M.S., He Zhang, Ph.D., George Hindy, M.D., Nicholas Masca, Ph.D., Kathleen Stirrups, Ph.D., Stavroula Kanoni, Ph.D., Ron Do, Ph.D., Goo Jun, Ph.D., Youna Hu, Ph.D., Hyun Min Kang, Ph.D., Chenyi Xue, M.S., Anuj Goel, M.Sc., Martin Farrall, F.R.C.Path., Stefano Duga, Ph.D., Pier Angelica Merlini, M.D., Rosanna Asselta, Ph.D., Domenico Girelli, M.D., Oliviero Olivieri, M.D., Nicola Martinelli, M.D., Ph.D., Wu Yin, Ph.D., Dermot Reilly, Ph.D., Elizabeth Speliotes, M.D., Ph.D., Caroline S. Fox, M.D., Kristian Hveem, M.D., Ph.D., Oddgeir L. Holmen, M.D., Majid Nikpay, Ph.D., Deborah N. Farlow, Ph.D., Themistocles L. Assimes, M.D., Ph.D., Nora Franceschini, M.D., M.P.H., Jennifer Robinson, M.D., Kari E. North, Ph.D., Lisa W. Martin, M.D., Mark DePristo, Ph.D., Namrata Gupta, Ph.D., Stefan A. Escher, Ph.D., Jan-Håkan Jansson, M.D., Natalie Van Zuydam, Ph.D., Colin N.A. Palmer, Ph.D., Nicholas Wareham, M.B., B.S., Ph.D., Werner Koch, M.D., Thomas Meitinger, M.D., Annette Peters, M.D., Wolfgang Lieb, M.D., Raimund Erbel, M.D., Inke R. Konig, Ph.D., Jochen Kruppa, M.Sc., Franziska Degenhardt, M.D., Omri Gottesman, M.D., Erwin P. Bottinger, M.D., Christopher J. O’Donnell, M.D., Bruce M. Psaty, M.D., Ph.D., Christie M. Ballantyne, M.D., Goncalo Abecasis, Ph.D., Jose M. Ordovas, Ph.D., Olle Melander, M.D., Ph.D., Hugh Watkins, F.R.C.P., Marju Orho-Melander, Ph.D., Diego Ardissino, M.D., Ruth J.F. Loos, Ph.D., Ruth McPherson, M.D., Ph.D., Cristen J. Willer, Ph.D., Jeanette Erdmann, Ph.D., Alistair S. Hall, F.R.C.P., Nilesh J. Samani, F.R.C.P., Panos Deloukas, Ph.D., Heribert Schunkert, M.D., James G. Wilson, M.D., Charles Kooperberg, Ph.D., Stephen S. Rich, Ph.D., Russell P. Tracy, Ph.D., Dan-Yu Lin, Ph.D., David Altshuler, M.D., Ph.D., Stacey Gabriel, Ph.D., Deborah A. Nickerson, Ph.D., Gail P. Jarvik, M.D., Ph.D., L. Adrienne Cupples, Ph.D., Alex P. Reiner, M.D., Eric Boerwinkle, Ph.D., and Sekar Kathiresan, M.D.
The authors’ affiliations are as follows: the Department of Biostatistics, Bioinformatics, and Systems Biology, University of Texas Graduate School of Biomedical Sciences at Houston (J.C.), the Human Genetics Center, University of Texas Health Science Center at Houston (J.C., E.B.), Baylor College of Medicine (C.M.B., E.B.), and Methodist DeBakey Heart and Vascular Center (C.M.B.) — all in Houston; the Center for Human Genetic Research and Cardiovascular Research Center, Massachusetts General Hospital (G.M.P., R.D., D. Altshuler, S. Kathiresan), the Department of Medicine, Harvard Medical School (G.M.P., R.D., D. Altshuler, S. Kathiresan), Merck Sharp & Dohme (W.Y., D.R.), Nutrition and Genomics Laboratory, Jean Mayer–USDA Human Nutrition Research Center on Aging at Tufts University (J.M.O.), and Department of Biostatistics, Boston University School of Public Health (L.A.C.) — all in Boston; the Program in Medical and Population Genetics, Broad Institute, Cambridge, MA (G.M.P., R.D., D.N.F., M.D., N.G., D. Altshuler, S.G., S. Kathiresan); the School of Public Health, University of Wisconsin–Milwaukee, Milwaukee (P.L.A.); the Departments of Genome Sciences (D.R.C., D.A.N., G.P.J.), Medicine (Medical Genetics) (D.R.C., G.P.J.), and Epidemiology (A.P.R.) and the Cardiovascular Health Research Unit, Departments of Medicine, Epidemiology, and Health Services (B.M.P.), University of Washington, the Group Health Research Institute, Group Health Cooperative (B.M.P.), and the Public Health Sciences Division, Fred Hutchinson Cancer Research Center (C.K., A.P.R.) — all in Seattle; the Cardiovascular Division, Department of Medicine, and the Division of Statistical Genomics, Washington University School of Medicine, St. Louis (N.O.S.); the Departments of Genetics (L.A.L.), Biostatistics (Z.T., D.-Y.L.), and Epidemiology (N.F., K.E.N.) and the Carolina Center for Genome Sciences (K.E.N.), University of North Carolina, Chapel Hill; the Charles Bronfman Institute for Personalized Medicine (Y.L., O.G., E.P.B., R.J.F.L.) and the Mindich Child Health and Development Institute (R.J.F.L.), Icahn School of Medicine at Mount Sinai, New York; the Departments of Internal Medicine (H.Z., E.S., C.J.W.), Computational Medicine and Bioinformatics (H.Z., C.X., E.S., C.J.W.), Human Genetics (H.Z., C.J.W.), and Biostatistics (G.J., Y.H., H.M.K., G.A.), the Division of Gastroenterology (E.S.), and the Center for Statistical Genetics (G.A.), University of Michigan, Ann Arbor; the Department of Clinical Sciences, Clinical Research Center (G.H., M.O.-M.), Department of Clinical Science, Genetic and Molecular Epidemiology Unit (S.A.E.), and Department of Clinical Sciences, Diabetes and Endocrinology (O.M.), Lund University and University Hospital Malmö, Malmö, the Department of Medicine, Skellefteå Hospital, Skellefteå (J.-H.J.), and the Department of Public Health and Clinical Medicine, Umeå University, Umeå, (J.-H.J.) — all in Sweden; the Department of Cardiovascular Sciences, University of Leicester, and National Institute for Health Research Leicester Cardiovascular Biomedical Research Unit, Leicester (N. Masca, N.J.S.), the William Harvey Research Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London (K.S., S. Kanoni, P.D.), the Division of Cardiovascular Medicine, Radcliffe Department of Medicine, Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford (A.G., M.F., H.W.), the Medical Research Institute, Ninewells Hospital and Medical School, Dundee (N.V.Z., C.N.A.P.), the Medical Research Council Epidemiology Unit, Institute of Metabolic Science, Addenbrooke’s Hospital (N.W., L.A.C.), and Wellcome Trust Sanger Institute (P.D.), Cambridge, and the Division of Epidemiology, School of Medicine, University of Leeds, Leeds (A.S.H.) — all in the United Kingdom; the Dipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano (S.D., R.A.) and the Division of Cardiology, Ospedale Niguarda (P.A.M.), Milan, the University of Verona School of Medicine, Department of Medicine, Verona (D.G., O.O., N. Martinelli), and the Division of Cardiology, Azienda Ospedaliero-Universitaria di Parma, Parma (D. Ardissino) — all in Italy; the National Heart, Lung and Blood Institute Framingham Heart Study (C.S.F., C.J.O.) and the Center for Population Studies (C.S.F.), Framingham, MA; the Nord-Trøndelag Health Study (HUNT) Research Center, Department of Public Health and General Practice, Norwegian University of Science and Technology (K.H., O.L.H.), and Levanger Hospital (K.H.), Levanger, and St. Olav Hospital, Trondheim University Hospital, Trondheim (O.L.H.) — both in Norway; the Division of Cardiology, University of Ottawa Heart Institute, Ottawa (M. Nikpay, R.M.); the Department of Medicine, Stanford University School of Medicine, Stanford, CA (T.L.A.); the Departments of Epidemiology and Medicine, College of Public Health, University of Iowa, Ames (J.R.); the Division of Cardiology, George Washington University School of Medicine and Health Sciences, Washington, DC (L.W.M.); Deutsches Herzzentrum München (W.K., H.S.), Medizinische Klinik, Klinikum rechts der Isar (W.K.), and Institute of Human Genetics (T.M.), Technische Universität München, and the German Center for Cardiovascular Research (W.K., A.P.), Munich, Helmholtz Zentrum München, German Research Center for Environmental Health, Institute of Epidemiology II, Neuherberg (A.P.), Institute of Epidemiology and Biobank popgen, Christian-Albrechts University Kiel, Kiel (W.L.), Department of Cardiology, West German Heart Center, Essen (R.E.), Institut für Medizinische Biometrie und Statistik (I.R.K., J.K.) and Institut für Integrative und Experimentelle Genomik (J.E.), Universität zu Lübeck, Universitätsklinikum Schleswig-Holstein (I.R.K., J.K.), and DZHK (German Research Center for Cardiovascular Research) (J.E.), Lübeck, and the Institute of Human Genetics, University of Bonn, Bonn (F.D.) — all in Germany; the Department of Cardiovascular Epidemiology and Population Genetics, National Center for Cardiovascular Investigation, and Instituto Madrileño de Estudios Avanzados–Alimentacion, Madrid (J.M.O.); King Abdulaziz University, Jeddah, Saudi Arabia (P.D.); the Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson (J.G.W.); the Center for Public Health Genomics, University of Virginia, Charlottesville (S.S.R.); and the Departments of Pathology and Biochemistry, University of Vermont College of Medicine, Burlington (R.P.T.).
Footnotes
The views expressed in this article are solely those of the authors and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute (NHLBI) or the National Institutes of Health (NIH).
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 2.Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–61. doi: 10.1093/eurheartj/ehr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Namboodiri KK, Kaplan EB, Heuch I, et al. The Collaborative Lipid Research Clinics Family Study: biological and cultural determinants of familial resemblance for plasma lipids and lipoproteins. Genet Epidemiol. 1985;2:227–54. doi: 10.1002/gepi.1370020302. [DOI] [PubMed] [Google Scholar]
- 4.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–9. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu W, O’Connor TD, Jun G, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–20. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The ARIC Investigators. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 12.Friedman GD, Cutter GR, Donahue RP, et al. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J Clin Epidemiol. 1988;41:1105–16. doi: 10.1016/0895-4356(88)90080-7. [DOI] [PubMed] [Google Scholar]
- 13.Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–76. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 14.Dawber TR, Meadors GF, Moore FE., Jr Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41:279–81. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor HA, Jr, Wilson JG, Jones DW, et al. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn Dis. 2005;15(Suppl 6):S4–S17. [PubMed] [Google Scholar]
- 16.Bild DE, Bluemke DA, Burke GL, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–81. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 17.The Women’s Health Initiative Study Group. Design of the Women’s Health Initiative clinical trial and observational study. Control Clin Trials. 1998;19:61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 18.Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–41. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families: the Framingham Offspring Study. Am J Epidemiol. 1979;110:281–90. doi: 10.1093/oxfordjournals.aje.a112813. [DOI] [PubMed] [Google Scholar]
- 21.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–67. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pennacchio LA, Olivier M, Hubacek JA, Krauss RM, Rubin EM, Cohen JC. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet. 2002;11:3031–8. doi: 10.1093/hmg/11.24.3031. [DOI] [PubMed] [Google Scholar]
- 24.MacArthur DG, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–8. doi: 10.1126/science.1215040. Erratum, Science 2012;336:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Labeur C, Xu CF, et al. Characterization of the lipid-binding properties and lipoprotein lipase inhibition of a novel apolipoprotein C-III variant Ala23Thr. J Lipid Res. 2000;41:1760–71. [PubMed] [Google Scholar]
- 26.Sundaram M, Zhong S, Bou Khalil M, et al. Functional analysis of the missense APOC3 mutation Ala23Thr associated with human hypotriglyceridemia. J Lipid Res. 2010;51:1524–34. doi: 10.1194/jlr.M005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.NHLBI Exome Sequencing Project. Exome Variant Server. ( http://evs.gs.washington.edu/EVS)
- 28.Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bochem AE, van Capelleveen JC, Dallinga-Thie GM, et al. Two novel mutations in apolipoprotein C3 underlie atheroprotective lipid profiles in families. Clin Genet. 2014;85:433–40. doi: 10.1111/cge.12201. [DOI] [PubMed] [Google Scholar]
- 30.Huff MW, Hegele RA. Apolipoprotein C-III: going back to the future for a lipid drug target. Circ Res. 2013;112:1405–8. doi: 10.1161/CIRCRESAHA.113.301464. [DOI] [PubMed] [Google Scholar]
- 31.Windler E, Havel RJ. Inhibitory effects of C apolipoproteins from rats and humans on the uptake of triglyceride-rich lipoproteins and their remnants by the perfused rat liver. J Lipid Res. 1985;26:556–65. [PubMed] [Google Scholar]
- 32.Zheng C, Khoo C, Furtado J, Sacks FM. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121:1722–34. doi: 10.1161/CIRCULATIONAHA.109.875807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74. doi: 10.1056/NEJMoa1001282. Erratum, N Engl J Med 2010;362:1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The ORIGIN Trial Investigators. n–3 Fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012;367:309–18. doi: 10.1056/NEJMoa1203859. [DOI] [PubMed] [Google Scholar]
- 35.Graham MJ, Lee RG, Bell TA, III, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90. doi: 10.1161/CIRCRESAHA.111.300367. [DOI] [PubMed] [Google Scholar]
- 36.Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 37.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 38.Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–18. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 39.Petersen KF, Dufour S, Hariri A, et al. Apolipoprotein C3 gene variants in non-alcoholic fatty liver disease. N Engl J Med. 2010;362:1082–9. doi: 10.1056/NEJMoa0907295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozlitina J, Boerwinkle E, Cohen JC, Hobbs HH. Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology. 2011;53:467–74. doi: 10.1002/hep.24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.